

Identification of the Candidate mGlu2 Allosteric Modulator THRX-195518 through In Silico Method and Evaluation of Its Neuroprotective Potential against Glutamate-Induced Neurotoxicity in SH-SY5Y Cell Line
 , , ,
, , , 
Abstract
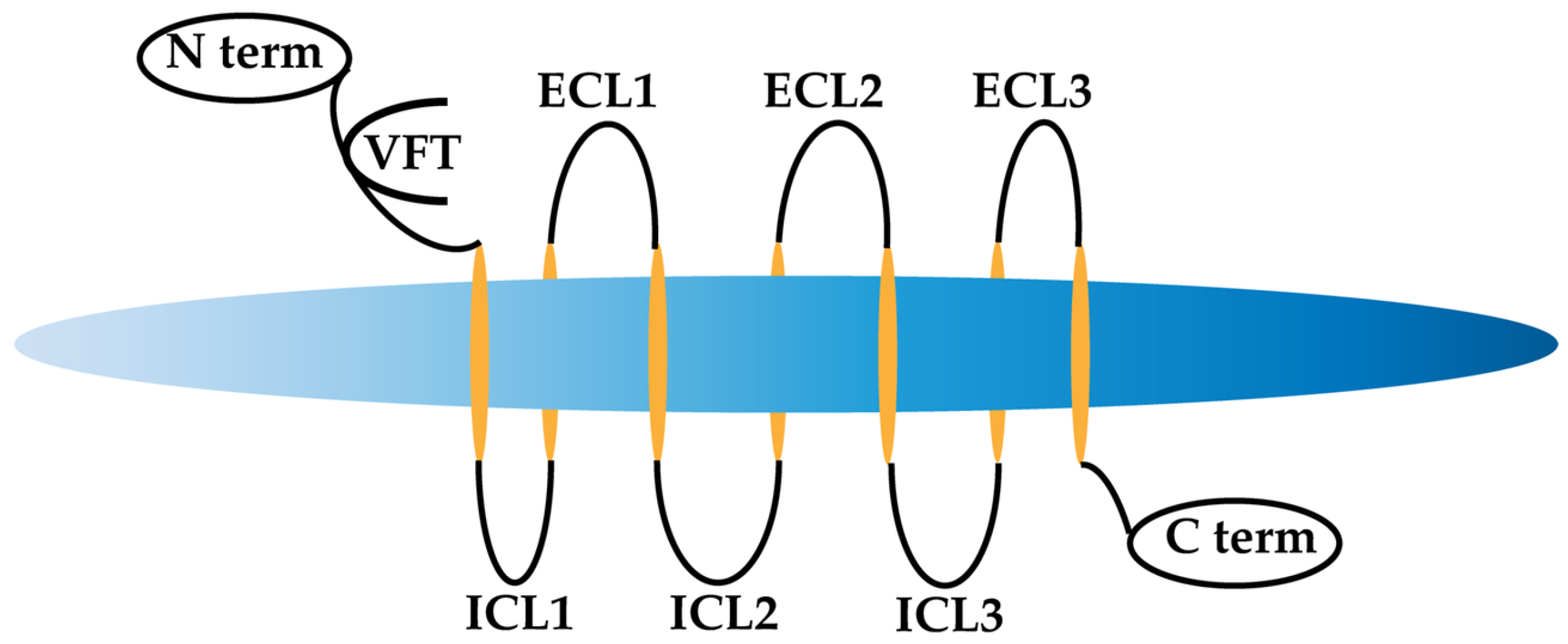
:1. Introduction
2. Materials and Method
2.1. In Silico Study
2.1.1. Protein Preparation
2.1.2. Ligand Preparation
2.1.3. Molecular Docking
2.1.4. Drug Likeness and Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Analysis
2.2. In Vitro Studies
2.2.1. Standard and Reagent
2.2.2. Cell Culture
2.2.3. Preparation of Drugs
2.2.4. Cell Treatment
2.2.5. 3-[4,5-Dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) Assay
2.3. Statistical Analysis
3. Results
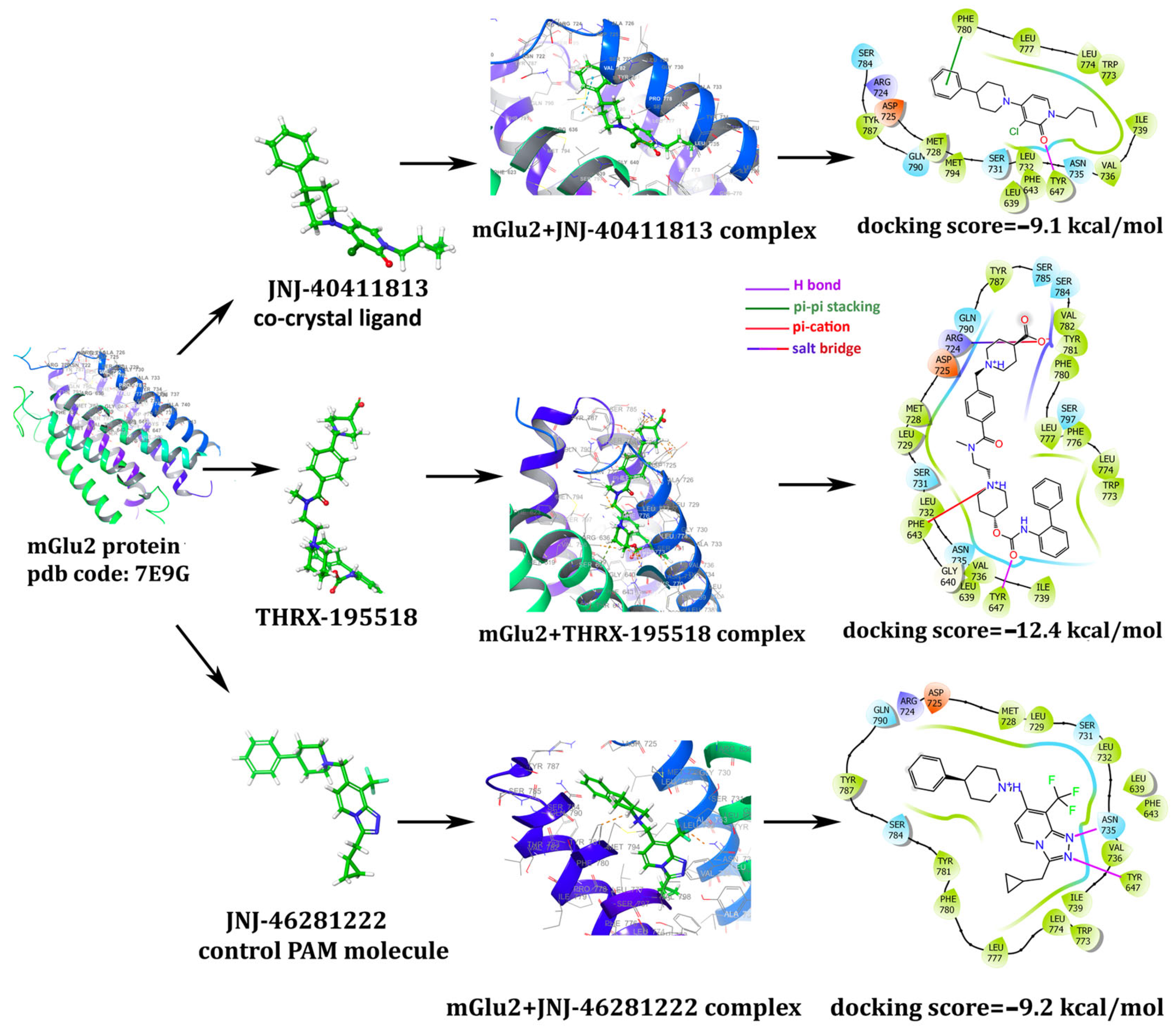
3.1. In Silico Studies: Library Generation, Virtual Screening, Molecular Docking, and ADMET
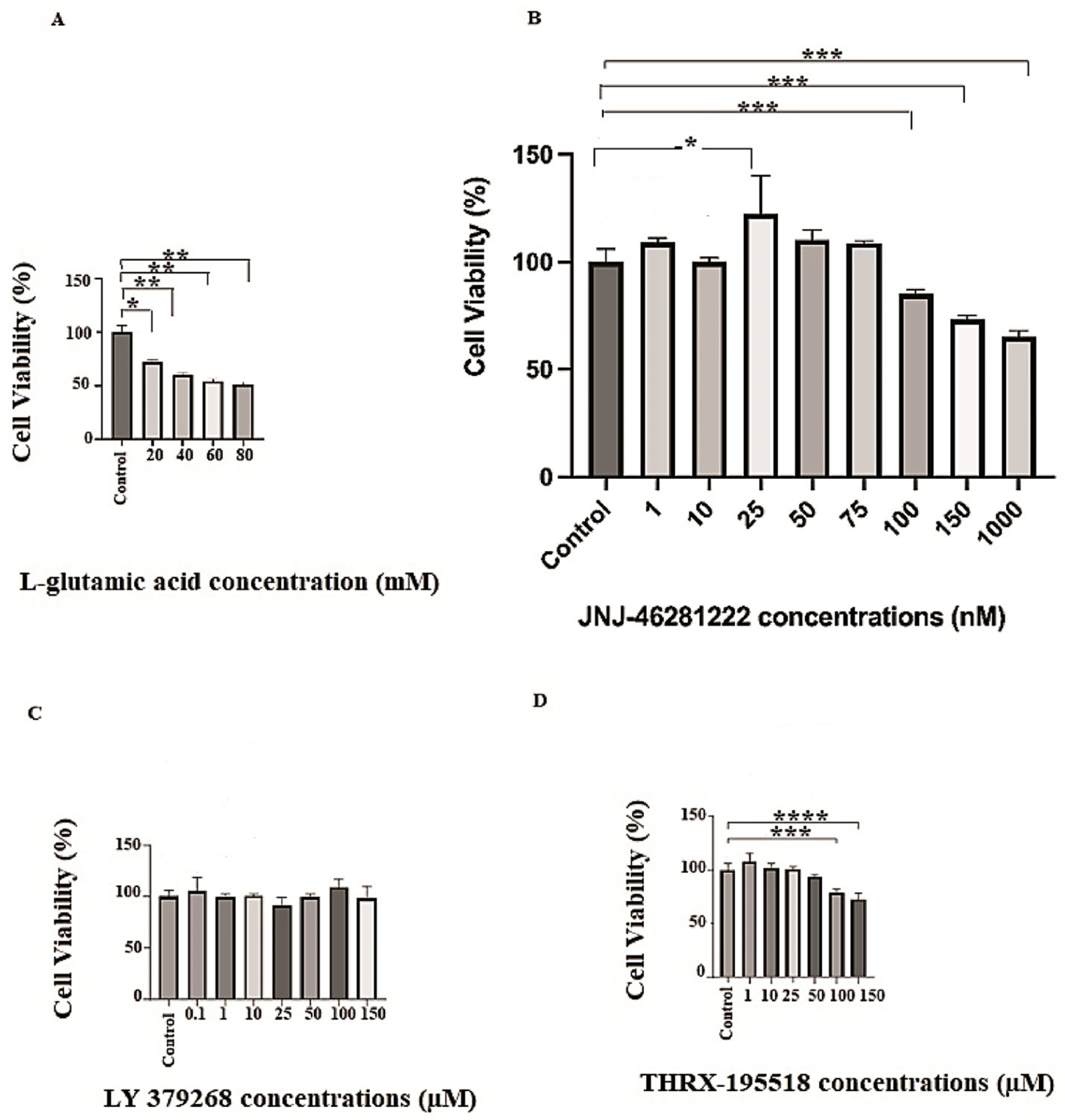
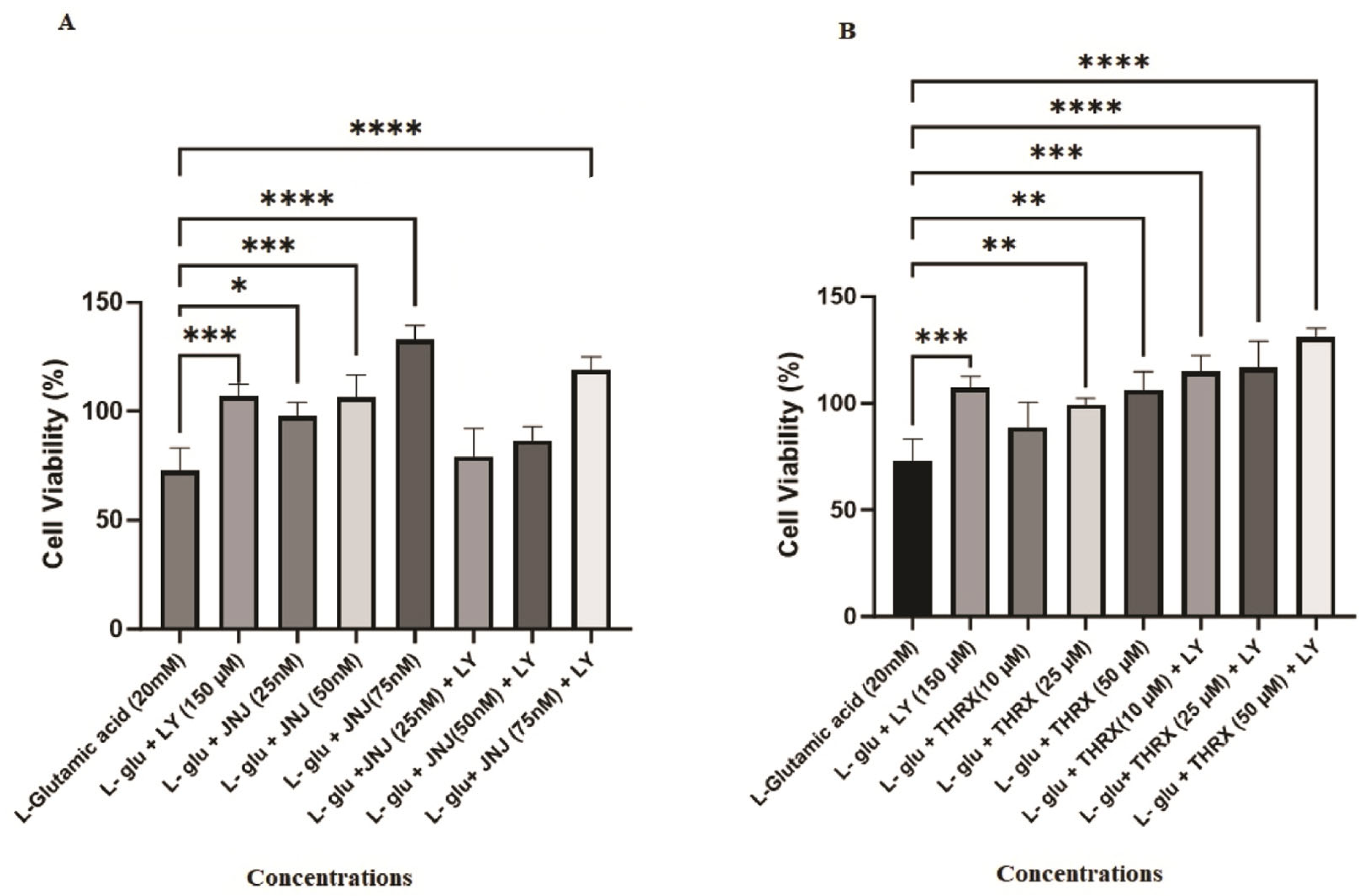
3.2. In Vitro Biological Evaluation: Effects of the Molecules on Cell Viability
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Canbolat, F. Role of metabotropic glutamate receptors in blocking excitotoxicity. In Health & Science 2022-III; EFE Academy publishing: Istanbul, Turkey, 2022; pp. 61–78. [Google Scholar]
- Yi, J.H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Melancon, B.J.; Hopkins, C.R.; Wood, M.R.; Emmitte, K.A.; Niswender, C.M.; Christopoulos, A.; Conn, P.J.; Lindsley, C.W. Allosteric modulation of seven transmembrane spanning receptors: Theory, practice, and opportunities for central nervous system drug discovery. J. Med. Chem. 2012, 55, 1445–1464. [Google Scholar] [CrossRef]
- Chun, L.; Zhang, W.H.; Liu, J.F. Structure and ligand recognition of class C GPCRs. Acta Pharmacol. Sin. 2012, 33, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Palucha, A.; Pilc, A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol. Ther. 2007, 115, 116–147. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, D.D.; Johnson, B.G.; Wright, R.A.; Salhoff, C.R.; Mayne, N.G.; Wu, S.; Cockerham, S.L.; Burnett, J.P.; Belegaje, R.; Bleakman, D.; et al. LY354740 is a potent and highly selective group II metabotropic glutamate receptor agonist in cells expressing human glutamate receptors. Neuropharmacology 1997, 36, 1–11. [Google Scholar] [CrossRef]
- Kingston, A.E.; O’Neill, M.J.; Lam, A.; Bales, K.R.; Monn, J.A.; Schoepp, D.D. Neuroprotection by metabotropic glutamate receptor agonists: LY354740, LY379268 and LY389795. Eur. J. Pharmacol. 1999, 377, 155–165. [Google Scholar] [CrossRef]
- Moldrich, R.X.; Jeffrey, M.; Talebi, A.; Beart, P.M.; Chapman, A.G.; Meldrum, B.S. Anti-epileptic activity of group II metabotropic glutamate receptor agonists (−)-2-oxa-4-aminobicyclo [3.1. 0] hexane-4, 6-dicarboxylate (LY379268) and (−)-2-thia-4-aminobicyclo [3.1. 0] hexane-4, 6-dicarboxylate (LY389795). Neuropharmacology 2001, 41, 8–18. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Copani, A.; D’Onofrio, M.; Di Iorio, P.; De Blasi, A.; Melchiorri, D.; Flor, P.J.; Nicoletti, F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow Metab. 2001, 21, 1013–1033. [Google Scholar] [CrossRef]
- Flor, P.J.; Battaglia, G.; Nicoletti, F.; Gasparini, F.; Bruno, V. Neuroprotective activity of metabotropic glutamate receptor ligands. In Molecular and Cellular Biology of Neuroprotection in the CNS; Springer: Berlin/Heidelberg, Germany, 2002; pp. 197–223. [Google Scholar] [CrossRef]
- Lavreysen, H.; Langlois, X.; Donck, L.V.; Nuñez, J.M.C.; Pype, S.; Lütjens, R.; Megens, A. Preclinical evaluation of the antipsychotic potential of the mGlu2-positive allosteric modulator JNJ-40411813. Pharmacol. Res. Perspect. 2015, 3, e00097. [Google Scholar] [CrossRef]
- Farinha, A.; Lavreysen, H.; Peeters, L.; Russo, B.; Masure, S.; Trabanco, A.A.; Cid, J.; Tresadern, G. Molecular determinants of positive allosteric modulation of the human metabotropic glutamate receptor 2. Br. J. Pharmacol. 2015, 172, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.L.; Pérez-Benito, L.; Tresadern, G.; Mulder-Krieger, T.; Biesmans, I.; Trabanco, A.A.; Cid, J.M.; Lavreysen, H.; Ijzerman, A.P.; Heitman, L.H. Molecular mechanism of positive allosteric modulation of the metabotropic glutamate receptor 2 by JNJ-46281222. Br. J. Pharmacol. 2016, 173, 588–600. [Google Scholar] [CrossRef]
- Pérez-Benito, L.; Doornbos, M.L.; Cordomí, A.; Peeters, L.; Lavreysen, H.; Pardo, L.; Tresadern, G. Molecular switches of allosteric modulation of the metabotropic glutamate 2 receptor. Structure 2017, 25, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, H.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc. Natl. Acad. Sci. USA 2004, 101, 7751–7756. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yao, H.H.; Wu, J.Y.; Yang, Y.J.; Ding, J.H.; Zhang, J.; Hu, G. Activation of Group II/III metabotropic glutamate receptors attenuates LPS-induced astroglial neurotoxicity via promoting glutamate uptake. J. Neurosci. Res. 2006, 84, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Greda, A.; Golda, S.; Korostynski, M.; Grygier, B.; Roman, A.; Pilc, A.; Lason, W. Neuroprotective effects of metabotropic glutamate receptor group II and III activators against MPP (+)-induced cell death in human neuroblastoma SH-SY5Y cells: The impact of cell differentiation state. Neuropharmacology 2014, 83, 36–53. [Google Scholar] [CrossRef]
- Naylor, S.; Kauppi, D.M.; Schonfeld, J.M. Therapeutic drug repurposing, repositioning and rescue. Drug Discov. 2015, 16, 57–72. [Google Scholar]
- Schrödinger. Small-Molecule Drug Discovery Suite (Version 2015-3); Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Schrödinger. Maestro (Version 2018-4); Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, 668–672. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2004, 58, 4066–4072. [Google Scholar] [CrossRef]
- Azzam, K.A. SwissADME and pkCSM webservers predictors: An integrated online platform for accurate and comprehensive predictions for in silico ADME/T properties of artemisinin and its derivatives. Complex Use Miner. Resour. 2023, 325, 14–21. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, 257–263. [Google Scholar] [CrossRef]
- Palanivel, V.; Gupta, V.; Mirshahvaladi, S.S.O.; Sharma, S.; Gupta, V.; Chitranshi, N.; Mirzai, M.; Graham, S.L.; Basavarajappa, D. Neuroprotective effects of neuropeptide Y on human neuroblastoma SH-SY5Y cells in glutamate excitotoxicity and ER stress conditions. Cells 2022, 11, 2–24. [Google Scholar] [CrossRef] [PubMed]
- Turati, J.; Ramírez, D.; Carniglia, L.; Saba, J.; Caruso, C.; Quarleri, J.; Durand, D.; Lasaga, M. Antioxidant and neuroprotective effects of mGlu3 receptor activation on astrocytes aged in vitro. Neurochem. Int. 2020, 140, 104837. [Google Scholar] [CrossRef]
- Wang, Y.; Song, J.H.; Denisova, J.V.; Park, W.M.; Fontes, J.D.; Belousov, A.B. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J. Neurosci. 2012, 32, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Durand, D.; Caruso, C.; Carniglia, L.; Lasaga, M. Metabotropic glutamate receptor 3 activation prevents nitric oxide-induced death in cultured rat astrocytes. J. Neurochem. 2010, 112, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.L.; Vermond, S.C.; Lavreysen, H.; Tresadern, G.; IJzerman, A.P.; Heitman, L.H. Impact of allosteric modulation: Exploring the binding kinetics of glutamate and other orthosteric ligands of the metabotropic glutamate receptor 2. Biochem. Pharmacol. 2018, 155, 356–365. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef]
- Bourdet, D.L.; Yeola, S.; Hegde, S.S.; Colson, P.J.; Barnes, C.N.; Borin, M.T. Revefenacin absorption, metabolism, and excretion in healthy subjects and pharmacological activity of its major metabolite. Drug Metab. Dispos. 2020, 48, 1312–1320. [Google Scholar] [CrossRef]
- de Lucas, A.I.; Vega, J.A.; Matesanz, E.; Linares, M.L.; García Molina, A.; Tresadern, G.; Lavreysen, H.; Trabanco, A.A.; Cid, J.M. Spiro-oxindole Piperidines and 3-(Azetidin-3-yl)-1 H-benzimidazol-2-ones as mGlu2 Receptor PAMs. ACS Med. Chem. Lett. 2019, 11, 303–308. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure–brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Matrisciano, F.; Zusso, M.; Panaccione, I.; Turriziani, B.; Caruso, A.; Iacovelli, L.; Togna, N.G.; Melchiorri, D.; Debetto, P.; Tatarelli, R.; et al. Synergism between fluoxetine and the mGlu2/3 receptor agonist, LY379268, in an in vitro model for antidepressant drug-induced neurogenesis. Neuropharmacology 2008, 54, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Abd-Elrahman, K.S.; Ferguson, S.S. Targeting mGluR2/3 for treatment of neurodegenerative and neuropsychiatric diseases. Pharmacol. Ther. 2022, 239, 108275. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Physicochemical Properties | Drug-likeness (Yes/No) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Molecular Formula | MW g/mol | TPSA Ų | H-Donor | H-Acceptor | Rotatable Bonds | MLOGP (Log Poctanol/water) | ESOL (Log S) | ||
| LY 379268 | C7H9NO5 | 187.15 | 109.85 | 3 | 4 | 2 | −3.65 | 1.83 | Yes |
| JNJ-46281222 | C23H25F3N4 | 414.47 | 33.43 | 0 | 4 | 5 | 4.30 | −5.87 | Yes |
| THRX-195518 | C35H42N4O5 | 598.73 | 102.42 | 2 | 6 | 10 | 3.24 | −4.54 | Yes |
| ADMET | |||||||||
| Molecule | GI Absorption (% Absorbed) | BBB Perm. (Log BB) | CNS Perm. (Log PS) | P450 Substrate | P450 Inhibitor | Carcinogenicity/ Ames Mutagenicity (Yes/No) | Predicted LD50 (mg/kg); Toxicity Class | ||
| LY 379268 | 23.39 | −0.56 | −3.59 | - | - | No/No | 50; 2 | ||
| JNJ-46281222 | 91.57 | 0.55 | −1.53 | CYP3A4, CYP2D6 | CYP1A2, CYP2C19, CYP2C9, CYP3A4 | No/No | 500; 4 | ||
| THRX-195518 | 62.54 | −1.10 | −2.47 | CYP3A4 | CYP2D6 | No/No | 700; 4 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canbolat, F.; Kantarci-Carsibasi, N.; Isik, S.; Shamshir, S.R.M.; Girgin, M. Identification of the Candidate mGlu2 Allosteric Modulator THRX-195518 through In Silico Method and Evaluation of Its Neuroprotective Potential against Glutamate-Induced Neurotoxicity in SH-SY5Y Cell Line. Curr. Issues Mol. Biol. 2024, 46, 788-807. https://doi.org/10.3390/cimb46010051
Canbolat F, Kantarci-Carsibasi N, Isik S, Shamshir SRM, Girgin M. Identification of the Candidate mGlu2 Allosteric Modulator THRX-195518 through In Silico Method and Evaluation of Its Neuroprotective Potential against Glutamate-Induced Neurotoxicity in SH-SY5Y Cell Line. Current Issues in Molecular Biology. 2024; 46(1):788-807. https://doi.org/10.3390/cimb46010051
Chicago/Turabian StyleCanbolat, Fadime, Nigar Kantarci-Carsibasi, Sevim Isik, Suhair Rami Mohammed Shamshir, and Münteha Girgin. 2024. "Identification of the Candidate mGlu2 Allosteric Modulator THRX-195518 through In Silico Method and Evaluation of Its Neuroprotective Potential against Glutamate-Induced Neurotoxicity in SH-SY5Y Cell Line" Current Issues in Molecular Biology 46, no. 1: 788-807. https://doi.org/10.3390/cimb46010051