
Repurposing of the Drug Tezosentan for Cancer Therapy



Abstract
1. Introduction
2. Structure of Tezosentan and Mechanism of Action
3. Clinical Use against Cancer
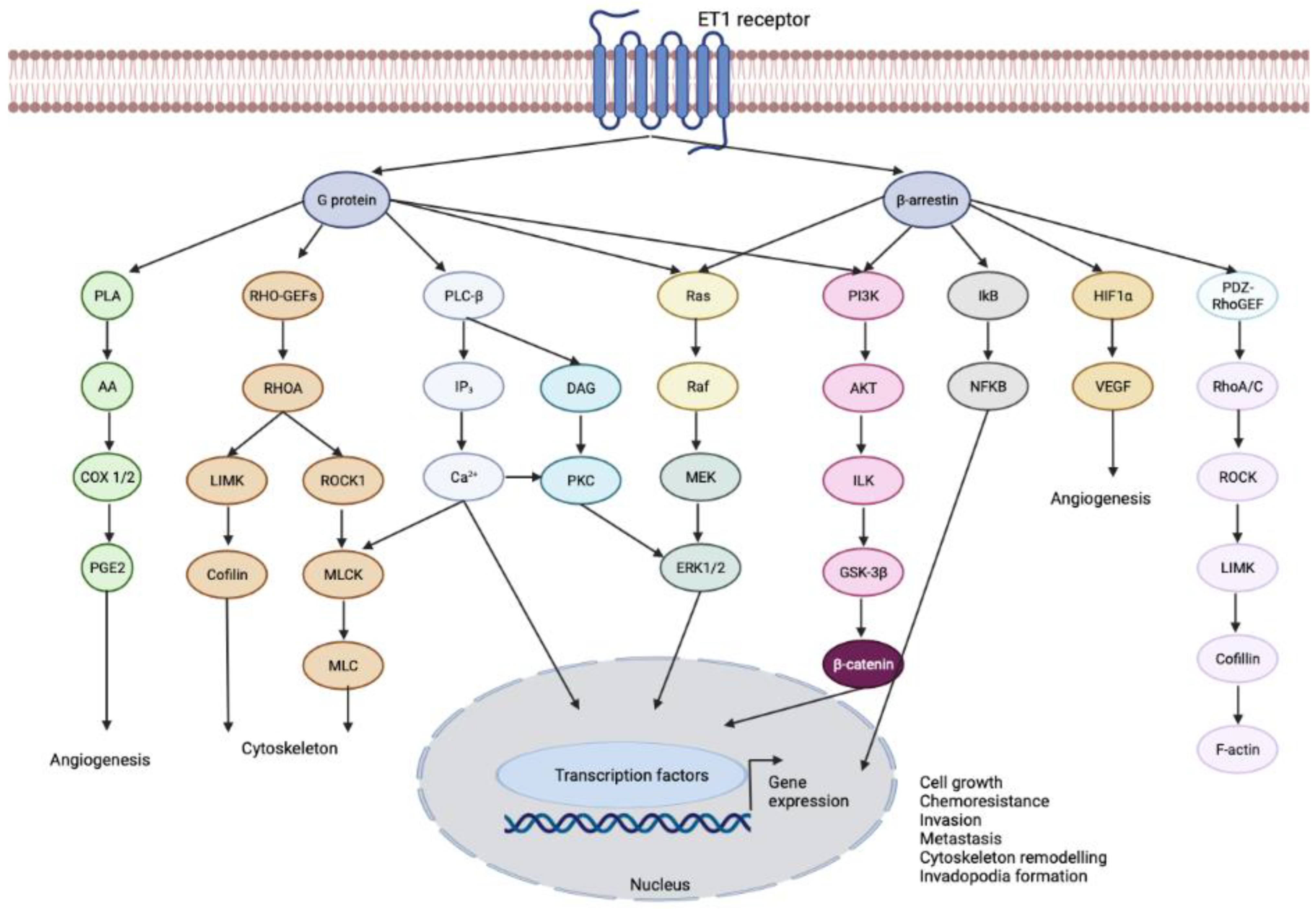
3.1. Endothelin-1 Receptor
3.2. Endothelin Receptor Type A
ATP-Binding Cassette Subfamily B Member 1 (ABCB1) Protein
3.3. Endothelin Receptor Type B
Nitric Oxide (NO) Release
3.4. METAP1 Methionyl Aminopeptidase 1
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell. Int. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Cho, W.C.S. Drug Repurposing for Cancer Therapy in the Era of Precision Medicine. Curr. Mol. Pharmacol. 2022, 15, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Khataniar, A.; Pathak, U.; Rajkhowa, S.; Jha, A.N. A Comprehensive Review of Drug Repurposing Strategies against Known Drug Targets of COVID-19. COVID 2022, 2, 148–167. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal. Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Sahragardjoonegani, B.; Beall, R.F.; Kesselheim, A.S.; Hollis, A. Repurposing existing drugs for new uses: A cohort study of the frequency of FDA-granted new indication exclusivities since 1997. J. Pharm. Policy Pract. 2021, 14, 3. [Google Scholar] [CrossRef]
- Ribeiro, E.; Costa, B.; Vasques-Nóvoa, F.; Vale, N. In Vitro Drug Repurposing: Focus on Vasodilators. Cells 2023, 12, 671. [Google Scholar] [CrossRef]
- Wang, R.; Dashwood, R.H. Endothelins and their receptors in cancer: Identification of therapeutic targets. Pharmacol. Res. 2011, 63, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; Clozel, M.; van Giersbergen, P.L. Pharmacokinetics and pharmacodynamics of tezosentan, an intravenous dual endothelin receptor antagonist, following chronic infusion in healthy subjects. Br. J. Clin. Pharmacol. 2002, 53, 355–362. [Google Scholar] [CrossRef]
- Cheng, J.W. Tezosentan in the management of decompensated heart failure. Cardiol. Rev. 2005, 13, 28–34. [Google Scholar] [CrossRef]
- Schalcher, C.; Cotter, G.; Reisin, L.; Bertel, O.; Kobrin, I.; Guyene, T.T.; Kiowski, W. The dual endothelin receptor antagonist tezosentan acutely improves hemodynamic parameters in patients with advanced heart failure. Am. Heart J. 2001, 142, 340–349. [Google Scholar] [CrossRef]
- Lebrec, D.; Bosch, J.; Jalan, R.; Dudley, F.J.; Jessic, R.; Moreau, R.; Garcia-Pagan, J.C.; Mookerjee, R.P.; Chiossi, E.; Van Giersbergen, P.L.; et al. Hemodynamics and pharmacokinetics of tezosentan, a dual endothelin receptor antagonist, in patients with cirrhosis. Eur. J. Clin. Pharmacol. 2012, 68, 533–541. [Google Scholar] [CrossRef]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.; Loizidou, M.; Taylor, I. Endothelin-1: A multifunctional molecule in cancer. Br. J. Cancer 2003, 88, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, M.; Yamaguchi, K.; Nagasaki, K.; Hayashi, C.; Suzaki, A.; Hori, S.; Handa, S.; Nakamura, Y.; Abe, K. Production of endothelin in human cancer cell lines. Cancer Res. 1990, 50, 3257–3261. [Google Scholar] [PubMed]
- Nakamuta, M.; Ohashi, M.; Tabata, S.; Tanabe, Y.; Goto, K.; Naruse, M.; Naruse, K.; Hiroshige, K.; Nawata, H. High plasma concentrations of endothelin-like immunoreactivities in patients with hepatocellular carcinoma. Am. J. Gastroenterol. 1993, 88, 248–252. [Google Scholar]
- Nelson, J.B.; Hedican, S.P.; George, D.J.; Reddi, A.H.; Piantadosi, S.; Eisenberger, M.A.; Simons, J.W. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat. Med. 1995, 1, 944–949. [Google Scholar] [CrossRef]
- Ferrari-Bravo, A.; Franciosi, C.; Lissoni, P.; Fumagalli, L.; Uggeri, F. Effects of oncological surgery on endothelin-1 secretion in patients with operable gastric cancer. Int. J. Biol. Mrk. 2000, 15, 56–57. [Google Scholar] [CrossRef]
- Morbidelli, L.; Orlando, C.; Maggi, C.A.; Ledda, F.; Ziche, M. Proliferation and migration of endothelial cells is promoted by endothelins via activation of ETB receptors. Am. J. Physiol. 1995, 269, H686–H695. [Google Scholar] [CrossRef]
- Alberts, G.F.; Peifley, K.A.; Johns, A.; Kleha, J.F.; Winkles, J.A. Constitutive endothelin-1 overexpression promotes smooth muscle cell proliferation via an external autocrine loop. J. Biol. Chem. 1994, 269, 10112–10118. [Google Scholar] [CrossRef] [PubMed]
- Salani, D.; Di Castro, V.; Nicotra, M.R.; Rosanò, L.; Tecce, R.; Venuti, A.; Natali, P.G.; Bagnato, A. Role of endothelin-1 in neovascularization of ovarian carcinoma. Am. J. Pathol. 2000, 157, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Spinella, F.; Rosanò, L. Emerging role of the endothelin axis in ovarian tumor progression. Endocr. Relat. Cancer 2005, 12, 761–772. [Google Scholar] [CrossRef]
- Rich, S.; McLaughlin, V.V. Endothelin Receptor Blockers in Cardiovascular Disease. Circulation 2003, 108, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.B.; Chan-Tack, K.; Hedican, S.P.; Magnuson, S.R.; Opgenorth, T.J.; Bova, G.S.; Simons, J.W. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996, 56, 663–668. [Google Scholar] [PubMed]
- Bagnato, A.; Salani, D.; Di Castro, V.; Wu-Wong, J.R.; Tecce, R.; Nicotra, M.R.; Venuti, A.; Natali, P.G. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: Evidence for an autocrine role in tumor growth. Cancer Res. 1999, 59, 720–727. [Google Scholar]
- Ali, H.; Dashwood, M.; Dawas, K.; Loizidou, M.; Savage, F.; Taylor, I. Endothelin receptor expression in colorectal cancer. J. Cardiovasc. Pharmacol. 2000, 36, S69–S71. [Google Scholar] [CrossRef]
- Gohji, K.; Kitazawa, S.; Tamada, H.; Katsuoka, Y.; Nakajima, M. Expression of endothelin receptor a associated with prostate cancer progression. J. Urol. 2001, 165, 1033–1036. [Google Scholar] [CrossRef]
- Akhavan, A.; McHugh, K.H.; Guruli, G.; Bies, R.R.; Zamboni, W.C.; Strychor, S.A.; Nelson, J.B.; Pflug, B.R. Endothelin receptor A blockade enhances taxane effects in prostate cancer. Neoplasia 2006, 8, 725–732. [Google Scholar] [CrossRef]
- Asham, E.; Shankar, A.; Loizidou, M.; Fredericks, S.; Miller, K.; Boulos, P.B.; Burnstock, G.; Taylor, I. Increased endothelin-1 in colorectal cancer and reduction of tumour growth by ET(A) receptor antagonism. Br. J. Cancer 2001, 85, 1759–1763. [Google Scholar] [CrossRef]
- Shankar, A.; Loizidou, M.; Aliev, G.; Fredericks, S.; Holt, D.; Boulos, P.B.; Burnstock, G.; Taylor, I. Raised endothelin 1 levels in patients with colorectal liver metastases. Br. J. Surg. 1998, 85, 502–506. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Q.; Zhang, Y.; Sun, L.; Xiao, H.; Luo, B. Epstein-Barr Virus Regulates Endothelin-1 Expression through the ERK/FOXO1 Pathway in EBV-Associated Gastric Cancer. Microbiol. Spectr. 2023, 11, e0089822. [Google Scholar] [CrossRef]
- Kappes, L.; Amer, R.L.; Sommerlatte, S.; Bashir, G.; Plattfaut, C.; Gieseler, F.; Gemoll, T.; Busch, H.; Altahrawi, A.; Al-Sbiei, A.; et al. Ambrisentan, an endothelin receptor type A-selective antagonist, inhibits cancer cell migration, invasion, and metastasis. Sci. Rep. 2020, 10, 15931. [Google Scholar] [CrossRef] [PubMed]
- Carducci, M.A.; Padley, R.J.; Breul, J.; Vogelzang, N.J.; Zonnenberg, B.A.; Daliani, D.D.; Schulman, C.C.; Nabulsi, A.A.; Humerickhouse, R.A.; Weinberg, M.A.; et al. Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: A randomized, phase II, placebo-controlled trial. J. Clin. Oncol. 2003, 21, 679–689. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Sharma, V. Tumour Angiogenesis and Angiogenic Inhibitors: A Review. J. Clin. Diagn. Res. 2015, 9, Xe01–Xe05. [Google Scholar] [CrossRef]
- Arabanian, L.S.; Johansson, P.; Staffas, A.; Nilsson, T.; Rouhi, A.; Fogelstrand, L.; Palmqvist, L. The endothelin receptor type A is a downstream target of Hoxa9 and Meis1 in acute myeloid leukemia. Leuk. Res. 2018, 75, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I. Functional significance of genetic polymorphisms in P-glycoprotein (MDR1, ABCB1) and breast cancer resistance protein (BCRP, ABCG2). Drug. Metab. Pharmacokinet. 2012, 27, 85–105. [Google Scholar] [CrossRef]
- Fung, K.L.; Gottesman, M.M. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. Acta 2009, 1794, 860–871. [Google Scholar] [CrossRef]
- Tulsyan, S.; Mittal, R.D.; Mittal, B. The effect of ABCB1 polymorphisms on the outcome of breast cancer treatment. Pharmgenomics Pers. Med. 2016, 9, 47–58. [Google Scholar] [CrossRef]
- Zou, F.; Seike, M.; Noro, R.; Kunugi, S.; Kubota, K.; Gemma, A. Prognostic significance of ABCB1 in stage I lung adenocarcinoma. Oncol. Lett. 2017, 14, 313–321. [Google Scholar] [CrossRef]
- Lei, Z.N.; Teng, Q.X.; Wu, Z.X.; Ping, F.F.; Song, P.; Wurpel, J.N.D.; Chen, Z.S. Overcoming multidrug resistance by knockout of ABCB1 gene using CRISPR/Cas9 system in SW620/Ad300 colorectal cancer cells. MedComm (2020) 2021, 2, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Amunni, G.; Villanucci, A.; Boddi, V.; Taddei, G.L. Increased cyclooxygenase-2 (COX-2) and P-glycoprotein-170 (MDR1) expression is associated with chemotherapy resistance and poor prognosis. Analysis in ovarian carcinoma patients with low and high survival. Int. J. Gynecol. Cancer 2005, 15, 255–260. [Google Scholar] [CrossRef]
- Yakirevich, E.; Sabo, E.; Naroditsky, I.; Sova, Y.; Lavie, O.; Resnick, M.B. Multidrug resistance-related phenotype and apoptosis-related protein expression in ovarian serous carcinomas. Gynecol. Oncol. 2006, 100, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hao, J.; Wang, L.; Li, Y. Coexpression of invasive markers (uPA, CD44) and multiple drug-resistance proteins (MDR1, MRP2) is correlated with epithelial ovarian cancer progression. Br. J. Cancer 2009, 101, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Sedláková, I.; Laco, J.; Caltová, K.; Červinka, M.; Tošner, J.; Řezáč, A.; Špaček, J. Clinical significance of the resistance proteins LRP, Pgp, MRP1, MRP3, and MRP5 in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2015, 25, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Lötsch, D.; Pirker, C.; Mohr, T.; van Schoonhoven, S.; Boidol, B.; Lardeau, C.H.; Spitzwieser, M.; Szabó, P.; Heffeter, P.; et al. Acquired nintedanib resistance in FGFR1-driven small cell lung cancer: Role of endothelin-A receptor-activated ABCB1 expression. Oncotarget 2016, 7, 50161–50179. [Google Scholar] [CrossRef]
- Eltze, E.; Wild, P.J.; Wülfing, C.; Zwarthoff, E.C.; Burger, M.; Stoehr, R.; Korsching, E.; Hartmann, A. Expression of the endothelin axis in noninvasive and superficially invasive bladder cancer: Relation to clinicopathologic and molecular prognostic parameters. Eur. Urol. 2009, 56, 837–845. [Google Scholar] [CrossRef]
- Demunter, A.; De Wolf-Peeters, C.; Degreef, H.; Stas, M.; van den Oord, J.J. Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch. 2001, 438, 485–491. [Google Scholar] [CrossRef]
- Blouquit-Laye, S.; Regnier, A.; Beauchet, A.; Zimmermann, U.; Devillier, P.; Chinet, T. Expression of endothelin receptor subtypes in bronchial tumors. Oncol. Rep. 2010, 23, 457–463. [Google Scholar] [CrossRef]
- Eltze, E.; Bertolin, M.; Korsching, E.; Wülfing, P.; Maggino, T.; Lellé, R. Expression and prognostic relevance of endothelin-B receptor in vulvar cancer. Oncol. Rep. 2007, 18, 305–311. [Google Scholar] [CrossRef]
- Wuttig, D.; Zastrow, S.; Füssel, S.; Toma, M.I.; Meinhardt, M.; Kalman, K.; Junker, K.; Sanjmyatav, J.; Boll, K.; Hackermüller, J.; et al. CD31, EDNRB and TSPAN7 are promising prognostic markers in clear-cell renal cell carcinoma revealed by genome-wide expression analyses of primary tumors and metastases. Int. J. Cancer 2012, 131, E693–E704. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Sho, M.; Takayama, T.; Wakatsuki, K.; Matsumoto, S.; Migita, K.; Ito, M.; Hamada, K.; Nakajima, Y. Endothelin B receptor expression correlates with tumour angiogenesis and prognosis in oesophageal squamous cell carcinoma. Br. J. Cancer 2014, 110, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.; Tsipras, G.; Landázuri, N.; Costa, H.; Wilhelmi, V.; Scicluna, P.; Cui, H.L.; Mohammad, A.A.; Davoudi, B.; Shang, M.; et al. Overexpression of endothelin B receptor in glioblastoma: A prognostic marker and therapeutic target? BMC Cancer 2018, 18, 154. [Google Scholar] [CrossRef]
- Pao, M.M.; Tsutsumi, M.; Liang, G.; Uzvolgyi, E.; Gonzales, F.A.; Jones, P.A. The endothelin receptor B (EDNRB) promoter displays heterogeneous, site specific methylation patterns in normal and tumor cells. Hum. Mol. Genet. 2001, 10, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Halaka, M.; Hired, Z.A.; Rutledge, G.E.; Hedgepath, C.M.; Anderson, M.P.; St John, H.; Do, J.M.; Majmudar, P.R.; Walker, C.; Alawawdeh, A.; et al. Differences in Endothelin B Receptor Isoforms Expression and Function in Breast Cancer Cells. J. Cancer 2020, 11, 2688–2701. [Google Scholar] [CrossRef]
- Wei, F.; Ge, Y.; Li, W.; Wang, X.; Chen, B. Role of endothelin receptor type B (EDNRB) in lung adenocarcinoma. Thorac. Cancer 2020, 11, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.W.; Edvinsson, L.; Xu, C.B. Role of ERK/MAPK in endothelin receptor signaling in human aortic smooth muscle cells. BMC Cell. Biol. 2009, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Theodorescu, D. Permissive role of endothelin receptors in tumor metastasis. Life Sci. 2012, 91, 522–527. [Google Scholar] [CrossRef]
- Fang, D.; Leishear, K.; Nguyen, T.K.; Finko, R.; Cai, K.; Fukunaga, M.; Li, L.; Brafford, P.A.; Kulp, A.N.; Xu, X.; et al. Defining the Conditions for the Generation of Melanocytes from Human Embryonic Stem Cells. Stem Cells 2006, 24, 1668–1677. [Google Scholar] [CrossRef]
- Berger, Y.; Bernasconi, C.C.; Juillerat-Jeanneret, L. Targeting the endothelin axis in human melanoma: Combination of endothelin receptor antagonism and alkylating agents. Exp. Biol. Med. 2006, 231, 1111–1119. [Google Scholar]
- Kefford, R.; Beith, J.M.; Van Hazel, G.A.; Millward, M.; Trotter, J.M.; Wyld, D.K.; Kusic, R.; Shreeniwas, R.; Morganti, A.; Ballmer, A.; et al. A Phase II study of bosentan, a dual endothelin receptor antagonist, as monotherapy in patients with stage IV metastatic melanoma. Investig. New Drugs 2007, 25, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Spinella, F.; Rosanò, L.; Di Castro, V.; Decandia, S.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cells. Cancer Res. 2007, 67, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Lahav, R.; Heffner, G.; Patterson, P.H. An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 11496–11500. [Google Scholar] [CrossRef]
- Bagnato, A.; Rosanò, L.; Spinella, F.; Di Castro, V.; Tecce, R.; Natali, P.G. Endothelin B receptor blockade inhibits dynamics of cell interactions and communications in melanoma cell progression. Cancer Res. 2004, 64, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Huang, J.; Rockey, D.C. Endothelin-1 activates endothelial cell nitric-oxide synthase via heterotrimeric G-protein betagamma subunit signaling to protein jinase B/Akt. J. Biol. Chem. 2003, 278, 49929–49935. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S. Nitric oxide for cancer therapy. Future Sci. OA 2015, 1, Fso44. [Google Scholar] [CrossRef]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell. Res. 2002, 12, 311–320. [Google Scholar] [CrossRef]
- Sang, J.; Chen, Y.; Tao, Y. Nitric oxide inhibits gastric cancer cell growth through the modulation of the Akt pathway. Mol. Med. Rep. 2011, 4, 1163–1167. [Google Scholar] [CrossRef]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): Potential role of cyclin D1. Proc. Natl. Acad. Sci. USA 2001, 98, 3583–3588. [Google Scholar] [CrossRef]
- Huguenin, S.; Fleury-Feith, J.; Kheuang, L.; Jaurand, M.C.; Bolla, M.; Riffaud, J.P.; Chopin, D.K.; Vacherot, F. Nitrosulindac (NCX 1102): A new nitric oxide-donating non-steroidal anti-inflammatory drug (NO-NSAID), inhibits proliferation and induces apoptosis in human prostatic epithelial cell lines. Prostate 2004, 61, 132–141. [Google Scholar] [CrossRef]
- Huguenin, S.; Vacherot, F.; Kheuang, L.; Fleury-Feith, J.; Jaurand, M.C.; Bolla, M.; Riffaud, J.P.; Chopin, D.K. Antiproliferative effect of nitrosulindac (NCX 1102), a new nitric oxide-donating non-steroidal anti-inflammatory drug, on human bladder carcinoma cell lines. Mol. Cancer Ther. 2004, 3, 291–298. [Google Scholar] [CrossRef]
- Peña-Altamira, E.; Petazzi, P.; Contestabile, A. Nitric oxide control of proliferation in nerve cells and in tumor cells of nervous origin. Curr. Pharm. Des. 2010, 16, 440–450. [Google Scholar] [CrossRef]
- Donia, M.; Mijatovic, S.; Maksimovic-Ivanic, D.; Miljkovic, D.; Mangano, K.; Tumino, S.; Biondi, A.; Basile, F.; Al-Abed, Y.; Stosic-Grujicic, S.; et al. The novel NO-donating compound GIT-27NO inhibits in vivo growth of human prostate cancer cells and prevents murine immunoinflammatory hepatitis. Eur. J. Pharmacol. 2009, 615, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Maksimovic-Ivanic, D.; Mijatovic, S.; Mojic, M.; Miljkovic, D.; Timotijevic, G.; Fagone, P.; Caponnetto, S.; Al-Abed, Y.; McCubrey, J.; et al. In vitro and in vivo anticancer action of Saquinavir-NO, a novel nitric oxide-derivative of the protease inhibitor saquinavir, on hormone resistant prostate cancer cells. Cell. Cycle 2011, 10, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Van de Wouwer, M.; Couzinié, C.; Serrano-Palero, M.; González-Fernández, O.; Galmés-Varela, C.; Menéndez-Antolí, P.; Grau, L.; Villalobo, A. Activation of the BRCA1/Chk1/p53/p21(Cip1/Waf1) pathway by nitric oxide and cell cycle arrest in human neuroblastoma NB69 cells. Nitric Oxide 2012, 26, 182–191. [Google Scholar] [CrossRef]
- Sonveaux, P.; Jordan, B.F.; Gallez, B.; Feron, O. Nitric oxide delivery to cancer: Why and how? Eur. J. Cancer 2009, 45, 1352–1369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cook, T.; Alber, S.; Liu, K.; Kovesdi, I.; Watkins, S.K.; Vodovotz, Y.; Billiar, T.R.; Blumberg, D. Adenoviral gene transfer of the human inducible nitric oxide synthase gene enhances the radiation response of human colorectal cancer associated with alterations in tumor vascularity. Cancer Res. 2004, 64, 1386–1395. [Google Scholar] [CrossRef]
- Cook, T.; Wang, Z.; Alber, S.; Liu, K.; Watkins, S.C.; Vodovotz, Y.; Billiar, T.R.; Blumberg, D. Nitric oxide and ionizing radiation synergistically promote apoptosis and growth inhibition of cancer by activating p53. Cancer Res. 2004, 64, 8015–8021. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, X.; Zhang, F.; Gabelli, S.B.; Wang, R.; Zhang, Y.; Bhat, S.; Chen, X.; Furlani, M.; Amzel, L.M.; et al. Pyridinylpyrimidines selectively inhibit human methionine aminopeptidase-1. Bioorg Med. Chem. 2013, 21, 2600–2617. [Google Scholar] [CrossRef]
- Hu, X.; Addlagatta, A.; Matthews, B.W.; Liu, J.O. Identification of pyridinylpyrimidines as inhibitors of human methionine aminopeptidases. Angew. Chem. Int. Ed. Engl. 2006, 45, 3772–3775. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, V.; Fijałkowska, D.; Van Damme, P. Omics Assisted N-terminal Proteoform and Protein Expression Profiling On Methionine Aminopeptidase 1 (MetAP1) Deletion. Mol. Cell. Proteom. 2018, 17, 694–708. [Google Scholar] [CrossRef]
- Frottin, F.; Bienvenut, W.V.; Bignon, J.; Jacquet, E.; Vaca Jacome, A.S.; Van Dorsselaer, A.; Cianferani, S.; Carapito, C.; Meinnel, T.; Giglione, C. MetAP1 and MetAP2 drive cell selectivity for a potent anti-cancer agent in synergy, by controlling glutathione redox state. Oncotarget 2016, 7, 63306–63323. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Addlagatta, A.; Lu, J.; Matthews, B.W.; Liu, J.O. Elucidation of the function of type 1 human methionine aminopeptidase during cell cycle progression. Proc. Natl. Acad. Sci. USA 2006, 103, 18148–18153. [Google Scholar] [CrossRef] [PubMed]
- Mauriz, J.L.; Martín-Renedo, J.; García-Palomo, A.; Tuñón, M.J.; González-Gallego, J. Methionine aminopeptidases as potential targets for treatment of gastrointestinal cancers and other tumours. Curr. Drug. Targets 2010, 11, 1439–1457. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chong, C.R.; Shi, L.; Yoshimoto, T.; Sullivan, D.J., Jr.; Liu, J.O. Inhibitors of Plasmodium falciparum methionine aminopeptidase 1b possess antimalarial activity. Proc. Natl. Acad. Sci. USA 2006, 103, 14548–14553. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Type of Cancer | Clinical Trial | Status | Identifier Trial Number (https://clinicaltrials.gov) Accessed on 25 April 2023 |
|---|---|---|---|---|---|
| Atrasentan | ETA | Prostate | Phase III | Completed (2021) | NCT00134056 |
| Prostate | Phase III | Completed (2006) | NCT00036543 | ||
| Prostate | Phase III | Completed (2011) | NCT00046943 | ||
| Prostate | Phase II | Completed (2010) | NCT00181558 | ||
| Prostate | Phase III | Completed (2007) | NCT00036556 | ||
| Prostate | Phase II | Completed (2006) | NCT00038662 | ||
| Kidney | Phase II | Completed (2010) | NCT00039429 | ||
| Ovarian | Phase II | Terminated (2012) | NCT00653328 | ||
| Prostate | Phase II/III | Completed (2007) | NCT00127478 | ||
| Brain | Phase I | Completed (2009) | NCT00017264 | ||
| Zibotentan (ZD4054) | ETA | Prostate | Phase III | Completed (2016) | NCT00554229 |
| Prostate | Phase III | Terminated (2012) | NCT00626548 | ||
| Prostate | Phase II | Terminated (2019) | NCT01119118 | ||
| Prostate | Phase III | Terminated (2012) | NCT00617669 | ||
| Prostate | NA | Unknown | NCT01168141 | ||
| Breast | Phase II | Withdrawn | NCT01134497 | ||
| Lung | Phase II | Completed (2012) | NCT00745875 | ||
| Prostate | Phase II | Completed (2013) | NCT00090363 | ||
| Prostate | Phase I | Completed (2013) | NCT00314782 | ||
| Ovarian | Phase II | Terminated (2012) | NCT00929162 | ||
| Prostate | Phase II | Completed (2012) | NCT00055471 | ||
| Colorectal | Phase II | Completed (2014) | NCT01205711 | ||
| BQ788 | ETB | Melanoma | Phase I | Terminated (2015) | NCT02442466 |
| Bosentan | ETA/ETB | Pancreatic | Phase I | Recruiting | NCT04158635 |
| Solid tumor | Phase I | Recruiting | NCT05072106 | ||
| Macitentan | ETA/ETB | Glioblastoma | Phase I | Terminated (2018) | NCT02254954 |
| Glioblastoma | Phase I | Terminated (2016) | NCT01499251 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, E.; Vale, N. Repurposing of the Drug Tezosentan for Cancer Therapy. Curr. Issues Mol. Biol. 2023, 45, 5118-5131. https://doi.org/10.3390/cimb45060325
Ribeiro E, Vale N. Repurposing of the Drug Tezosentan for Cancer Therapy. Current Issues in Molecular Biology. 2023; 45(6):5118-5131. https://doi.org/10.3390/cimb45060325
Chicago/Turabian StyleRibeiro, Eduarda, and Nuno Vale. 2023. "Repurposing of the Drug Tezosentan for Cancer Therapy" Current Issues in Molecular Biology 45, no. 6: 5118-5131. https://doi.org/10.3390/cimb45060325
APA StyleRibeiro, E., & Vale, N. (2023). Repurposing of the Drug Tezosentan for Cancer Therapy. Current Issues in Molecular Biology, 45(6), 5118-5131. https://doi.org/10.3390/cimb45060325