
An Unusual Presentation of Novel Missense Variant in PAX6 Gene: NM_000280.4:c.341A>G, p.(Asn114Ser)
 , , , , ,
, , , , ,
Abstract
:1. Introduction
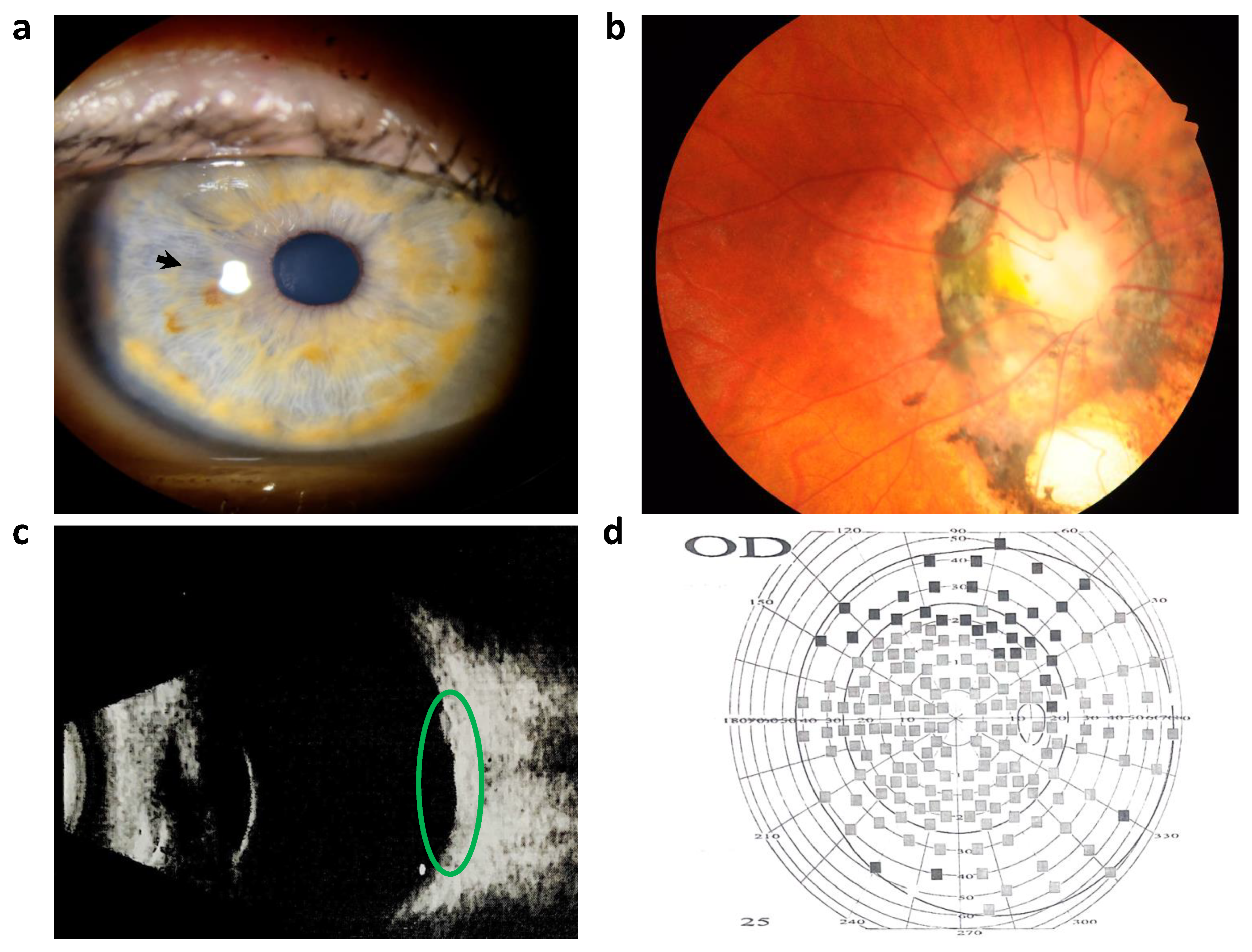
2. Case Presentation
Materials and Methods
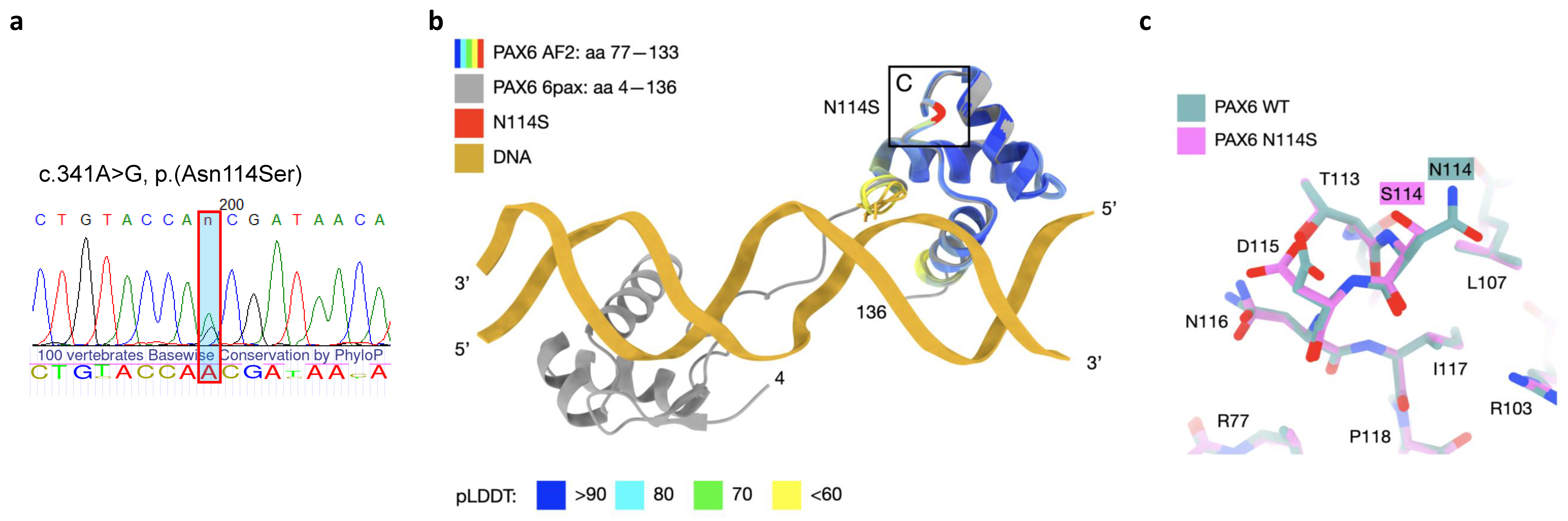
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hingorani, M.; Hanson, I.; van Heyningen, V. Aniridia. Eur. J. Hum. Genet. 2012, 20, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; White, I.M.; Hanson, I.M. Pax6 mutations: Genotype-phenotype correlations. BMC Genet. 2005, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Tadokoro, K.; Asaka, A.; Kawase, E.; Yamada, M. Mutations of the pax6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 2003, 72, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, I.; Izumi, H.; Ueno, S.; Hayashi, T.; Fujinami, K.; Tsunoda, K.; Iwata, T.; Kiuchi, Y.; Kondo, H. Functional characteristics of diverse pax6 mutations associated with isolated foveal hypoplasia. Genes 2023, 14, 1483. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, B.K.; Yang, Y.; Cveklova, K.; Cvekl, A. Functional properties of natural human pax6 and pax6(5a) mutants. Investig. Ophthalmol. Vis. Sci. 2004, 45, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Kelsey, K.J.; Wolfner, M.F.; Clark, A.G. Candidate genetic modifiers of retinitis pigmentosa identified by exploiting natural variation in drosophila. Hum. Mol. Genet. 2015, 25, 651–659. [Google Scholar] [CrossRef]
- Vu, V.; Verster, A.J.; Schertzberg, M.; Chuluunbaatar, T.; Spensley, M.; Pajkic, D.; Hart, G.T.; Moffat, J.; Fraser, A.G. Natural variation in gene expression modulates the severity of mutant phenotypes. Cell 2015, 162, 391–402. [Google Scholar] [CrossRef]
- Hussain, A.S.; Ali, S.R.; Mohammad, N.; Ali, N.; Ahmed, S.; Ahmad, T. Aniridia: A rare manifestation of congenital rubella syndrome. J. Ayub Med. Coll. Abbottabad 2019, 31, 131–133. [Google Scholar]
- Alibés, A.; Nadra, A.D.; De Masi, F.; Bulyk, M.L.; Serrano, L.; Stricher, F. Using protein design algorithms to understand the molecular basis of disease caused by protein-DNA interactions: The pax6 example. Nucleic Acids Res. 2010, 38, 7422–7431. [Google Scholar] [CrossRef]
- Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Kadyshev, V.V.; Sukhanova, N.V.; Minzhenkova, M.E.; Shilova, N.V.; Latyshova, A.A.; Ginter, E.K.; Kutsev, S.I.; et al. Epidemiology of pax6 gene pathogenic variants and expected prevalence of pax6-associated congenital aniridia across the russian federation: A nationwide study. Genes 2023, 14, 2041. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. Colabfold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. Ucsf chimerax: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, T.A.; Marakhonov, A.V.; Kutsev, S.I.; Zinchenko, R.A. Relative frequencies of pax6 mutational events in a russian cohort of aniridia patients in comparison with the world’s population and the human genome. Int. J. Mol. Sci. 2022, 23, 6690. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Khmelkova, D.N.; Pomerantseva, E.A.; Slepchenkov, A.V.; Zubashenko, N.A.; Mironova, I.V.; Kaimonov, V.S.; Polev, D.E.; Tsay, V.V.; Glotov, A.S.; et al. Expanding the russian allele frequency reference via cross-laboratory data integration: Insights from 7,452 exome samples. medRxiv 2022. [Google Scholar] [CrossRef]
- Huang, X.; Pearce, R.; Zhang, Y. Faspr: An open-source tool for fast and accurate protein side-chain packing. Bioinformatics 2020, 36, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. Dynamut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C. Conservation and prediction of solvent accessibility in protein families. Proteins 1994, 20, 216–226. [Google Scholar] [CrossRef]
- Xu, H.E.; Rould, M.A.; Xu, W.; Epstein, J.A.; Maas, R.L.; Pabo, C.O. Crystal structure of the human pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes. Dev. 1999, 13, 1263–1275. [Google Scholar] [CrossRef]
- Filatova, A.Y.; Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Zinchenko, R.A.; Skoblov, M.Y. Functional reassessment of pax6 single nucleotide variants by in vitro splicing assay. Eur. J. Hum. Genet. 2019, 27, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.A.; Hall, H.N.; Owen, L.J.; Livesey, B.J.; Hanson, I.M.; Adams, G.G.W.; Bodek, S.; Calvas, P.; Castle, B.; Clarke, M.; et al. Recurrent heterozygous pax6 missense variants cause severe bilateral microphthalmia via predictable effects on DNA-protein interaction. Genet. Med. 2020, 22, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.I.; Price, D.J. Pax6; a pleiotropic player in development. Bioessays 2002, 24, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Ruzickova, J.; Piatigorsky, J.; Kozmik, Z. Eye-specific expression of an ancestral jellyfish paxb gene interferes with pax6 function despite its conserved pax6/pax2 characteristics. Int. J. Dev. Biol. 2009, 53, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.; Mirzayans, F.; Footz, T.; Walter, M.; Levin, A.V. Elliptical anterior iris stromal defects associated with pax6 gene sequence changes. J. AAPOS 2008, 12, 340–343. [Google Scholar] [CrossRef]
- Shaham, O.; Menuchin, Y.; Farhy, C.; Ashery-Padan, R. Pax6: A multi-level regulator of ocular development. Prog. Retin. Eye Res. 2012, 31, 351–376. [Google Scholar] [CrossRef]
- Schedl, A.; Ross, A.; Lee, M.; Engelkamp, D.; Rashbass, P.; van Heyningen, V.; Hastie, N.D. Influence of pax6 gene dosage on development: Overexpression causes severe eye abnormalities. Cell 1996, 86, 71–82. [Google Scholar] [CrossRef]
- Aalfs, C.M.; Fantes, J.A.; Wenniger-Prick, L.J.; Sluijter, S.; Hennekam, R.C.; van Heyningen, V.; Hoovers, J.M. Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: Dose effect of the pax6 gene product? Am. J. Med. Genet. 1997, 73, 267–271. [Google Scholar] [CrossRef]
- Strobel, R.J.; Riccardi, V.M.; Ledbetter, D.H.; Hittner, H.M. Duplication 11p11.3 leads to 14.1 to meiotic crossing--over. Am. J. Med. Genet. 1980, 7, 15–20. [Google Scholar] [CrossRef]
- Schilter, K.F.; Reis, L.M.; Schneider, A.; Bardakjian, T.M.; Abdul-Rahman, O.; Kozel, B.A.; Zimmerman, H.H.; Broeckel, U.; Semina, E.V. Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin. Genet. 2013, 84, 473–481. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Eye Structure | Left Eye (Subatrophy) | Right Eye |
|---|---|---|
| Visual acuity | 0 | 0.1 |
| Cornea | Reduced in size and presented an old scar with vascularization | Transparent cornea, corneal thickness at pachymetry of 562 µm |
| Anterior chamber | Shallow and irregular | Depth was within normal range |
| Iris | Irregular shape of the pupil | Structurally changed in the upper outer quadrant, slightly hypopigmented with radial elliptical anterior iris stromal defects |
| Lens | n/a * | Transparent |
| Intraocular pressure | n/a * | 15 mm Hg |
| Optic nerve | Ophthalmoscopy was not possible | Enlarged optic nerve disc with a deep funnel-shaped excavation, clearly demarcated borders, and surrounded by a ring of damaged hyperpigmented choroid and retina, persistent hyaloid remnants |
| Macular zone | n/a * | Lacks differentiation and no reflex |
| Patient | This Study | Sharan et al. [25] |
|---|---|---|
| Age | 28 y.o. | 11 y.o. |
| Gender | Female | Female |
| Visual acuity (Snellen chart) | OD 20/200 OS proectio lucis certa | OD 20/240-20/480 OS 20/240-20/480 |
| Anterior segment | elliptical iris defects, bilateral iris stromal hypoplasia, corectopia | bilateral elliptical anterior stromal iris defects, superonasal corectopia |
| Posterior segment | enlarged optic nerve disc with a deep funnel-shaped excavation, surrounded by a ring of damaged hyperpigmented choroid and retina, foveal hypoplasia, nystagmus appeared at 6 mo | hypopigmented fundus, severe photophobia, bilateral nystagmus |
| Other findings | optic nerve coloboma | |
| PAX6 mutation (NM_000280.4) | c.341A>G, p.(Asn114Ser). | c.107G>A, p.(Gly36Glu) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilyeva, T.A.; Sukhanova, N.V.; Khalanskaya, O.V.; Marakhonov, A.V.; Prokhorov, N.S.; Kadyshev, V.V.; Skryabin, N.A.; Kutsev, S.I.; Zinchenko, R.A. An Unusual Presentation of Novel Missense Variant in PAX6 Gene: NM_000280.4:c.341A>G, p.(Asn114Ser). Curr. Issues Mol. Biol. 2024, 46, 96-105. https://doi.org/10.3390/cimb46010008
Vasilyeva TA, Sukhanova NV, Khalanskaya OV, Marakhonov AV, Prokhorov NS, Kadyshev VV, Skryabin NA, Kutsev SI, Zinchenko RA. An Unusual Presentation of Novel Missense Variant in PAX6 Gene: NM_000280.4:c.341A>G, p.(Asn114Ser). Current Issues in Molecular Biology. 2024; 46(1):96-105. https://doi.org/10.3390/cimb46010008
Chicago/Turabian StyleVasilyeva, Tatyana A., Natella V. Sukhanova, Olga V. Khalanskaya, Andrey V. Marakhonov, Nikolai S. Prokhorov, Vitaly V. Kadyshev, Nikolay A. Skryabin, Sergey I. Kutsev, and Rena A. Zinchenko. 2024. "An Unusual Presentation of Novel Missense Variant in PAX6 Gene: NM_000280.4:c.341A>G, p.(Asn114Ser)" Current Issues in Molecular Biology 46, no. 1: 96-105. https://doi.org/10.3390/cimb46010008