
2-Methoxyestradiol as an Antiproliferative Agent for Long-Term Estrogen-Deprived Breast Cancer Cells
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture, Chemical Treatments, and Cell Morphology Analysis
2.3. Preparation of Total RNA and Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.4. Ponceau Staining and Western Blot Analysis
2.5. Cell Proliferation Analysis (MTS Assay) and Cell Counting
2.6. Cell Cycle Analysis Using Flow Cytometry
2.7. Gene Silencing
2.8. Data Analysis
3. Results
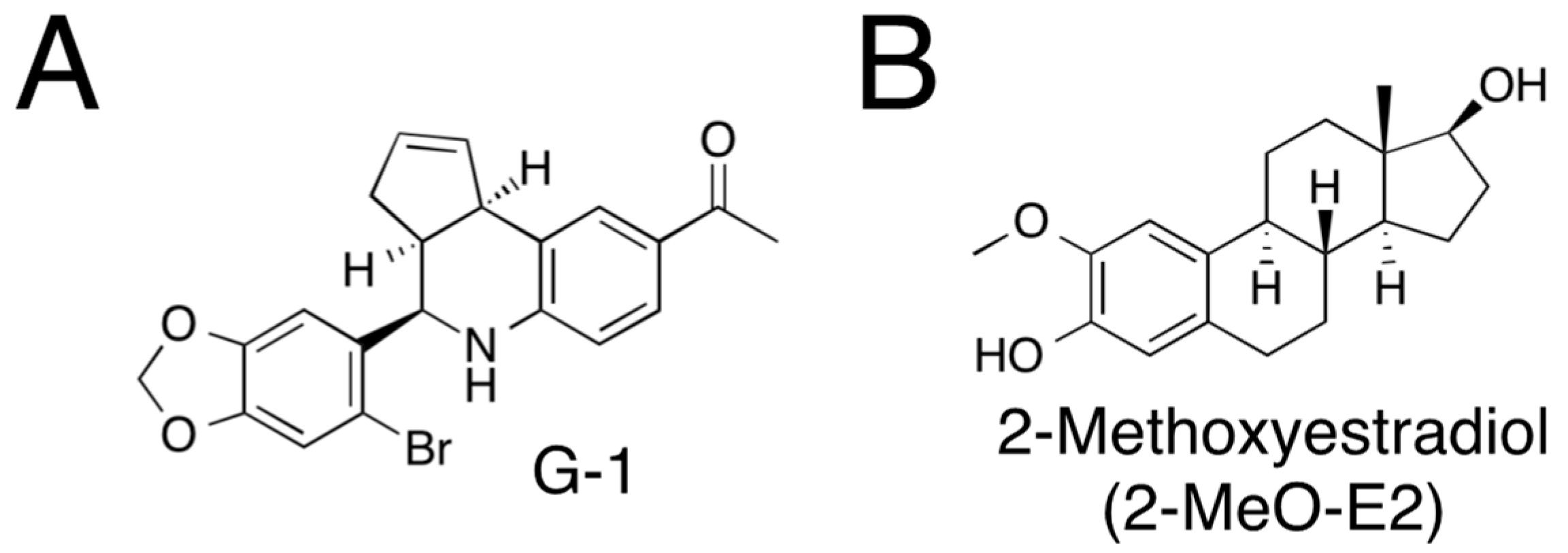
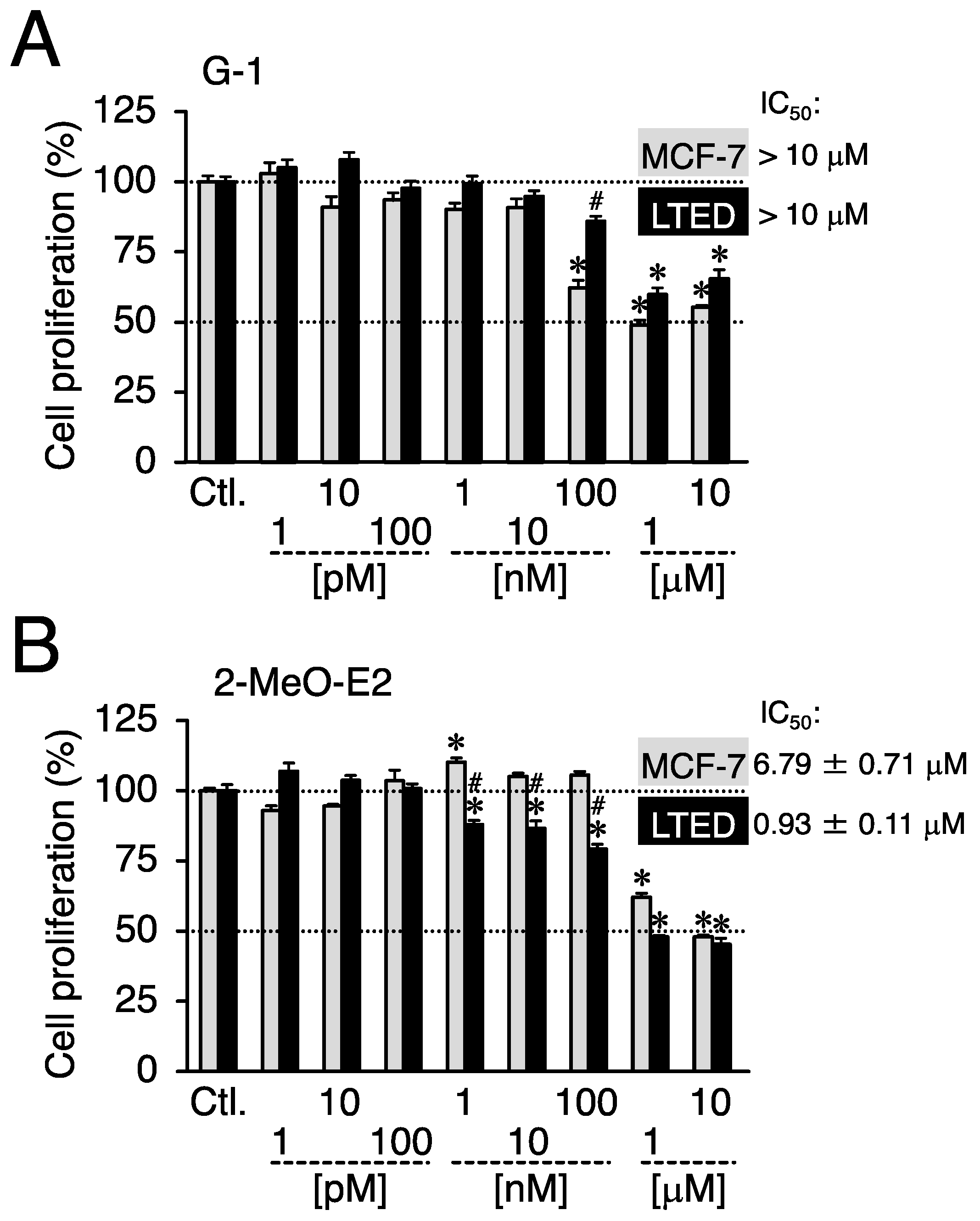
3.1. On LTED Cells, 2-MeO-E2 Exerts Selective Antiproliferation
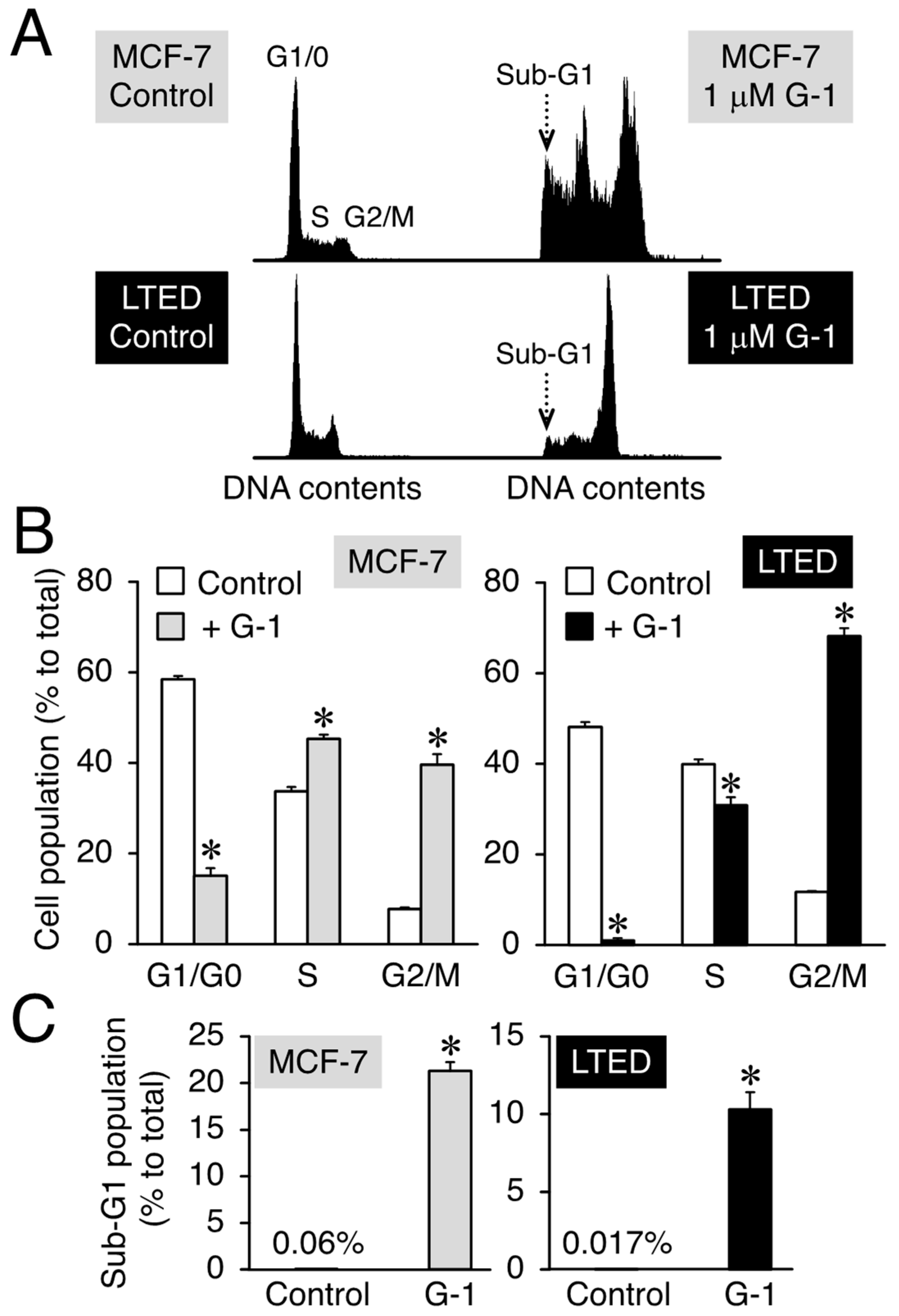
3.2. G-1 Arrests LTED Cells at the G2/M Phase with Accompanying Sub-G1
3.3. 2-MeO-E2 Arrests LTED Cells at the G2/M Phase without Accompanying Sub-G1
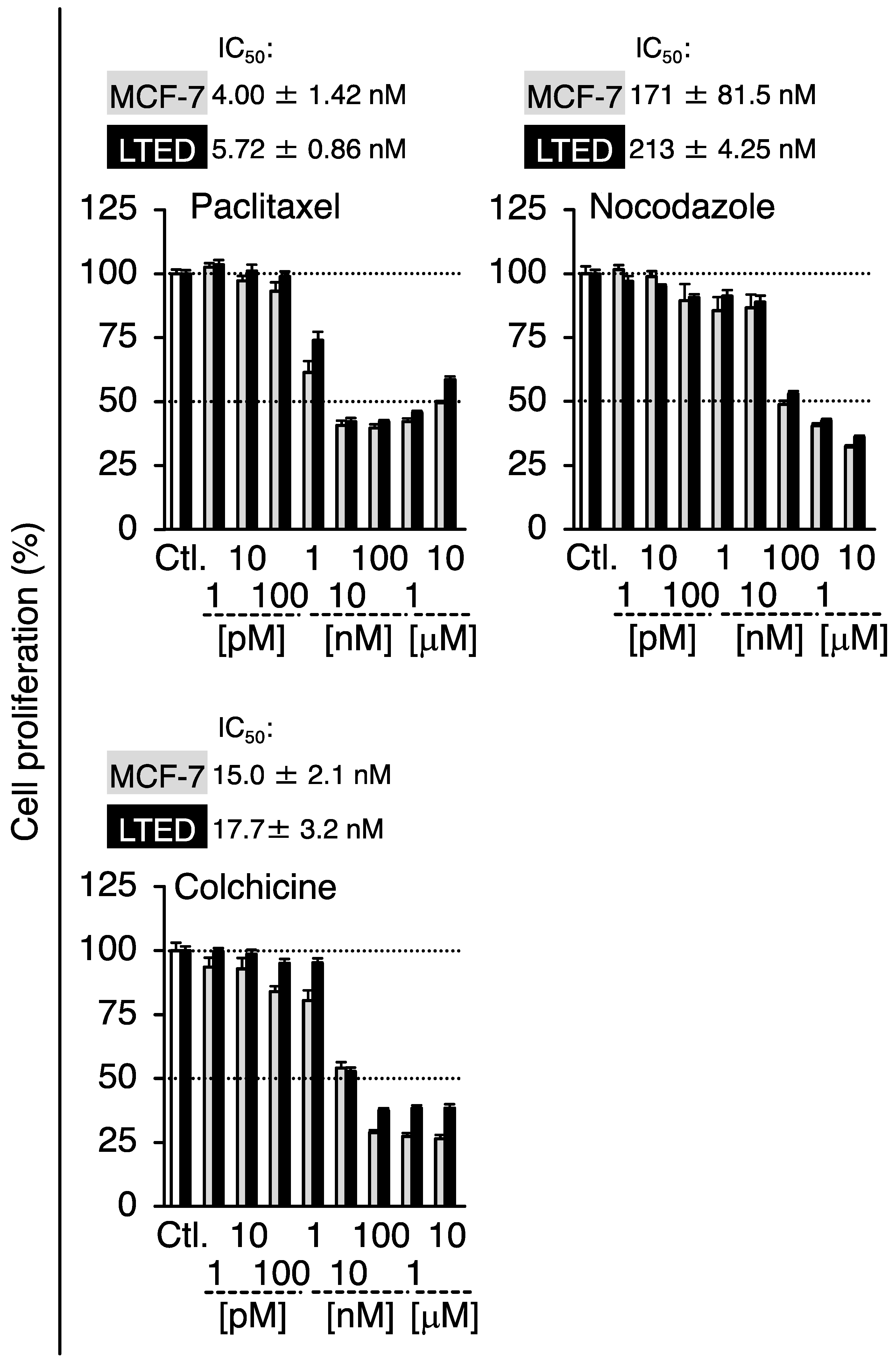
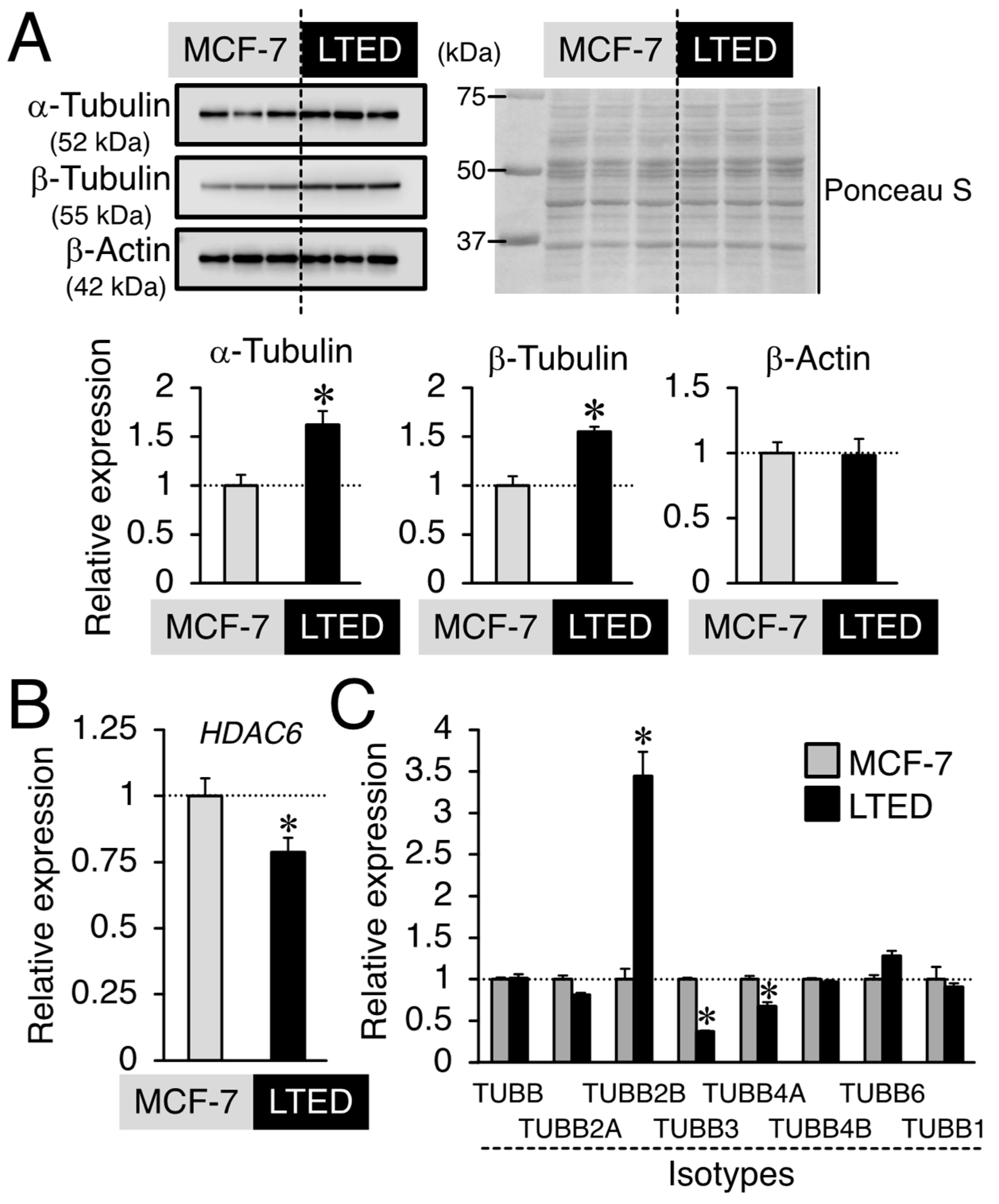
3.4. Expression Profiles of β-Tubulin and Its Isotype in MCF-7 and LTED Cells
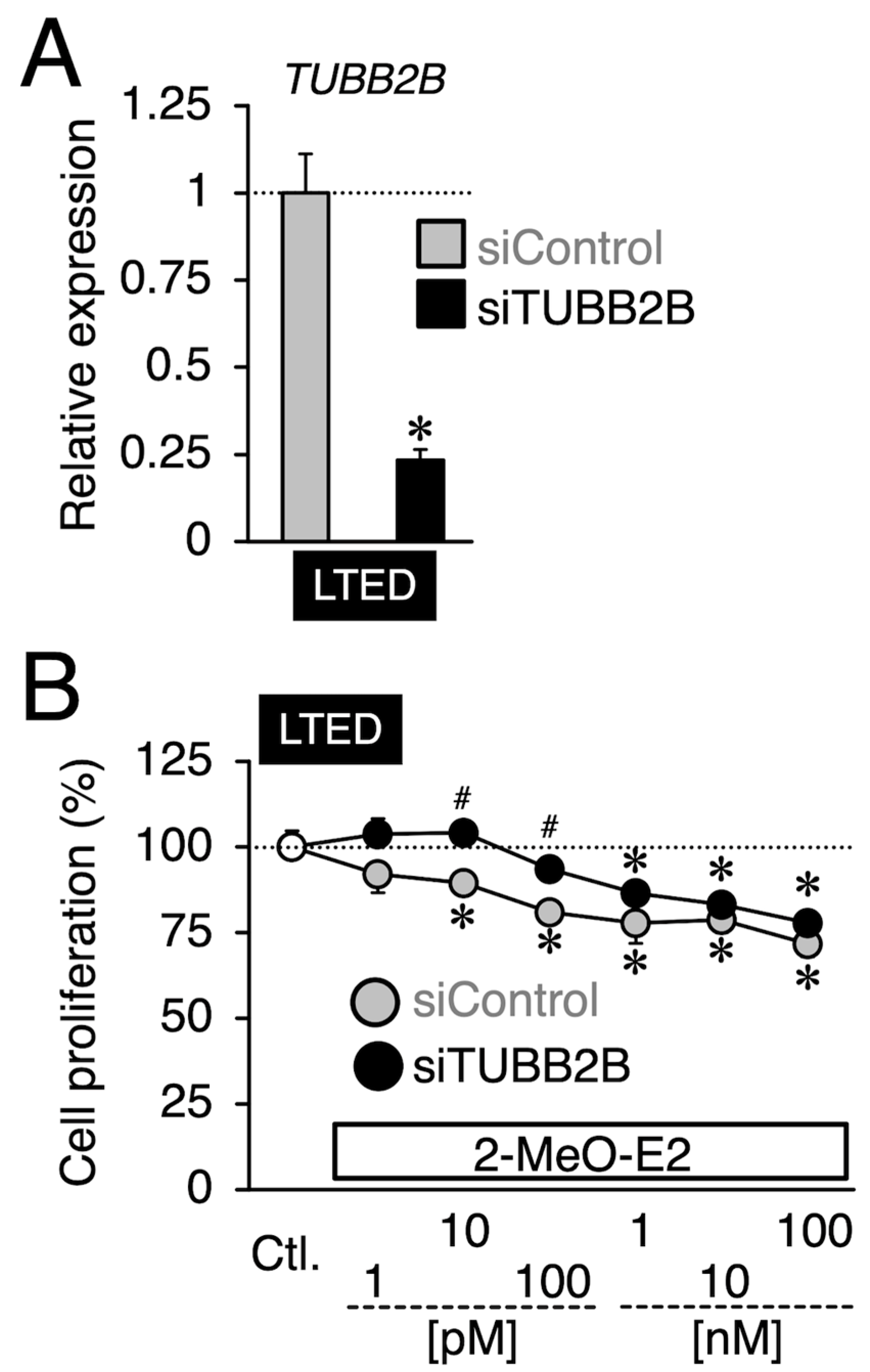
3.5. Effects of siTUBB2B on the Antiproliferation by 2-MeO-E2 in LTED Cells
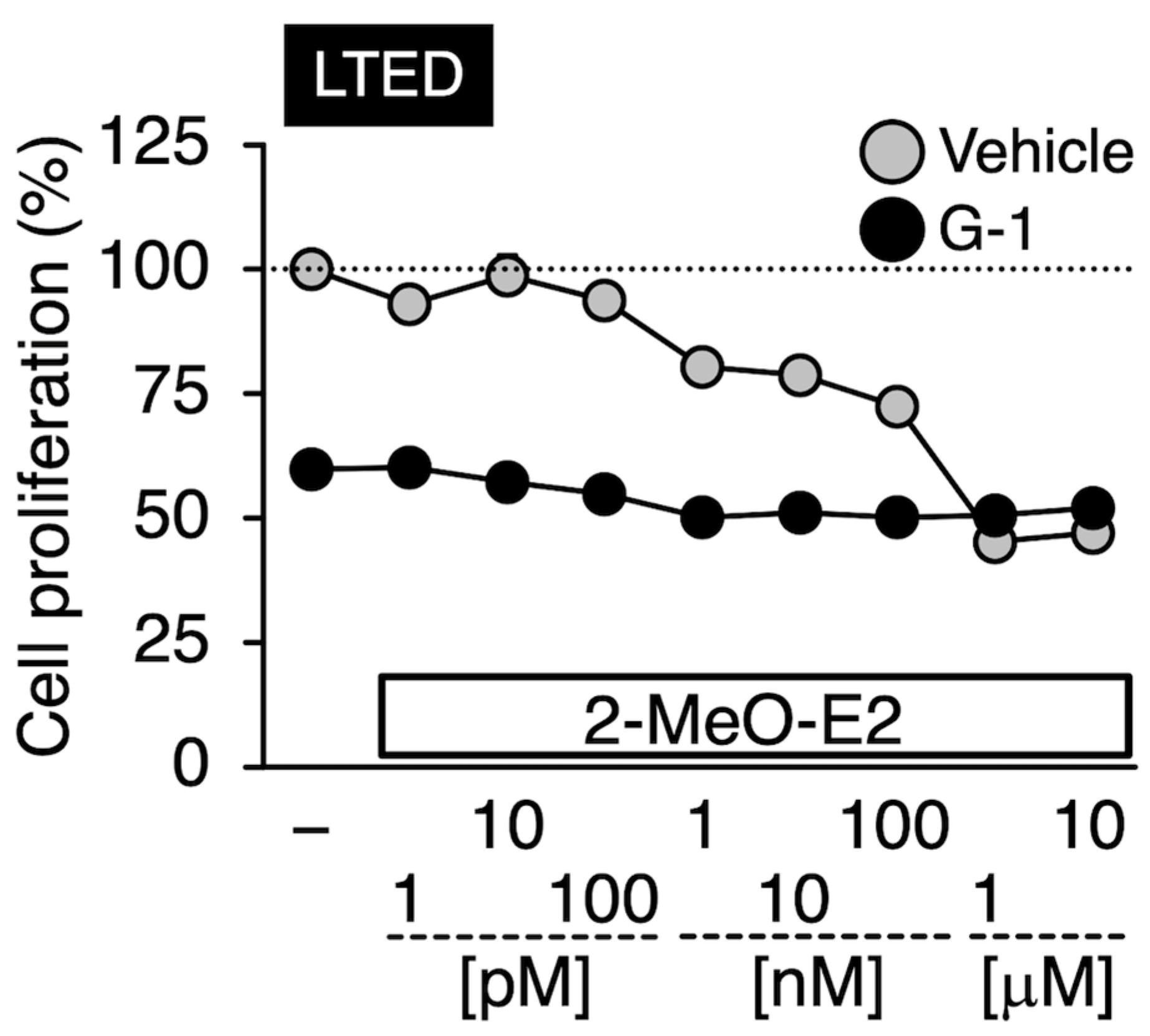
3.6. Effects of G-1 on the 2-MeO-E2-Mediated Antiproliferation in LTED Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Katzenellenbogen, B.S.; Kendra, K.L.; Norman, M.J.; Berthois, Y. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res. 1987, 47, 4355–4360. [Google Scholar] [PubMed]
- Jeng, M.H.; Shupnik, M.A.; Bender, T.P.; Westin, E.H.; Bandyopadhyay, D.; Kumar, R.; Masamura, S.; Santen, R.J. Estrogen receptor expression and function in long-term estrogen-deprived human breast cancer cells. Endocrinology 1998, 139, 4164–4174. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.W.; Martin, L.-A.; Johnston, S.R.D.; Ali, S.; Dowsett, M. Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. J. Steroid Biochem. Mol. Biol. 2002, 81, 333–341. [Google Scholar] [CrossRef]
- Martin, L.A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat. Commun. 2017, 8, 1865. [Google Scholar] [CrossRef]
- Hirao-Suzuki, M.; Takeda, S.; Kodama, Y.; Takiguchi, M.; Toda, A.; Ohara, M. Metalloestrogenic effects of cadmium are absent in long-term estrogen-deprived MCF-7 cells: Evidence for the involvement of constitutively activated estrogen receptor α and very low expression of G protein-coupled estrogen receptor 1. Toxicol. Lett. 2020, 319, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Ando, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef]
- Lucki, N.C.; Sewer, M.B. Genistein stimulates MCF-7 breast cancer cell growth by inducing acid ceramidase (ASAH1) gene expression. J. Biol. Chem. 2011, 286, 19399–19409. [Google Scholar] [CrossRef]
- Vivacqua, A.; Romeo, E.; De Marco, P.; De Francesco, E.M.; Abonante, S.; Maggiolini, M. GPER mediates the egr-1 expression induced by 17β-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Res. Treat. 2012, 133, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Scaling, A.L.; Prossnitz, E.R.; Hathaway, H.J. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 2014, 5, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Avino, S.; Pellegrino, M.; De Francesco, E.M.; De Marco, P.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Rigiracciolo, D.C.; Scarpelli, A.; et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis. 2015, 6, e1834. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; He, C.; Huang, C.; Hua, G.; Wang, Z.; Remmenga, S.W.; Rodabough, K.J.; Karpf, A.R.; Dong, J.; Davis, J.S.; et al. G-1 inhibits breast cancer cell growth via targeting colchicine-binding site of tubulin to interfere with microtubule assembly. Mol. Cancer Ther. 2017, 16, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lv, X.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis. 2013, 4, e869. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Lin, C.M.; Flynn, E.; Folkman, J.; Hamel, E. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc. Natl. Acad. Sci. USA 1994, 91, 3964–3968. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B. International union of basic and clinical pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.; Chakravarti, D.; Guttenplan, J.; Hart, E.; Ingle, J.; Jankowiak, R.; Muti, P.; Rogan, E.; Russo, J.; Santen, R.; et al. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta 2006, 1766, 63–78. [Google Scholar] [CrossRef]
- Mueck, A.O.; Seeger, H.; Huober, J. Chemotherapy of breast cancer-additive anticancerogenic effects by 2-methoxyestradiol? Life Sci. 2004, 75, 1205–1210. [Google Scholar] [CrossRef]
- Zhu, B.T.; Conney, A.H. Is 2-methoxyestradiol an endogenous estrogen metabolite that inhibits mammary carcinogenesis? Cancer Res. 1998, 58, 2269–2277. [Google Scholar]
- Ludueńa, R.F.; Shooter, E.M.; Wilson, L. Structure of the tubulin dimer. J. Biol. Chem. 1977, 252, 7006–7014. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Benetatos, C.A.; Colarusso, P.J.; Dexter, D.W.; Hudes, G.R. Altered β-tubulin isotype expression in paclitaxel-resistant human prostate carcinoma cells. Br. J. Cancer 1998, 77, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M.; Burkhart, C.A.; Horwitz, S.B. Antisense oligonucleotides to class III β-tubulin sensitize drug-resistant cells to taxol. Br. J. Cancer 1999, 80, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Hirao-Suzuki, M.; Yamagishi, Y.; Sugihara, T.; Kaneko, M.; Sakai, G.; Nakamura, T.; Hieda, Y.; Takiguchi, M.; Okada, M.; et al. Effects of the ethanol extract of Neopyropia Yezoensis, cultivated in the Seto Inland Sea (Setonaikai), on the viability of 10 human cancer cells including endocrine therapy-resistant breast cancer cells. Fundam. Toxicol. Sci. 2021, 8, 75–80. [Google Scholar] [CrossRef]
- Sakai, G.; Hirao-Suzuki, M.; Koga, T.; Kobayashi, T.; Kamishikiryo, J.; Tanaka, M.; Fujii, K.; Takiguchi, M.; Sugihara, N.; Toda, A.; et al. Perfluorooctanoic acid (PFOA) as a stimulator of estrogen receptor-negative breast cancer MDA-MB-231 cell aggressiveness: Evidence for involvement of fatty acid 2-hydroxylase (FA2H) in the stimulated cell migration. J. Toxicol. Sci. 2022, 47, 159–168. [Google Scholar] [CrossRef]
- Takeda, S.; Hirao-Suzuki, M.; Shindo, M.; Aramaki, H. (–)-Xanthatin as a killer of human breast cancer MCF-7 mammosphere cells: A comparative study with salinomycin. Curr. Issues Mol. Biol. 2022, 44, 3849–3858. [Google Scholar] [CrossRef]
- Duong, V.; Bret, C.; Altucci, L.; Mai, A.; Duraffourd, C.; Loubersac, J.; Harmand, P.O.; Bonnet, S.; Valente, S.; Maudelonde, T.; et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol. Cancer Res. 2008, 6, 1908–1919. [Google Scholar] [CrossRef]
- Narvi, E.; Jaakkola, K.; Winsel, S.; Oetken-Lindholm, C.; Halonen, P.; Kallio, L.; Kallio, M.J. Altered TUBB3 expression contributes to the epothilone response of mitotic cells. Br. J. Cancer 2013, 108, 82–90. [Google Scholar] [CrossRef]
- Saussede-Aim, J.; Matera, E.-L.; Ferlini, C.; Dumontet, C. β3-Tubulin is induced by estradiol in human breast carcinoma cells through an estrogen-receptor dependent pathway. Cell Motil. Cytoskelet. 2009, 66, 378–388. [Google Scholar] [CrossRef]
- Azumi, M.; Yoshie, M.; Nakachi, N.; Tsuru, A.; Kusama, K.; Tamura, K. Stathmin expression alters the antiproliferative effect of eribulin in leiomyosarcoma cells. J. Pharmacol. Sci. 2022, 150, 259–266. [Google Scholar] [CrossRef]
- Li, W.; Zhai, B.; Zhi, H.; Li, Y.; Jia, L.; Ding, C.; Zhang, B.; You, W. Association of ABCB1, β tubulin I, and III with multidrug resistance of MCF7/DOC subline from breast cancer cell line MCF7. Tumour Biol. 2014, 35, 8883–8891. [Google Scholar] [CrossRef] [PubMed]
- Hirao-Suzuki, M.; Takiguchi, M.; Yoshihara, S.; Takeda, S. Repeated exposure to 4-methyl-2,4-bis(4-hydroxyphenyl)pent-1-ene (MBP) accelerates ligand-independent activation of estrogen receptors in long-term estradiol-deprived MCF-7 cells. Toxicol. Lett. 2023, 378, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chen, Z.-J.; Zhang, K.-S.; Yang, X.-L.; Wu, Y.-M.; Chen, X.-H.; Huang, H.-B.; Liu, H.-L.; Cai, S.-H.; Du, J.; et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1428. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Gründker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205. [Google Scholar] [CrossRef]
- Weißenborn, C.; Ignatov, T.; Ochel, H.-J.; Costa, S.D.; Zenclussen, A.C.; Ignatova, Z.; Ignatov, A. GPER functions as a tumor suppressor in triple-negative breast cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 713–723. [Google Scholar] [CrossRef]
- Weißenborn, C.; Ignatov, T.; Poehlmann, A.; Wege, A.K.; Costa, S.D.; Zenclussen, A.C.; Ignatov, A. GPER functions as a tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 663–671. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Mills, J.C.; Stone, N.L.; Erhardt, J.; Pittman, R.N. Apoptotic membrane blebbing is regulated by myosin light vhain phosphorylation. J. Cell Biol. 1998, 140, 627–636. [Google Scholar] [CrossRef]
- van den Brand, A.D.; Villevoye, J.; Nijmeijer, S.M.; van den Berg, M.; van Duursen, M.B.M. Anti-tumor properties of methoxylated analogues of resveratrol in malignant MCF-7 but not in non-tumorigenic MCF-10A mammary epithelial cell lines. Toxicology 2019, 422, 35–43. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Azuma, K.; Urano, T.; Horie-Inoue, K.; Hayashi, S.; Sakai, R.; Ouchi, Y.; Inoue, S. Association of estrogen receptor α and histone deacetylase 6 causes rapid deacetylation of tubulin in breast cancer cells. Cancer Res. 2009, 69, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Omoto, Y.; Inoue, A.; Eguchi, H.; Kobayashi, Y.; Kurosumi, M.; Saji, S.; Suemasu, K.; Okazaki, T.; Nakachi, K.; et al. Prediction of prognosis of estrogen receptor-positive breast cancer with combination of selected estrogen-regulated genes. Cancer Sci. 2004, 95, 496–502. [Google Scholar] [CrossRef] [PubMed]
- El-Zein, R.; Thaiparambil, J.; Abdel-Rahman, S.Z. 2-Methoxyestradiol sensitizes breast cancer cells to taxanes by targeting centrosomes. Oncotarget 2020, 11, 4479–4489. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Wang, X.; Shi, J.; Huang, M.; Chen, J.; Dan, J.; Tang, Y.; Guo, Z.; He, X.; Zhao, Q. TUBB2B facilitates progression of hepatocellular carcinoma by regulating cholesterol metabolism through targeting HNF4A/CYP27A1. Cell Death Dis. 2023, 14, 179. [Google Scholar] [CrossRef]
- Mooberry, S.L. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr. Opin. Oncol. 2003, 15, 425–430. [Google Scholar] [CrossRef]
- Sutherland, T.E.; Schuliga, M.; Harris, T.; Eckhardt, B.L.; Anderson, R.L.; Quan, L.; Stewart, A.G. 2-Methoxyestradiol is an estrogen receptor agonist that supports tumor growth in murine xenograft models of breast cancer. Clin. Cancer Res. 2005, 11, 1722–1732. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirao-Suzuki, M.; Kanameda, K.; Takiguchi, M.; Sugihara, N.; Takeda, S. 2-Methoxyestradiol as an Antiproliferative Agent for Long-Term Estrogen-Deprived Breast Cancer Cells. Curr. Issues Mol. Biol. 2023, 45, 7336-7351. https://doi.org/10.3390/cimb45090464
Hirao-Suzuki M, Kanameda K, Takiguchi M, Sugihara N, Takeda S. 2-Methoxyestradiol as an Antiproliferative Agent for Long-Term Estrogen-Deprived Breast Cancer Cells. Current Issues in Molecular Biology. 2023; 45(9):7336-7351. https://doi.org/10.3390/cimb45090464
Chicago/Turabian StyleHirao-Suzuki, Masayo, Koki Kanameda, Masufumi Takiguchi, Narumi Sugihara, and Shuso Takeda. 2023. "2-Methoxyestradiol as an Antiproliferative Agent for Long-Term Estrogen-Deprived Breast Cancer Cells" Current Issues in Molecular Biology 45, no. 9: 7336-7351. https://doi.org/10.3390/cimb45090464