Heat Shock Protein 90 and Role of Its Chemical Inhibitors in Treatment of Hematologic Malignancies

Abstract
:1. Introduction
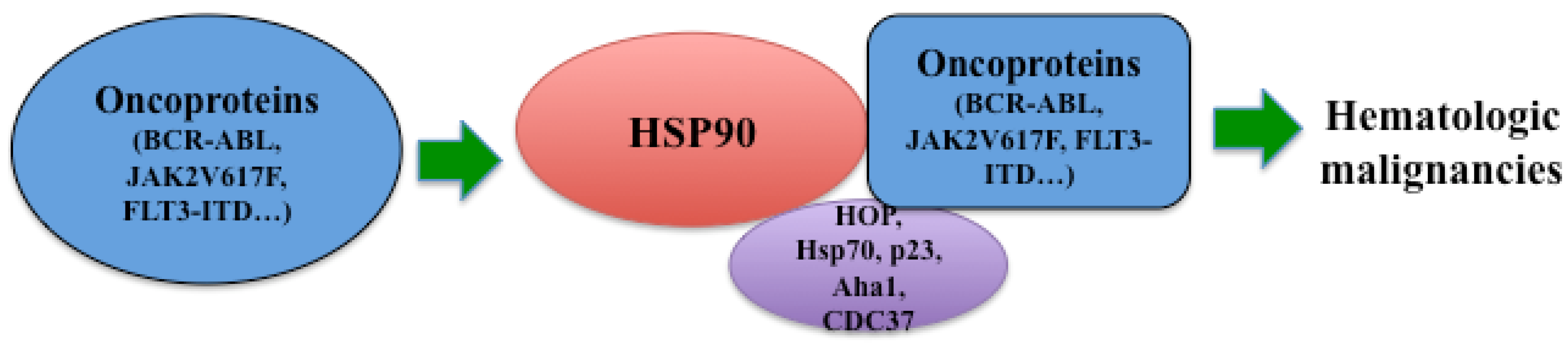
2. Structure and Functional Regulation of Hsp90
2.1. Structure of Hsp90
2.2. Functional Regulation of Hsp90

{kind=link}
{kind=link}
{kind=link}
| Co-chaperone | Function |
|---|---|
| Cdc37 | Interacts with protein kinases |
| p23 | Facilitates the maturation of client proteins |
| Aha1 | Stimulates Hsp90 ATPase activity |
| SGT1 | Binds to Hsp90 N-terminal domain, and inhibits Hsp90 ATPase activity |
| HOP | Delivers steroid hormone receptor clients to Hsp90, and also mediates the binding of Hsp90 and Hsp70 |
| TAH1 | TPR containing protein, inhibits Hsp90 ATPase activity by forming cochaperone complex with PIH1 |
| CHIP | Is an E3 ubiquitin ligase, and regulates the balance of folding/degradation for Hsp90 clients |
| FKBP51/52 | Mediates the interaction of steroid receptor with Hsp90 |
| Post-translational modification | Function |
|---|---|
| Phosphorylation | Hsp90 has been identified as a substrate of prtoein kinases such as BRAF, CK2, Src, PP5, WEE1. The phosphorylation status of Hsp90 affects its function |
| Acetylation | About 11 lysine residues in Hsp90 have been found to be acetylated |
| Nitrosylation | Nitrosylation of Cys597 inhibits the ATPase activity of Hsp90 |
3. Hsp90 in Hematologic Malignancies

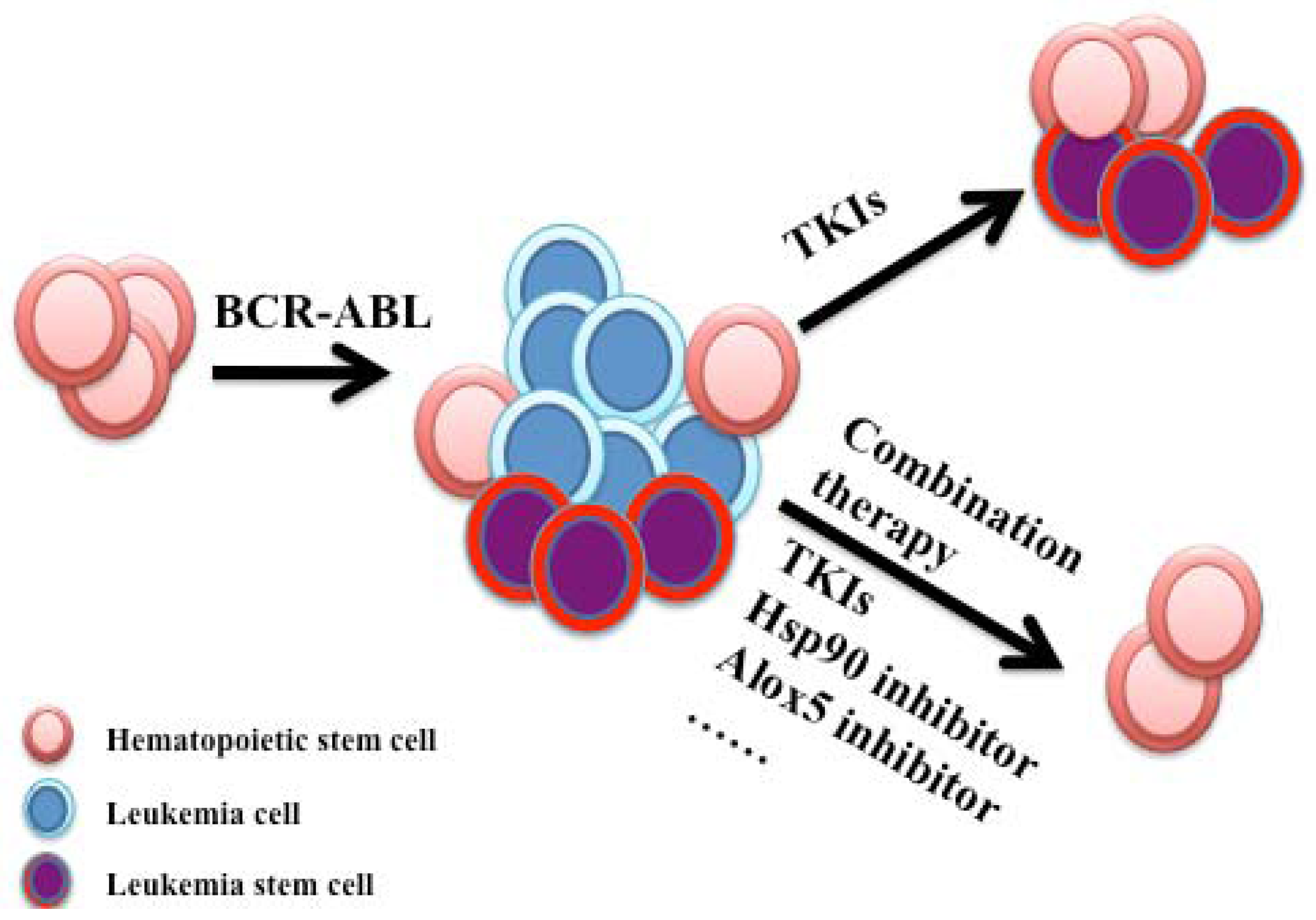
3.1. Hsp90 and Philadelphia Chromosome-Positive Leukemia
3.2. Hsp90 and Philadelphia Chromosome-Negative Myeloproliferative Neoplasms
3.3. Hsp90 and Acute Myeloid Leukemia
3.4. Hsp90 and Other Blood Cancers
| Hematologic malignancies | Hsp90 clients |
|---|---|
| CML, B-ALL | BCR-ABL |
| MPN | JAK2V617F |
| AML | FLT3-ITD |
| Multiple myeloma | CCND1, RAS, MYC, NF-kB pathway, STAT3 |
| B-chronic lymphocytic leukemia | Lyn, BCR pathway |
| Mantle cell lymphoma | CCND1 |
| Diffuse large B-cell lymphoma | BCL6, BCL2, MYC, P53 |
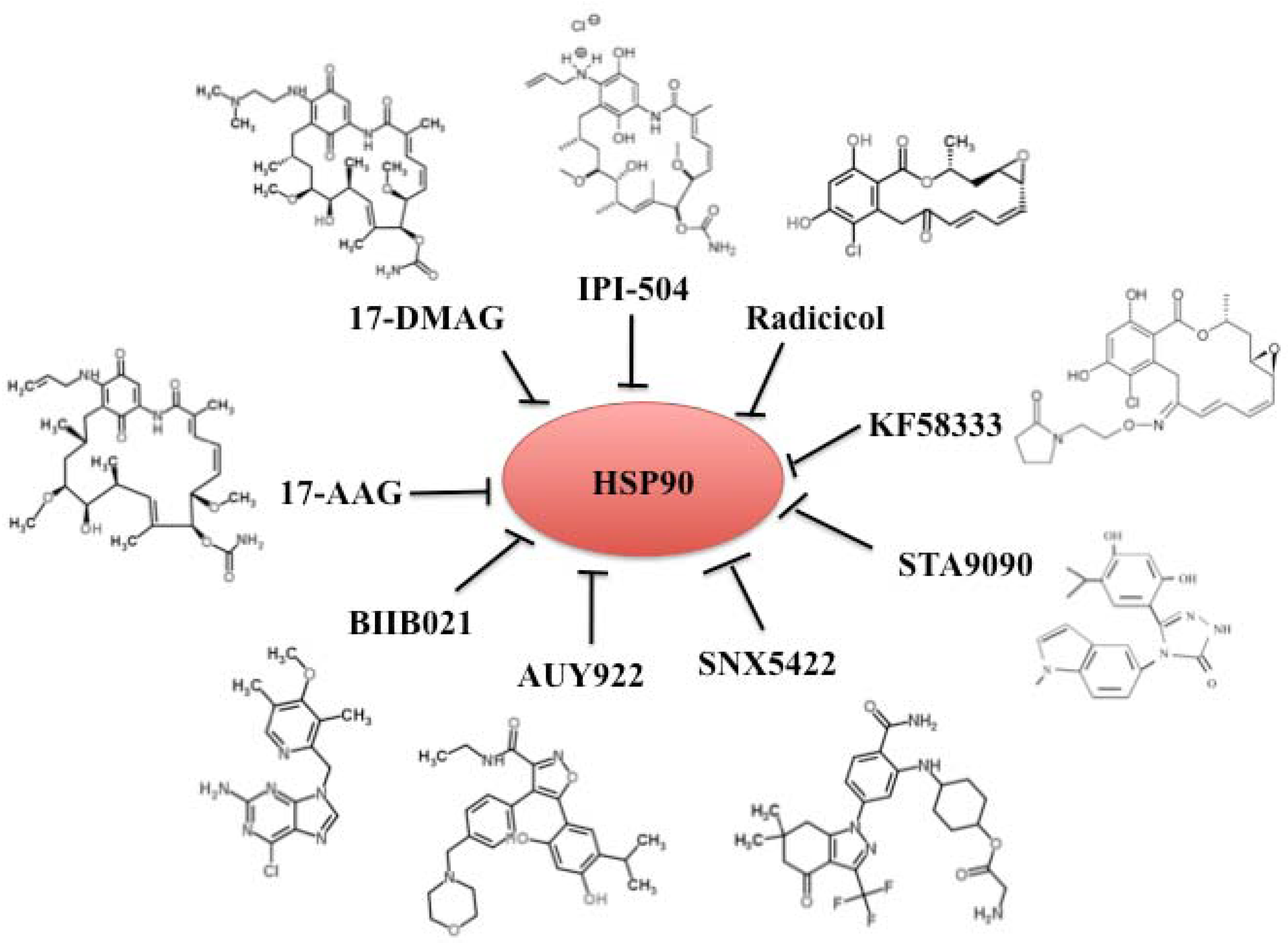
4. Chemical Inhibitors of Hsp90
| Inhibitors | Properties | Group | Clinical trial phase |
|---|---|---|---|
| 17-AAG (tanespimycin) | Well tolerated; limited oral bioavailability and solubility | Benzoquinone ansamycin | II/III |
| 17-DMAG (alvespimycin) | Well tolerated; soluble | Benzoquinone ansamycin | I |
| IPI-504 (retaspimycin) | Highly soluble and well tolerated | Benzoquinone ansamycin | III |
| IPI493 | Primary active, long-lived metabolite of 17-AAG; low solubility | Benzoquinone ansamycin | I |
| Radicicol | Macrocycli antibotic; poorly soluble and unstable | Radicicol | None |
| KF58333 | Highly soluble and stable | Radicicol | None |
| BIIB021 (CNF2024) | An oral purine scaffold compound | Small molecular inhibitor | II |
| AUY922 | An isoxazole resorcinol derivative | Small molecular inhibitor | II |
| STA-9090 | A resorcinol-containing triazole compound; highly soluble | Small molecular inhibitor | II |
| SNX-5422/SNX-2112 | A pyrazole-containing compound; highly soluble | Small molecular inhibitor | I |
| KW-2478 | A resorcinol analog; highly soluble | Small molecular inhibitor | I/II |
| AT13387 | A resorcinol-containing compound | Small molecular inhibitor | I |
| XL888 | Highly soluble | Small molecular inhibitor | I |
| NVP-HSP990 | An isoxazole resorcinol derivative | Small molecular inhibitor | I |
| MPC-3100 | An oral purine scaffold compound | Small molecular inhibitor | I |
| ABI-010 | Developed using nanoparticle albumin-bound (nab) technology | Small molecular inhibitor | I |

4.1. Benzoquinone Ansamycins 17-AAG and Its Derivatives 17-DMAG and IPI-504
4.2. Radicicol and Its Derivates
4.3. Synthetic Small-Molecular Inhibitors
5. Conclusions

Acknowledgments
Conflict of Interest
References
- Taipale, M.; Jarosz, D.F.; Lindquist, S. Hsp90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic Hsp90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- An, W.G.; Schulte, T.W.; Neckers, L.M. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ. 2000, 11, 355–360. [Google Scholar]
- Peng, C.; Brain, J.; Hu, Y.; Goodrich, A.; Kong, L.; Grayzel, D.; Pak, R.; Read, M.; Li, S. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood 2007, 110, 678–685. [Google Scholar]
- Weigert, O.; Lane, A.A.; Bird, L.; Kopp, N.; Chapuy, B.; van Bodegom, D.; Toms, A.V.; Marubayashi, S.; Christie, A.L.; McKeown, M.; et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by Hsp90 inhibition. J. Exp. Med. 2012, 209, 259–273. [Google Scholar] [CrossRef]
- Kurokawa, M.; Zhao, C.; Reya, T.; Kornbluth, S. Inhibition of apoptosome formation by suppression of Hsp90beta phosphorylation in tyrosine kinase-induced leukemias. Mol. Cell. Biol. 2008, 28, 5494–5506. [Google Scholar]
- Prodromou, C.; Piper, P.W.; Pearl, L.H. Expression and crystallization of the yeast Hsp82 chaperone, and preliminary X-ray diffraction studies of the amino-terminal domain. Proteins 1996, 25, 517–522. [Google Scholar]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural analysis of E. Coli Hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the Hsp90 chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar]
- Hainzl, O.; Lapina, M.C.; Buchner, J.; Richter, K. The charged linker region is an important regulator of Hsp90 function. J. Biol. Chem. 2009, 284, 22559–22567. [Google Scholar]
- Minami, Y.; Kimura, Y.; Kawasaki, H.; Suzuki, K.; Yahara, I. The carboxy-terminal region of mammalian Hsp90 is required for its dimerization and function in vivo. Mol. Cell. Biol. 1994, 14, 1459–1464. [Google Scholar]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the atpase activity of Hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar]
- Shao, J.; Grammatikakis, N.; Scroggins, B.T.; Uma, S.; Huang, W.; Chen, J.J.; Hartson, S.D.; Matts, R.L. Hsp90 regulates p50(CDC37) function during the biogenesis of the activeconformation of the heme-regulated eIF2 alpha kinase. J. Biol. Chem. 2001, 276, 206–214. [Google Scholar]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef]
- Miyata, Y.; Nishida, E. Evaluating ck2 activity with the antibody specific for the ck2-phosphorylated form of a kinase-targeting cochaperone cdc37. Mol. Cell. Biochem. 2008, 316, 127–134. [Google Scholar] [CrossRef]
- Cintron, N.S.; Toft, D. Defining the requirements for Hsp40 and Hsp70 in the Hsp90 chaperone pathway. J. Biol. Chem. 2006, 281, 26235–26244. [Google Scholar] [CrossRef]
- Yang, Y.; Rao, R.; Shen, J.; Tang, Y.; Fiskus, W.; Nechtman, J.; Atadja, P.; Bhalla, K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008, 68, 4833–4842. [Google Scholar]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 2007, 25, 151–159. [Google Scholar]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. Hdac6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar]
- Martinez-Ruiz, A.; Villanueva, L.; Gonzalez de Orduna, C.; Lopez-Ferrer, D.; Higueras, M.A.; Tarin, C.; Rodriguez-Crespo, I.; Vazquez, J.; Lamas, S. S-nitrosylation of Hsp90 promotes the inhibition of its atpase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. USA 2005, 102, 8525–8530. [Google Scholar]
- George, P.; Bali, P.; Cohen, P.; Tao, J.; Guo, F.; Sigua, C.; Vishvanath, A.; Fiskus, W.; Scuto, A.; Annavarapu, S.; et al. Cotreatment with 17-allylamino-demethoxygeldanamycin and flt-3 kinase inhibitor pkc412 is highly effective against human acute myelogenous leukemia cells with mutant flt-3. Cancer Res. 2004, 64, 3645–3652. [Google Scholar]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gonen, M.; Gozman, A.; et al. Hsp90 is a therapeutic target in jak2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Invest. 2010, 120, 3578–3593. [Google Scholar]
- Wong, S.; Witte, O.N. The BCR-ABL story: Bench to bedside and back. Annu. Rev. Immunol. 2004, 22, 247–306. [Google Scholar] [CrossRef]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of BCR-ABL positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar]
- Gorre, M.E.; Ellwood-Yen, K.; Chiosis, G.; Rosen, N.; Sawyers, C.L. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood 2002, 100, 3041–3044. [Google Scholar]
- Li, S.; Ilaria, R.L., Jr.; Million, R.P.; Daley, G.Q.; van Etten, R.A. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J. Exp. Med. 1999, 189, 1399–1412. [Google Scholar]
- Huntly, B.J.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 2005, 5, 311–321. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Wang, J.C.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 2005, 15, 494–501. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Hu, Y.; Swerdlow, S.; Duffy, T.M.; Weinmann, R.; Lee, F.Y.; Li, S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of ph+ leukemia in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 16870–16875. [Google Scholar]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. Hif1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef]
- Mo, J.H.; Choi, I.J.; Jeong, W.J.; Jeon, E.H.; Ahn, S.H. HIF-1alpha and Hsp90: Target molecules selected from a tumorigenic papillary thyroid carcinoma cell line. Cancer Sci. 2012, 103, 464–471. [Google Scholar]
- Bohonowych, J.E.; Peng, S.; Gopal, U.; Hance, M.W.; Wing, S.B.; Argraves, K.M.; Lundgren, K.; Isaacs, J.S. Comparative analysis of novel and conventional Hsp90 inhibitors on HIF activity and angiogenic potential in clear cell renal cell carcinoma: Implications for clinical evaluation. BMC Cancer 2011, 11, 520. [Google Scholar]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar]
- Knapper, S. FLT3 inhibition in acute myeloid leukaemia. Br. J. Haematol. 2007, 138, 687–699. [Google Scholar] [CrossRef]
- Daver, N.; Cortes, J. Molecular targeted therapy in acute myeloid leukemia. Hematology 2012, 17, 59–62. [Google Scholar]
- Boyer, S.W.; Schroeder, A.V.; Smith-Berdan, S.; Forsberg, E.C. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 2011, 9, 64–73. [Google Scholar] [CrossRef]
- Gotze, K.S.; Ramirez, M.; Tabor, K.; Small, D.; Matthews, W.; Civin, C.I. Flt3high and flt3low CD34+ progenitor cells isolated from human bone marrow are functionally distinct. Blood 1998, 91, 1947–1958. [Google Scholar]
- Lavagna-Sevenier, C.; Marchetto, S.; Birnbaum, D.; Rosnet, O. The CBL-related protein CBLB participates in FLT3 and interleukin-7 receptor signal transduction in pro-B cells. J. Biol. Chem. 1998, 273, 14962–14967. [Google Scholar]
- Lavagna-Sevenier, C.; Marchetto, S.; Birnbaum, D.; Rosnet, O. FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia 1998, 12, 301–310. [Google Scholar] [CrossRef]
- Rosnet, O.; Buhring, H.J.; Marchetto, S.; Rappold, I.; Lavagna, C.; Sainty, D.; Arnoulet, C.; Chabannon, C.; Kanz, L.; Hannum, C.; et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 1996, 10, 238–248. [Google Scholar]
- Kottaridis, P.D.; Gale, R.E.; Frew, M.E.; Harrison, G.; Langabeer, S.E.; Belton, A.A.; Walker, H.; Wheatley, K.; Bowen, D.T.; Burnett, A.K.; et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the united kingdom medical research council aml 10 and 12 trials. Blood 2001, 98, 1752–1759. [Google Scholar]
- Flandrin, P.; Guyotat, D.; Duval, A.; Cornillon, J.; Tavernier, E.; Nadal, N.; Campos, L. Significance of heat-shock protein (Hsp) 90 expression in acute myeloid leukemia cells. Cell Stress Chaperones 2008, 13, 357–364. [Google Scholar] [CrossRef]
- Reikvam, H.; Ersvaer, E.; Bruserud, O. Heat shock protein 90—A potential target in the treatment of human acute myelogenous leukemia. Curr. Cancer Drug Targets 2009, 9, 761–776. [Google Scholar] [CrossRef]
- Reikvam, H.; Hatfield, K.J.; Ersvaer, E.; Hovland, R.; Skavland, J.; Gjertsen, B.T.; Petersen, K.; Bruserud, O. Expression profile of heat shock proteins in acute myeloid leukaemia patients reveals a distinct signature strongly associated with FLT3 mutation status—Consequences and potentials for pharmacological intervention. Br. J. Haematol. 2012, 156, 468–480. [Google Scholar] [CrossRef]
- Minami, Y.; Kiyoi, H.; Yamamoto, Y.; Yamamoto, K.; Ueda, R.; Saito, H.; Naoe, T. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia 2002, 16, 1535–1540. [Google Scholar] [CrossRef]
- Oshikawa, G.; Nagao, T.; Wu, N.; Kurosu, T.; Miura, O. C-CBL and CBL-B ligases mediate 17-allylaminodemethoxygeldanamycin-induced degradation of autophosphorylated FLT3 kinase with internal tandem duplication through the ubiquitin proteasome pathway. J. Biol. Chem. 2011, 286, 30263–30273. [Google Scholar]
- Fredly, H.; Ersvaer, E.; Gjertsen, B.T.; Bruserud, O. Immunogenic apoptosis in human acute myeloid leukemia (AML): Primary human aml cells expose calreticulin and release heat shock protein (Hsp) 70 and Hsp90 during apoptosis. Oncol. Rep. 2011, 25, 1549–1556. [Google Scholar]
- Yeh, C.H.; Tseng, R.; Hannah, A.; Estrov, Z.; Estey, E.; Kantarjian, H.; Albitar, M. Clinical correlation of circulating heat shock protein 70 in acute leukemia. Leuk. Res. 2010, 34, 605–609. [Google Scholar] [CrossRef]
- Fredly, H.; Reikvam, H.; Gjertsen, B.T.; Bruserud, O. Disease-stabilizing treatment with all-trans retinoic acid and valproic acid in acute myeloid leukemia: Serum Hsp70 and Hsp90 levels and serum cytokine profiles are determined by the disease, patient age, and anti-leukemic treatment. Am. J. Hematol. 2012, 87, 368–376. [Google Scholar] [CrossRef]
- Valbuena, J.R.; Rassidakis, G.Z.; Lin, P.; Atwell, C.; Georgakis, G.V.; Younes, A.; Jones, D.; Medeiros, L.J. Expression of heat-shock protein-90 in non-hodgkin’s lymphomas. Mod. Pathol. 2005, 18, 1343–1349. [Google Scholar]
- Stuhmer, T.; Zollinger, A.; Siegmund, D.; Chatterjee, M.; Grella, E.; Knop, S.; Kortum, M.; Unzicker, C.; Jensen, M.R.; Quadt, C.; et al. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 2008, 22, 1604–1612. [Google Scholar]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar]
- Georgakis, G.V.; Li, Y.; Younes, A. The heat shock protein 90 inhibitor 17-aag induces cell cycle arrest and apoptosis in mantle cell lymphoma cell lines by depleting cyclin D1, Akt, Bid and activating caspase 9. Br. J. Haematol. 2006, 135, 68–71. [Google Scholar] [CrossRef]
- Azoitei, N.; Hoffmann, C.M.; Ellegast, J.M.; Ball, C.R.; Obermayer, K.; Gossele, U.; Koch, B.; Faber, K.; Genze, F.; Schrader, M.; et al. Targeting of kras mutant tumors by Hsp90 inhibitors involves degradation of STK33. J. Exp. Med. 2012, 209, 697–711. [Google Scholar]
- Wang, X.; Ju, W.; Renouard, J.; Aden, J.; Belinsky, S.A.; Lin, Y. 17-allylamino-17-demethoxygeldanamycin synergistically potentiates tumor necrosis factor-induced lung cancer cell death by blocking the nuclear factor-kappab pathway. Cancer Res. 2006, 66, 1089–1095. [Google Scholar]
- Chatterjee, M.; Jain, S.; Stuhmer, T.; Andrulis, M.; Ungethum, U.; Kuban, R.J.; Lorentz, H.; Bommert, K.; Topp, M.; Kramer, D.; et al. STAT3 and mapk signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728. [Google Scholar]
- Kuppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Cerchietti, L.C.; Lopes, E.C.; Yang, S.N.; Hatzi, K.; Bunting, K.L.; Tsikitas, L.A.; Mallik, A.; Robles, A.I.; Walling, J.; Varticovski, L.; et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat. Med. 2009, 15, 1369–1376. [Google Scholar]
- Zenz, T.; Mertens, D.; Kuppers, R.; Dohner, H.; Stilgenbauer, S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2010, 10, 37–50. [Google Scholar]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 2012. [Google Scholar]
- Trentin, L.; Frasson, M.; Donella-Deana, A.; Frezzato, F.; Pagano, M.A.; Tibaldi, E.; Gattazzo, C.; Zambello, R.; Semenzato, G.; Brunati, A.M. Geldanamycin-induced lyn dissociation from aberrant Hsp90-stabilized cytosolic complex is an early event in apoptotic mechanisms in B-chronic lymphocytic leukemia. Blood 2008, 112, 4665–4674. [Google Scholar]
- Nimmanapalli, R.; O'Bryan, E.; Bhalla, K. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers BCR-ABL levels and induces apoptosis and differentiation of BCR-ABL-positive human leukemic blasts. Cancer Res. 2001, 61, 1799–1804. [Google Scholar]
- Richardson, P.G.; Chanan-Khan, A.A.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Mitsiades, C.S.; Anderson, K.C. Tanespimycin monotherapy in relapsed multiple myeloma: Results of a phase 1 dose-escalation study. Br. J. Haematol. 2010, 150, 438–445. [Google Scholar]
- Solit, D.B.; Osman, I.; Polsky, D.; Panageas, K.S.; Daud, A.; Goydos, J.S.; Teitcher, J.; Wolchok, J.D.; Germino, F.J.; Krown, S.E.; et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin. Cancer Res. 2008, 14, 8302–8307. [Google Scholar]
- Ronnen, E.A.; Kondagunta, G.V.; Ishill, N.; Sweeney, S.M.; Deluca, J.K.; Schwartz, L.; Bacik, J.; Motzer, R.J. A phase II trial of 17-(allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest. New Drugs 2006, 24, 543–546. [Google Scholar]
- Heath, E.I.; Hillman, D.W.; Vaishampayan, U.; Sheng, S.; Sarkar, F.; Harper, F.; Gaskins, M.; Pitot, H.C.; Tan, W.; Ivy, S.P.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 2008, 14, 7940–7946. [Google Scholar]
- Kaufmann, S.H.; Karp, J.E.; Litzow, M.R.; Mesa, R.A.; Hogan, W.; Steensma, D.P.; Flatten, K.S.; Loegering, D.A.; Schneider, P.A.; Peterson, K.L.; et al. Phase I and pharmacological study of cytarabine and tanespimycin in relapsed and refractory acute leukemia. Haematologica 2011, 96, 1619–1626. [Google Scholar]
- Hollingshead, M.; Alley, M.; Burger, A.M.; Borgel, S.; Pacula-Cox, C.; Fiebig, H.H.; Sausville, E.A. In vivo antitumor efficacy of 17-dmag (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother. Pharmacol. 2005, 56, 115–125. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Rahmani, M.; Gao, N.; Kramer, L.; Corbin, A.S.; Druker, B.J.; Dent, P.; Grant, S. Synergistic interactions between dmag and mitogen-activated protein kinase kinase 1/2 inhibitors in bcr/abl+ leukemia cells sensitive and resistant to imatinib mesylate. Clin. Cancer Res. 2006, 12, 2239–2247. [Google Scholar]
- Hertlein, E.; Wagner, A.J.; Jones, J.; Lin, T.S.; Maddocks, K.J.; Towns, W.H., 3rd; Goettl, V.M.; Zhang, X.; Jarjoura, D.; Raymond, C.A.; et al. 17-dmag targets the nuclear factor-kappab family of proteins to induce apoptosis in chronic lymphocytic leukemia: Clinical implications of Hsp90 inhibition. Blood 2010, 116, 45–53. [Google Scholar]
- Kobayashi, N.; Toyooka, S.; Soh, J.; Yamamoto, H.; Dote, H.; Kawasaki, K.; Otani, H.; Kubo, T.; Jida, M.; Ueno, T.; et al. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-dmag, on non-small-cell lung cancers being resistant to egfr tyrosine kinase inhibitor. Lung Cancer 2012, 75, 161–166. [Google Scholar]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar]
- Roue, G.; Perez-Galan, P.; Mozos, A.; Lopez-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Saborit-Villarroya, I.; Normant, E.; Campo, E.; Colomer, D. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival er chaperone bip/grp78. Blood 2011, 117, 1270–1279. [Google Scholar]
- Abramson, J.S.; Chen, W.; Juszczynski, P.; Takahashi, H.; Neuberg, D.; Kutok, J.L.; Takeyama, K.; Shipp, M.A. The heat shock protein 90 inhibitor IPI-504 induces apoptosis of akt-dependent diffuse large B-cell lymphomas. Br. J. Haematol. 2009, 144, 358–366. [Google Scholar] [CrossRef]
- Sequist, L.V.; Gettinger, S.; Senzer, N.N.; Martins, R.G.; Janne, P.A.; Lilenbaum, R.; Gray, J.E.; Iafrate, A.J.; Katayama, R.; Hafeez, N.; et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4953–4960. [Google Scholar]
- Soga, S.; Shiotsu, Y.; Akinaga, S.; Sharma, S.V. Development of radicicol analogues. Curr. Cancer Drug Targets 2003, 3, 359–369. [Google Scholar] [CrossRef]
- Shiotsu, Y.; Neckers, L.M.; Wortman, I.; An, W.G.; Schulte, T.W.; Soga, S.; Murakata, C.; Tamaoki, T.; Akinaga, S. Novel oxime derivatives of radicicol induce erythroid differentiation associated with preferential g(1) phase accumulation against chronic myelogenous leukemia cells through destabilization of BCR-ABL with Hsp90 complex. Blood 2000, 96, 2284–2291. [Google Scholar]
- Janin, Y.L. Heat shock protein 90 inhibitors. A text book example of medicinal chemistry? J. Med. Chem. 2005, 48, 7503–7512. [Google Scholar] [CrossRef]
- Chiosis, G.; Lucas, B.; Huezo, H.; Solit, D.; Basso, A.; Rosen, N. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr. Cancer Drug Targets 2003, 3, 371–376. [Google Scholar] [CrossRef]
- Boll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.J.; Lundgren, K.; Hansen, H.P.; Engert, A.; et al. Heat shock protein 90 inhibitor BIIB021 (Cnf2024) depletes nf-kappab and sensitizes hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. 2009, 15, 5108–5116. [Google Scholar]
- Lundgren, K.; Zhang, H.; Brekken, J.; Huser, N.; Powell, R.E.; Timple, N.; Busch, D.J.; Neely, L.; Sensintaffar, J.L.; Yang, Y.C.; et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol. Cancer Ther. 2009, 8, 921–929. [Google Scholar]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef]
- Okawa, Y.; Hideshima, T.; Steed, P.; Vallet, S.; Hall, S.; Huang, K.; Rice, J.; Barabasz, A.; Foley, B.; Ikeda, H.; et al. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and Erk. Blood 2009, 113, 846–855. [Google Scholar]
- Rajan, A.; Kelly, R.J.; Trepel, J.B.; Kim, Y.S.; Alarcon, S.V.; Kummar, S.; Gutierrez, M.; Crandon, S.; Zein, W.M.; Jain, L.; et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin. Cancer Res. 2011, 17, 6831–6839. [Google Scholar]
- Proia, D.A.; Foley, K.P.; Korbut, T.; Sang, J.; Smith, D.; Bates, R.C.; Liu, Y.; Rosenberg, A.F.; Zhou, D.; Koya, K.; et al. Multifaceted intervention by the Hsp90 inhibitor ganetespib (STA-9090) in cancer cells with activated JAK/STAT signaling. PLoS One 2011, 6, e18552. [Google Scholar]
- Lin, T.Y.; Bear, M.; Du, Z.; Foley, K.P.; Ying, W.; Barsoum, J.; London, C. The novel Hsp90 inhibitor STA-9090 exhibits activity against KIT-dependent and -independent malignant mast cell tumors. Exp. Hematol. 2008, 36, 1266–1277. [Google Scholar] [CrossRef]
- Gaspar, N.; Sharp, S.Y.; Pacey, S.; Jones, C.; Walton, M.; Vassal, G.; Eccles, S.; Pearson, A.; Workman, P. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 2009, 69, 1966–1975. [Google Scholar]
- Guo, W.; Reigan, P.; Siegel, D.; Zirrolli, J.; Gustafson, D.; Ross, D. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:Quinone oxidoreductase 1: Role of 17-aag hydroquinone in heat shock protein 90 inhibition. Cancer Res. 2005, 65, 10006–10015. [Google Scholar] [CrossRef]
- Prodromou, C.; Nuttall, J.M.; Millson, S.H.; Roe, S.M.; Sim, T.S.; Tan, D.; Workman, P.; Pearl, L.H.; Piper, P.W. Structural basis of the radicicol resistance displayed by a fungal Hsp90. ACS Chem. Biol. 2009, 4, 289–297. [Google Scholar]
- McDowell, C.L.; Bryan Sutton, R.; Obermann, W.M. Expression of Hsp90 chaperone proteins in human tumor tissue. Int. J. Biol. Macromol. 2009, 45, 310–314. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ho, N.; Li, A.; Li, S.; Zhang, H. Heat Shock Protein 90 and Role of Its Chemical Inhibitors in Treatment of Hematologic Malignancies. Pharmaceuticals 2012, 5, 779-801. https://doi.org/10.3390/ph5080779
Ho N, Li A, Li S, Zhang H. Heat Shock Protein 90 and Role of Its Chemical Inhibitors in Treatment of Hematologic Malignancies. Pharmaceuticals. 2012; 5(8):779-801. https://doi.org/10.3390/ph5080779
Chicago/Turabian StyleHo, Ngoc, Adam Li, Shaoguang Li, and Haojian Zhang. 2012. "Heat Shock Protein 90 and Role of Its Chemical Inhibitors in Treatment of Hematologic Malignancies" Pharmaceuticals 5, no. 8: 779-801. https://doi.org/10.3390/ph5080779