
5-AZA Upregulates SOCS3 and PTPN6/SHP1, Inhibiting STAT3 and Potentiating the Effects of AG490 against Primary Effusion Lymphoma Cells
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Trypan Blue Assay
2.3. Sub G1 Analysis
2.4. Western Blot Analysis
2.5. Antibodies
2.6. Chemiluminescent Immunometric Assay
2.7. Densitometric Analysis
2.8. Statistical Analysis
3. Results
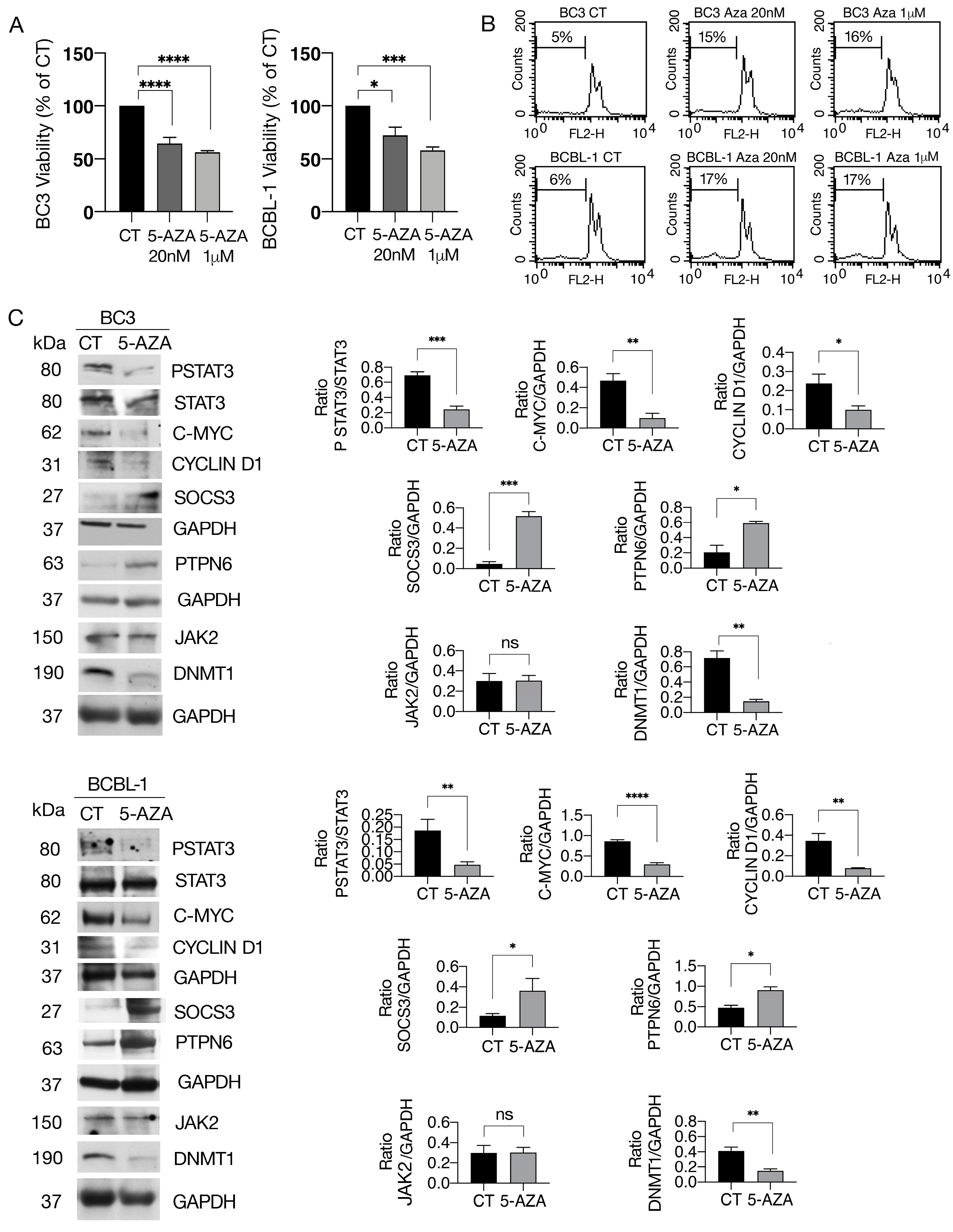
3.1. 5-AZA Reduces Cell Survival and STAT3 Y705 Tyrosine Phosphorylation and c-Myc, Cyclin D1 and HSP27 Expression Levels While Upregulating SOCS3 and SHP1 in PEL Cells
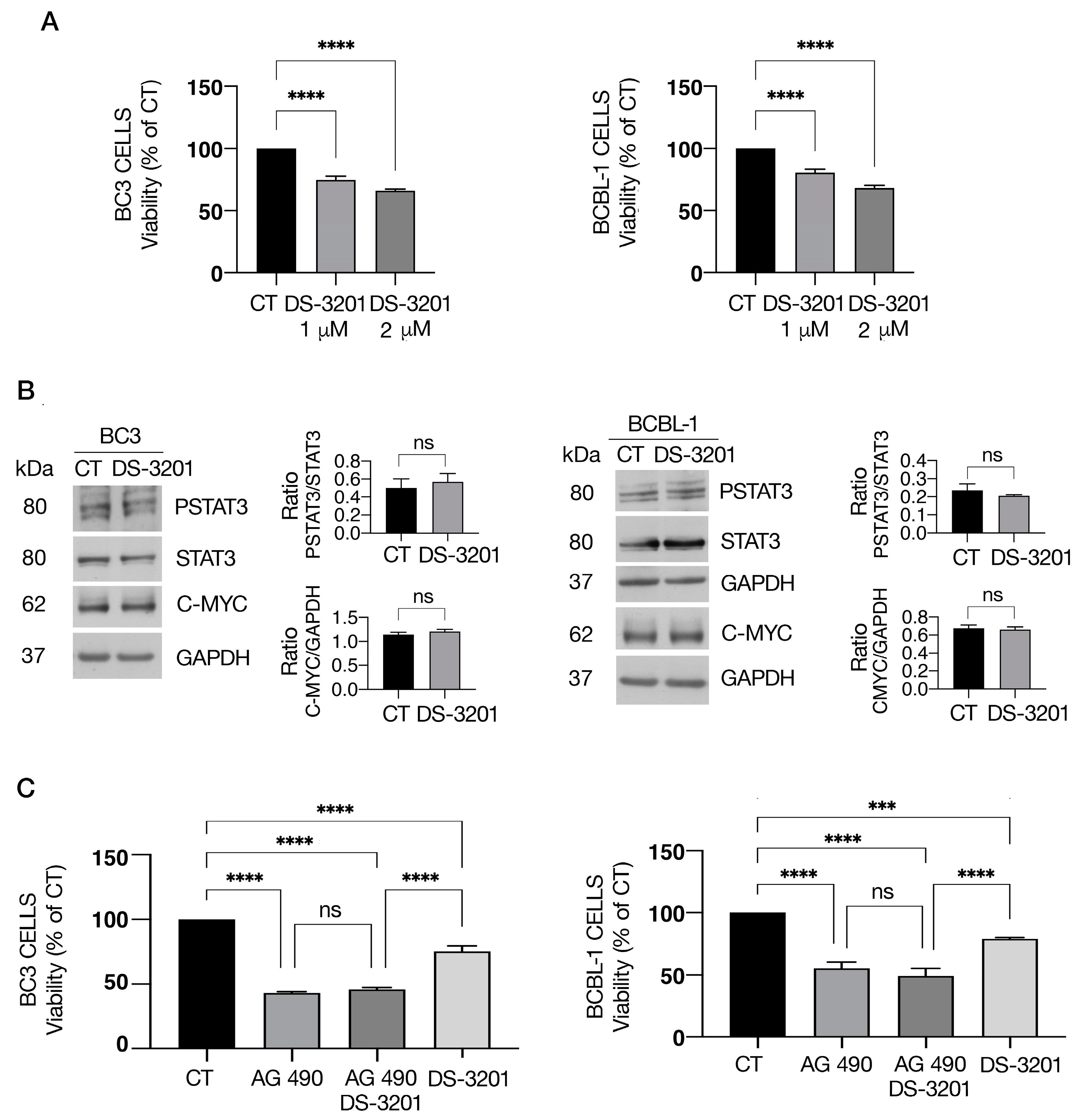
3.2. 5-AZA Potentiates the Anti-Cancer Effects of AG490 Y705 Tyrosine STAT3 Inhibitor
3.3. 5-AZA/AG490 Combination Strongly Impairs the Release of Pro-Survival Cytokines by PEL Cells
3.4. EZH1/2 Inhibitor Valemetostat (DS-3201) Slightly Influences PEL Cell Survival and STAT3 Activation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef]
- Liu, C.Y.; Chen, B.J.; Chuang, S.S. Primary Effusion Lymphoma: A Timely Review on the Association with HIV, HHV8, and EBV. Diagnostics 2022, 12, 713. [Google Scholar] [CrossRef]
- Okada, S.; Goto, H.; Yotsumoto, M. Current status of treatment for primary effusion lymphoma. Intractable Rare Dis Res. 2014, 3, 65–74. [Google Scholar] [CrossRef]
- Adam, N.; Rabe, B.; Suthaus, J.; Grotzinger, J.; Rose-John, S.; Scheller, J. Unraveling viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways independently of the interleukin-6 receptor. J. Virol. 2009, 83, 5117–5126. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Chiozzi, B.; Filardi, M.R.; Lotti, L.V.; Di Renzo, L.; Faggioni, A.; Cirone, M. Tyrosine kinase inhibitor tyrphostin AG490 triggers both apoptosis and autophagy by reducing HSF1 and Mcl-1 in PEL cells. Cancer Lett. 2015, 366, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Arena, A.; Di Crosta, M.; Gonnella, R.; Zarrella, R.; Romeo, M.A.; Benedetti, R.; Gilardini Montani, M.S.; Santarelli, R.; D’Orazi, G.; Cirone, M. NFE2L2 and STAT3 Converge on Common Targets to Promote Survival of Primary Lymphoma Cells. Int. J. Mol. Sci. 2023, 24, 11598. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Santarelli, R.; D’Orazi, G.; Cirone, M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front. Oncol. 2020, 10, 1102. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef]
- Granato, M.; Gilardini Montani, M.S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef]
- Belo, Y.; Mielko, Z.; Nudelman, H.; Afek, A.; Ben-David, O.; Shahar, A.; Zarivach, R.; Gordan, R.; Arbely, E. Unexpected implications of STAT3 acetylation revealed by genetic encoding of acetyl-lysine. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1343–1350. [Google Scholar] [CrossRef]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Hu, Q.; Chen, Y.; Shen, N.; Zhang, N.; Li, A.; Li, L.; Li, J. PRMT6 methylation of STAT3 regulates tumor metastasis in breast cancer. Cell Death Dis. 2023, 14, 655. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar] [CrossRef] [PubMed]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.S.; Cho, Y.Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, H.A.; Mat Jusoh, S.A.; Hassan, R.; Johan, M.F. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 2015, 15, 869. [Google Scholar] [CrossRef]
- Chim, C.S.; Fung, T.K.; Cheung, W.C.; Liang, R.; Kwong, Y.L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef]
- Momparler, R.L. Epigenetic therapy of cancer with 5-aza-2′-deoxycytidine (decitabine). Semin. Oncol. 2005, 32, 443–451. [Google Scholar] [CrossRef]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef]
- Arena, A.; Gilardini Montani, M.S.; Romeo, M.A.; Benedetti, R.; Gaeta, A.; Cirone, M. DNA damage triggers an interplay between wtp53 and c-Myc affecting lymphoma cell proliferation and Kaposi sarcoma herpesvirus replication. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119168. [Google Scholar] [CrossRef]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef]
- Williams, J.J.; Munro, K.M.; Palmer, T.M. Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression. Cells 2014, 3, 546–562. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Liu, X.; Liu, J. Aberrant promoter 2 methylation-mediated downregulation of protein tyrosine phosphatase, non-receptor type 6, is associated with progression of esophageal squamous cell carcinoma. Mol. Med. Rep. 2019, 19, 3273–3282. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Dickson, J.; Din, S.; Macleod, K.; Jodrell, D.; Ramsahoye, B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010, 38, 4313–4324. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, R.; Arena, A.; Zarrella, R.; Gilardini Montani, M.S.; Santarelli, R.; Cirone, M. HSPs/STAT3 Interplay Sustains DDR and Promotes Cytokine Release by Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2023, 24, 3933. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, P.; Sakakibara, S.; Tosato, G. Contribution of viral and cellular cytokines to Kaposi’s sarcoma-associated herpesvirus pathogenesis. J. Leukoc. Biol. 2008, 84, 994–1000. [Google Scholar] [CrossRef]
- Dheilly, E.; Battistello, E.; Katanayeva, N.; Sungalee, S.; Michaux, J.; Duns, G.; Wehrle, S.; Sordet-Dessimoz, J.; Mina, M.; Racle, J.; et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell 2020, 37, 674–689.e12. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K.; et al. Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep. 2019, 29, 2321–2337.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kim, M.Y. Cancer epigenetics: Past, present and future. Semin. Cancer Biol. 2022, 83, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Man, Q.W.; Huo, F.Y.; Gao, X.; Lin, H.; Li, S.R.; Wang, J.; Su, F.C.; Cai, L.; Shi, Y.; et al. STAT3 pathway in cancers: Past, present, and future. MedComm 2022, 3, e124. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Lemonnier, F.; Dupuis, J.; Sujobert, P.; Tournillhac, O.; Cheminant, M.; Sarkozy, C.; Pelletier, L.; Marcais, A.; Robe, C.; Fataccioli, V.; et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood 2018, 132, 2305–2309. [Google Scholar] [CrossRef]
- Karahoca, M.; Momparler, R.L. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin. Epigenetics 2013, 5, 3. [Google Scholar] [CrossRef]
- Guan, H.J.; Li, X.X.; Guo, Y.P.; Dong, J.; Rong, S.Z.; Niu, Y.Y.; Meng, L.L.; Zhao, F.Y.; Fan, X.J.; Zhang, Y.S.; et al. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in bladder cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 11326–11334. [Google Scholar]
- Dai, L.; Li, Z.; Tao, Y.; Liang, W.; Hu, W.; Zhou, S.; Fu, X.; Wang, X. Emerging roles of suppressor of cytokine signaling 3 in human cancers. Biomed. Pharmacother. 2021, 144, 112262. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Lindeman, G.J.; Choong, D.Y.; Wittlin, S.; Brentzell, L.; Phillips, W.; Campbell, I.G.; Visvader, J.E. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 2004, 23, 7726–7733. [Google Scholar] [CrossRef]
- Chu, Q.; Shen, D.; He, L.; Wang, H.; Liu, C.; Zhang, W. Prognostic significance of SOCS3 and its biological function in colorectal cancer. Gene 2017, 627, 114–122. [Google Scholar] [CrossRef]
- Yu, Y.; Sung, S.K.; Lee, C.H.; Ha, M.; Kang, J.; Kwon, E.J.; Kang, J.W.; Kim, Y.; Kim, G.H.; Heo, H.J.; et al. SOCS3 is Related to Cell Proliferation in Neuronal Tissue: An Integrated Analysis of Bioinformatics and Experiments. Front. Genet. 2021, 12, 743786. [Google Scholar] [CrossRef]
- Galm, O.; Yoshikawa, H.; Esteller, M.; Osieka, R.; Herman, J.G. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 2003, 101, 2784–2788. [Google Scholar] [CrossRef]
- Gonnella, R.; Gilardini Montani, M.S.; Guttieri, L.; Romeo, M.A.; Santarelli, R.; Cirone, M. IRE1 Alpha/XBP1 Axis Sustains Primary Effusion Lymphoma Cell Survival by Promoting Cytokine Release and STAT3 Activation. Biomedicines 2021, 9, 118. [Google Scholar] [CrossRef]
- Tigu, A.B.; Bancos, A. The Role of Epigenetic Modifier Mutations in Peripheral T-Cell Lymphomas. Curr. Issues Mol. Biol. 2023, 45, 8974–8988. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Kuang, H.; Yue, C.; Yang, Q.; Wu, A.; Li, Y.; Assaraf, Y.G.; Yang, D.H.; Wu, S. Targeting metabolism to overcome cancer drug resistance: A promising therapeutic strategy for diffuse large B cell lymphoma. Drug Resist. Updat. 2022, 61, 100822. [Google Scholar] [CrossRef] [PubMed]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; George, A.M.; Keshwara, S.M.; Sideris, M.; Szabados, B. Cytokine-based Cancer Immunotherapy: Challenges and Opportunities for IL-10. Anticancer Res. 2021, 41, 3247–3252. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Crosta, M.; Arena, A.; Benedetti, R.; Gilardini Montani, M.S.; Cirone, M. 5-AZA Upregulates SOCS3 and PTPN6/SHP1, Inhibiting STAT3 and Potentiating the Effects of AG490 against Primary Effusion Lymphoma Cells. Curr. Issues Mol. Biol. 2024, 46, 2468-2479. https://doi.org/10.3390/cimb46030156
Di Crosta M, Arena A, Benedetti R, Gilardini Montani MS, Cirone M. 5-AZA Upregulates SOCS3 and PTPN6/SHP1, Inhibiting STAT3 and Potentiating the Effects of AG490 against Primary Effusion Lymphoma Cells. Current Issues in Molecular Biology. 2024; 46(3):2468-2479. https://doi.org/10.3390/cimb46030156
Chicago/Turabian StyleDi Crosta, Michele, Andrea Arena, Rossella Benedetti, Maria Saveria Gilardini Montani, and Mara Cirone. 2024. "5-AZA Upregulates SOCS3 and PTPN6/SHP1, Inhibiting STAT3 and Potentiating the Effects of AG490 against Primary Effusion Lymphoma Cells" Current Issues in Molecular Biology 46, no. 3: 2468-2479. https://doi.org/10.3390/cimb46030156