
Inflammation and Liver Cell Death in Patients with Hepatitis C Viral Infection


Abstract
:1. Introduction
1.1. Viral Hepatitis C
1.2. Liver Disease and the Cytokine Storm Syndrome
2. Aims
3. Materials and Methods
3.1. Patient Cohort and Clinical Data
3.2. Special Viral and Cytokine Analysis
3.3. Apoptosis
3.4. Histological Analysis
3.5. Statistical Analysis
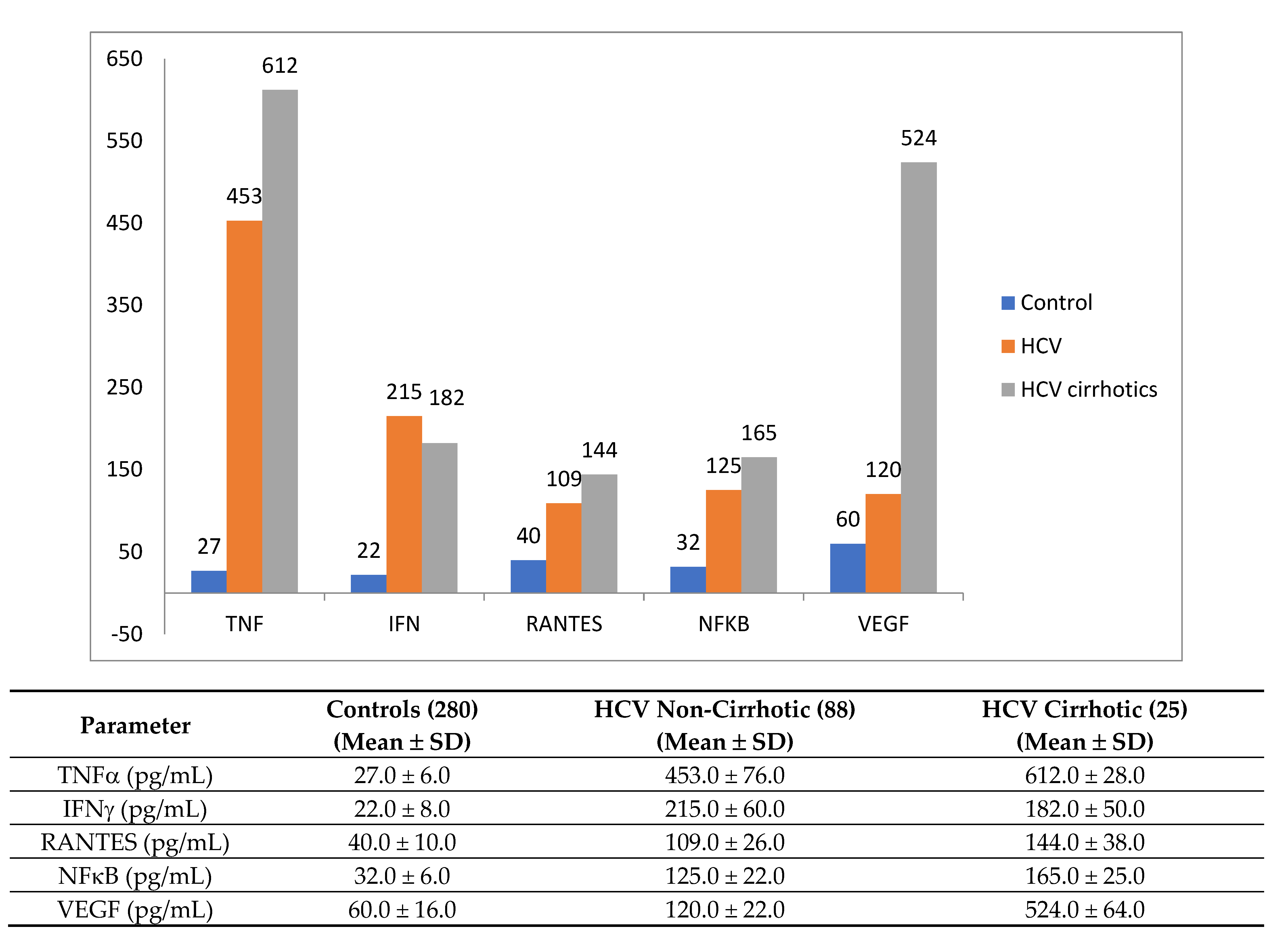
4. Results
4.1. Patients Characteristics
4.1.1. Apoptosis and Necrosis and Inflammation
4.1.2. Correlation of Serum Biomarkers of Apoptosis, Necrosis and Inflammation with Histology
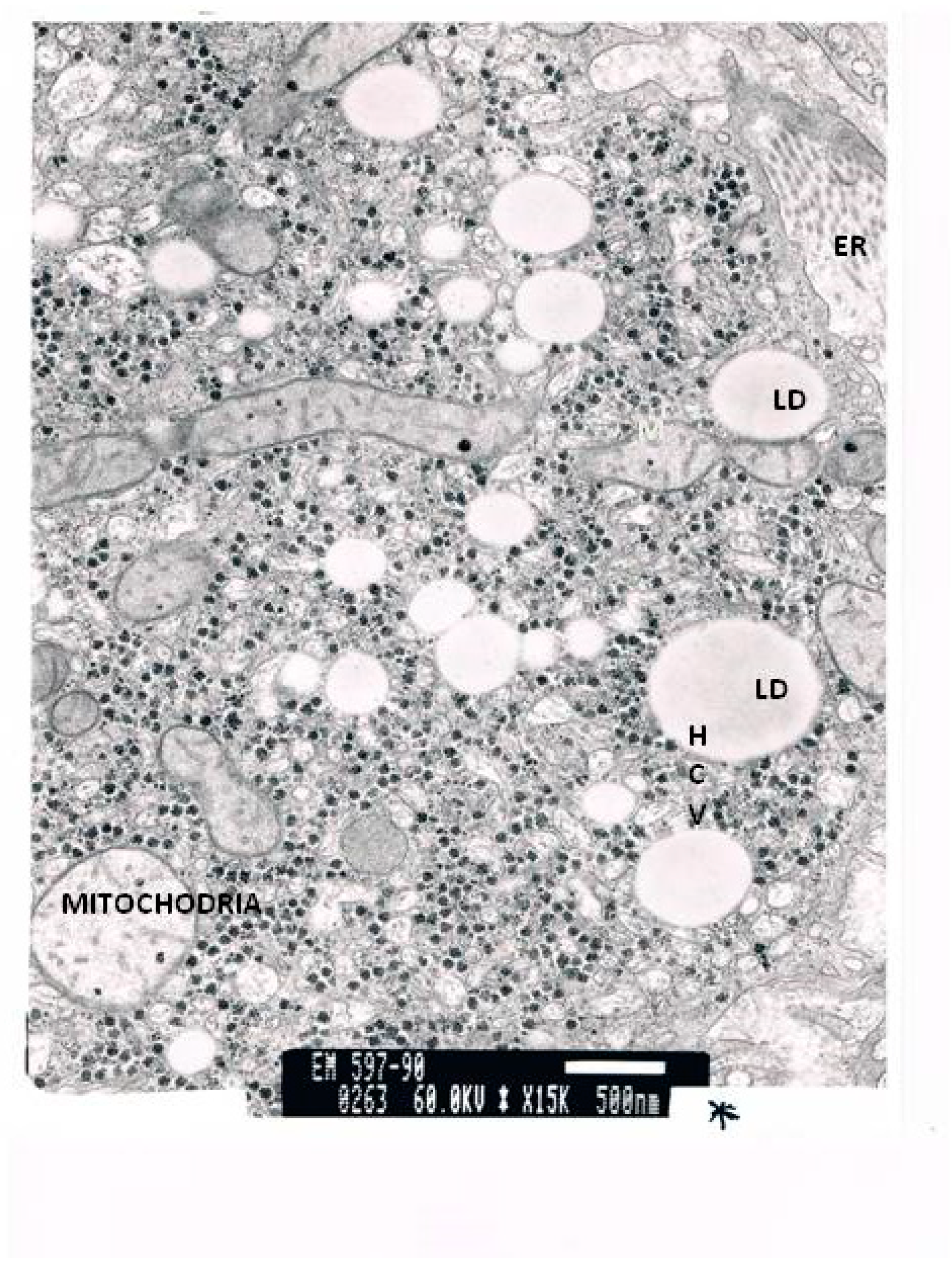
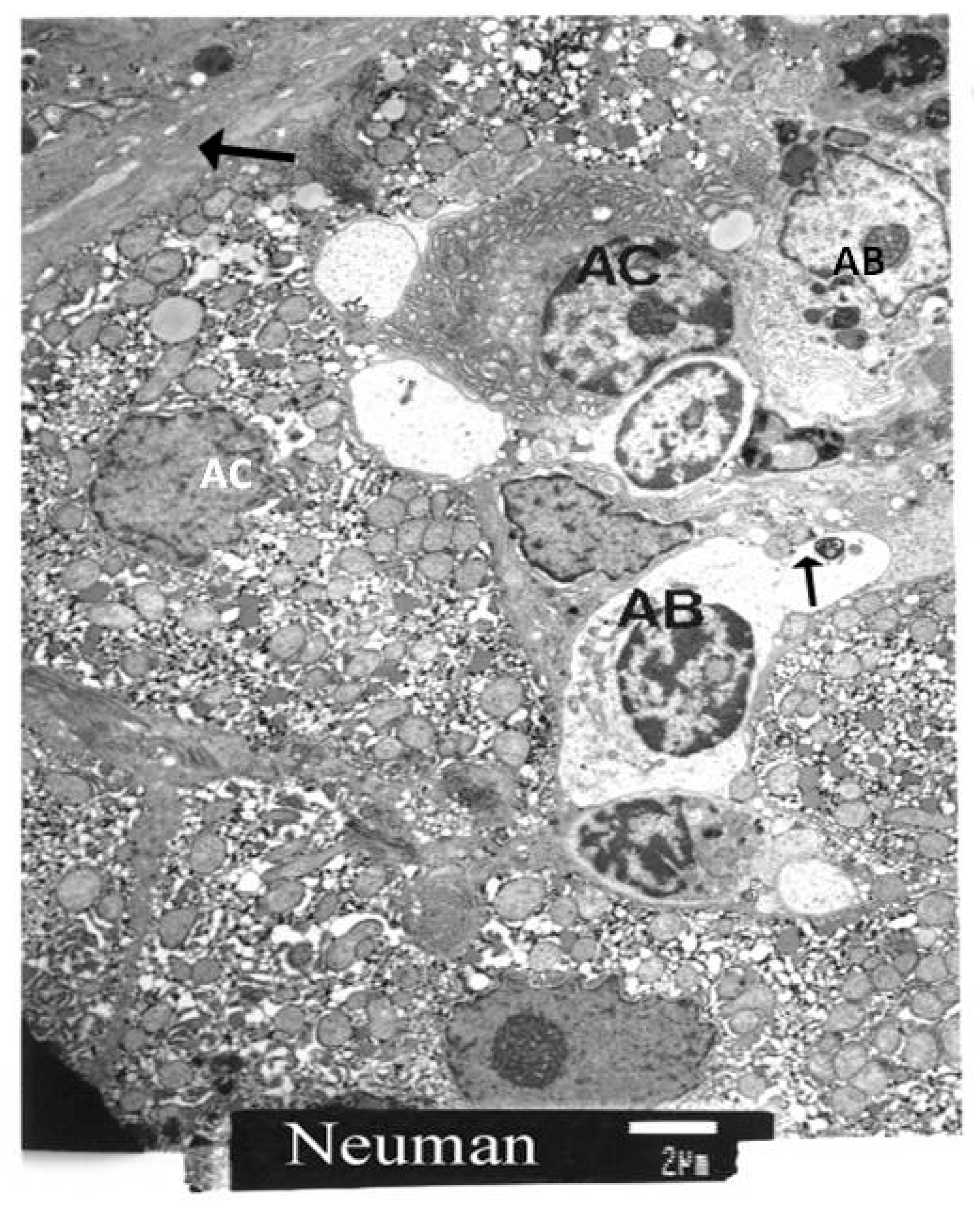
4.2. Histology
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALT | alanine aminotransferase (glutamic pyruvic transaminase, GPT) |
| ALP | alkaline phosphatase |
| AST | aspartate aminotransferase (glutamic oxalic transaminase, GOT) |
| CCK | Caspase-cleaved cytokeratin (8 and 18) M-30 and M-65 |
| DAMPs | danger-associated molecular patterns |
| EBV | Epstein–Barr virus |
| EGF | endothelial growth factor |
| ELISA | enzyme-linked immunosorbent assay; |
| FasL/TNFSF6 | factor-related apoptosis ligand |
| Fas | ligand, a member of tumor necrosis factor super-family |
| FDA | Food and Drug Administration |
| GGT | γ-glutamyl transferase |
| HBV | hepatitis virus B |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis virus C |
| HCV-RNA | hepatitis C virus ribonucleic acid |
| HHV6 | human herpes virus 6 |
| IFN | interferon |
| IL | interleukin |
| INR | international normalized ratio |
| IRF | interferon regulatory factor |
| ISGs | IFN-stimulated genes |
| ISRE | IFN-stimulated response element |
| IU/L | international units/liter |
| Jak | Janus kinase |
| mean ± st. d | mean ± standard deviation |
| MIP | macrophage inflammatory protein-1 (CCR5 ligand) |
| MMP | matrix metalloprotease |
| NFκB | nuclear factor-κB |
| NLRP | NOD-like receptor pyrin domain |
| PAI -1 | plasminogen activator inhibitor 1 |
| PAMPs | pathogen-associated molecular patterns |
| pro-CASP-1 | procaspase 1 |
| PICP | C-terminal procollagen I peptide |
| PIIINP | amino-terminal propeptide of procollagen type III |
| PRRs | pattern recognition receptors |
| PYCARD | apoptosis-associated speck-like protein containing a CARD ASC |
| RANTES (CCL5) | regulated upon activation normal T cell expressed and secreted |
| RNA | ribonucleic acid |
| TGF | β-transforming growth factor beta |
| TIMP | tissue inhibitor of matrix metalloproteinase |
| TNF-α | tumor necrosis factor alpha |
| TLR | Toll-like receptors |
| VEGF | vascular endothelial growth factor |
References
- Hoofnagle, J.H.; Hepatitis, C. The clinical spectrum of disease Hepatology 1997 26 International conference on harmonisation expert working group. In ICH Harmonised Tripartite The Guideline for Good Clinical Practice, Proceedings of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; CFR & ICH Guidelines; Barnett International: Needham, MA, USA, 1997; Volume 1, pp. 19063–22043. [Google Scholar] [CrossRef] [Green Version]
- Seeff, L.B.; Miller, R.N.; Rabkin, C.S.; Buskell, B.Z.; Straley-Eason, K.D.; Smoak, B.L. 45-year follow-up of hepatitis C virus infection in healthy young adults. Ann. Intern. Med. 2000, 132, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Seeff, L.B.; Buskell-Bales, Z.; Wright, E.C.; Durako, S.J.; Alter, H.J.; Iber, F.L. Long-term mortality after transfusion-associated non-A, non-B hepatitis. The National Heart, Lung, and Blood Institute Study Group. N. Engl. J. Med. 1992, 327, 1906–1911. [Google Scholar] [CrossRef]
- Seeff, L.B. Natural history of hepatitis C. Am. J. Med. 1999, 107, 10–15. [Google Scholar] [CrossRef]
- Asselah, T.; Bouejema, H.; Francoz, C.; Sobesky, R.; Valla, D.; Belghiti, J.; Marcellin, P.; Durand, F. Hepatitis C and liver transplantation: Fibrosis progression and treatment. Or how to improve management. Gastroent. Clin. Biol. 2006, 30, 1281–1295. [Google Scholar] [CrossRef]
- Zimmerman, J.E.; Wagner, D.P.; Draper, E.A. Evaluation of acute physiology and chronic health evaluation III predictions of hospital mortality in an independent database. Crit. Care Med. 1998, 26, 1317–1326. [Google Scholar] [CrossRef]
- Wiesner, R.; Edwards, E.; Freeman, R. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology 2003, 124, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, M.G.; Benhamou, J.P.; Marcellin, P.; Valla, D.C.; Malkiewicz, I.M.; Katz, G.G.; Trepo, C.; Bourliere, M.; Cameron, R.; Cohen, L.; et al. Cytokine-chemokine signature in chronic hepatitis C patients. Transl. Res. 2007, 149, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Benhamou, J.P.; Malkiewicz, I.M.; Akremi, R.; Shear, N.H.; Asselah, T.; Ibrahim, B.N.; Martinot, P.M.; Jacobson, B.P.; Katz, G.G.; et al. Cytokines as predictors for sustained response and as markers for immunomodulation in patients with chronic hepatitis C. Clin. Biochem. 2001, 34, 173–182. [Google Scholar] [CrossRef]
- Neuman, M.G.; Benhamou, J.P.; Malkiewicz, I.M.; Ibrahim, A.; Valla, D.C.; Martinot, P.M.; Asselah, T.; Bourliere, M.; Katz, G.G.; Shear, N.H.; et al. Kinetics of serum cytokines reflect changes in the severity of chronic hepatitis C presenting minimal fibrosis. J. Viral Hepat. 2002, 9, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Schmilovitz-Weiss, H.; Hilzenrat, N.; Bourliere, M.; Marcellin, P.; Trepo, C.; Mazulli, T.; Moussa, G.; Patel, A.; Baig, A.A.; et al. Markers of Inflammation and Fibrosis in Alcoholic Hepatitis and Viral Hepatitis C. Int. J. Hepatol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.M.; Thimme, R.; Melpolder, J.; Oldach, D.; Pemberton, J.; Moorhead, L.J.; McHutchison, J.G.; Alter, G.H.; Chisari, F. Differential CD4 and CDS T cell responsiveness in hepatitis C virus infection. Hepatology 2001, 33, 267–276. [Google Scholar] [CrossRef]
- Cerny, A.; McHutchison, J.G.; Pasquinelli, C.; Brown, M.E.; Brothers, M.A.; Grabscheid, B.; Fowler, P.; Houghton, M.; Chisari, F.V. Cytotoxic T lymphocyte response to hepatitis C virus-derived peptides containing the HLA A2.1 binding motif. J. Clin. Investig. 1995, 95, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B.; Chang, K.M.; McHutchison, J.G.; Kokka, R.; Houghton, M.; Chisari, F.V. Quantitative analysis of the peripheral blood cytotoxic T lymphocyte response, disease activity and viral load in patients with chronic hepatitis C virus infection. J. Clin. Investig. 1996, 98, 1432–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, M.G.; Monteiro, M.; Rehm, J. Drug Interactions Between Psychoactive Substances and Antiretroviral Therapy in Individuals Infected With Human Immunodeficiency and Hepatitis Viruses. Subst. Use Misuse 2006, 41, 1395–1463. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.; Patel, K.; Bostrom, A.; Pianko, S.; Pockros, P.; McHutchison, J.G.; Wright, T.L. Risks of a range of alcohol intake on hepatitis C-related fibrosis. Hepatology 2004, 39, 826–834. [Google Scholar] [CrossRef]
- Neuman, M.G. Apoptosis in diseases of the liver. Crit. Rev. Clin. Lab. Sci. 2001, 38, 109–166. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Benhamou, J.-P.; Martinot, M.; Boyer, N.; Shear, N.H.; Malkiewicz, I.; Katz, G.G.; Suneja, A.; Singh, S.; Marcellin, P. Predictors of sustained response to alpha interferon therapy in chronic hepatitis C. Clin. Biochem. 1999, 32, 537–545. [Google Scholar] [CrossRef]
- Neuman, M.G.; Blendis, L.M.; Shear, N.H.; Malkiewicz, I.M.; Ibrahim, A.; Katz, G.G.; Sapir, D.; Halpern, Z.; Brill, S.; Peretz, H.; et al. Cytokine network in non responding chronic hepatitis C patients with genotype 1: Role of triple therapy with interferon ribavirin and ursodeoxycholate. Clin. Biochem. 2001, 34, 183–188. [Google Scholar] [CrossRef]
- Neuman, M.G.; Benhamou, J.P.; Bourliere, M.; Ibrahim, A.; Malkiewicz, I.; Asselah, T.; Martinot, P.M.; Shear, N.H.; Katz, G.G.; Akremi, R.; et al. Serum tumour necrosis factor-alpha and transforming growth factor-beta levels in chronic hepatitis C patients are immunomodulated by therapy. Cytokine 2002, 17, 108–117. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Appel, N.T.; Pietschmann, N.; Bartenschlager, R. Mutational analysis of hepatitis C virus nonstructural protein 5A: Potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 2005, 79, 3187–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, W.C.; Yeow, W.S.; Pitha, P. Analysis of Functional Domains of Interferon Regulatory Factor 7 and Its Association with IRF-3. Virology 2001, 280, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepat. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.; Pisetsky, D.S.; Schwartz, A.D. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 2004, 5, 975–979. [Google Scholar] [CrossRef]
- De Francesco, R.; Steinkühler, C. Structure and Function of the Hepatitis C Virus NS3-NS4A Serine Proteinase. Hepat. C Viruses 2000, 242, 149–169. [Google Scholar]
- Der, S.D.; Zhou, A.; Williams, B.R.G.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [Green Version]
- Foy, E.; Li, K.; Sumpter, R., Jr.; Loo, Y.M.; Loo, C.L.; Johnson, C.; Wang, P.M.; Fish, M.; Yoneyama, T.; Fujita, S.M.; et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-l signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 2986–2991. [Google Scholar] [CrossRef] [Green Version]
- McCormick, C.; Challinor, L.; Macdonald, A.; Rowlands, D.J.; Harris, M. Introduction of replication-competent hepatitis C virus transcripts using a tetracycline-regulable baculovirus delivery system. J. Gen. Virol. 2004, 85, 429–439. [Google Scholar] [CrossRef]
- Sumpter, R.Y.; Loo, M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M.J. Regulating intracellular anti-viral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R.; Ikeda, M.; Lemon, S.M.; Gale, M.J. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredericksen, B.; Akkaraju, G.; Foy, E.; Wang, C.; Pflugheber, J.; Chen, Z.J.; Gale, M., Jr. Activation of the inteferon-p promoter during hepatitis C virus RNA replication. Viral Immunol. 2001, 15, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sztark, F.; Dubroca, J.; Latry, P.; Quinton, A.; Balabaud, C.; Bioulac-Sage, P. Perisinusoidal cells in patients with normal liver histology: A morphometric study. J. Hepatol. 1986, 2, 358–369. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Katze, M.G. Molecular mechanisms of interferon resistance mediated by viral directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 1998, 78, 29–46. [Google Scholar] [CrossRef]
- Gale, M.; Korth, M.J.; Tang, N.M.; Tan, S.-L.; Hopkins, D.A.; Dever, T.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence That Hepatitis C Virus Resistance to Interferon Is Mediated through Repression of the PKR Protein Kinase by the Nonstructural 5A Protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Gale, M., Jr.; Kwieciszewski, B.; Dossett, M.; Nakao, H.; Katze, M.G. Anti-apoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J. Virol. 1999, 73, 6506–6516. [Google Scholar] [CrossRef] [Green Version]
- Lana, J.P.; Martins, L.B.; Oliveira, M.; Menezes-Garcia, Z.; Yamada, L.T.P.; Vieira, L.; Teixeira, M.M.; Ferreira, A. TNF and IL-18 cytokines may regulate liver fat storage under homeostasis conditions. Appl. Physiol. Nutr. Metab. 2016, 41, 1295–1302. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.; Shafran, S.; Greenbloom, S.; Enns, R.; Farley, F.; Hilzerat, N.; Williams, M.; Elkashab, M.; Abadir, N.; Neuman, M.G. Single Dose Infliximab in Hepatitis C Genotype 1 Naïve Patients with High Serum tumor necrosis factor alpha does not Influence Efficacy of Pegylated Interferon alpha-2b/Ribavirin Therapy. Can. J. Gastroenterol. Hepatol. 2014, 28, 367131. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Mueller, J.; Mueller, S. Non-invasive Biomarkers of Liver Inflammation and Cell Death in Response to Alcohol Detoxification. Front. Physiol. 2021, 12, 678118. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory Parameters | Cirrhotic | Non-Cirrhotic | p |
|---|---|---|---|
| AST (U/L) | 175 ± 62 | 72 ± 24 | <0.05 |
| ALT (U/L) | 174 ± 67 | 54 ± 15 | <0.05 |
| GGT (U/L) | 202 ± 88 | 133 ± 12 | <0.001 |
| AP (U/L) | 195 ± 71 | 73 ± 21 | <0.001 |
| Bilirubin total (mg/dL) | 1.76 ± 0.25 | 1.3 ± 0.4 | <0.05 |
| Parameter | Normal Values Controls (280) (Mean ± SD) | HCV Patients (88) (Mean ± SD) | p Value |
|---|---|---|---|
| IL-1 (pg/mL) | 24.0 ± 6.0 | 85.3 ± 28.0 | 0.003 |
| IL-6 (pg/mL) | 30.0 ± 10.0 | 65.0 ± 20.0 | 0.005 |
| IL-8 (pg/mL) | 44.0 ± 10.0 | 182.6 ± 43.0 | <0.001 |
| IL-10 (pg/mL) | 30.0 ± 5.0 | 45.0 ± 20.0 | NS |
| IL-12 (pg/mL) | 45.0 ± 10.0 | 58.0 ± 30.0 | NS |
| IL-13 (pg/mL) | 12.0 ± 4.0 | 75.0 ± 10.0 | <0.001 |
| IL-17 (pg/mL) | 45.0 ± 5.0 | 98.0 ± 12.0 | 0.005 |
| TGF beta (ng/mL) | 25.0 ± 5.0 | 88.0 ± 22.0 | 0.005 |
| M-30 (U/L) | 80.0 ± 25.0 | 159.0 ± 37.0 | <0.0001 |
| M-65 (U/L) | 120.0 ± 60.0 | 476.0 ± 118.0 | <0.0001 |
| Spearman Correlation | M30 | M65 | TNFα | TGFβ |
|---|---|---|---|---|
| lobular inflammation 0–3 | 0.506 * | 0.489 * | 0.441 * | 0.159 |
| ballooning 0–2 | 0.419 * | 0.449 * | 0.557 ** | 0.136 |
| steatohepatitis 0–2 | 0.340 | 0.510 * | 0.466 * | −0.044 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuman, M.G.; Cohen, L.B. Inflammation and Liver Cell Death in Patients with Hepatitis C Viral Infection. Curr. Issues Mol. Biol. 2021, 43, 2022-2035. https://doi.org/10.3390/cimb43030139
Neuman MG, Cohen LB. Inflammation and Liver Cell Death in Patients with Hepatitis C Viral Infection. Current Issues in Molecular Biology. 2021; 43(3):2022-2035. https://doi.org/10.3390/cimb43030139
Chicago/Turabian StyleNeuman, Manuela G., and Lawrence B. Cohen. 2021. "Inflammation and Liver Cell Death in Patients with Hepatitis C Viral Infection" Current Issues in Molecular Biology 43, no. 3: 2022-2035. https://doi.org/10.3390/cimb43030139
APA StyleNeuman, M. G., & Cohen, L. B. (2021). Inflammation and Liver Cell Death in Patients with Hepatitis C Viral Infection. Current Issues in Molecular Biology, 43(3), 2022-2035. https://doi.org/10.3390/cimb43030139