
Autosomal Recessive Rod–Cone Dystrophy with Mild Extra-Ocular Manifestations Due to a Splice-Affecting Variant in BBS9
 , , and
, , and
Abstract
:1. Introduction
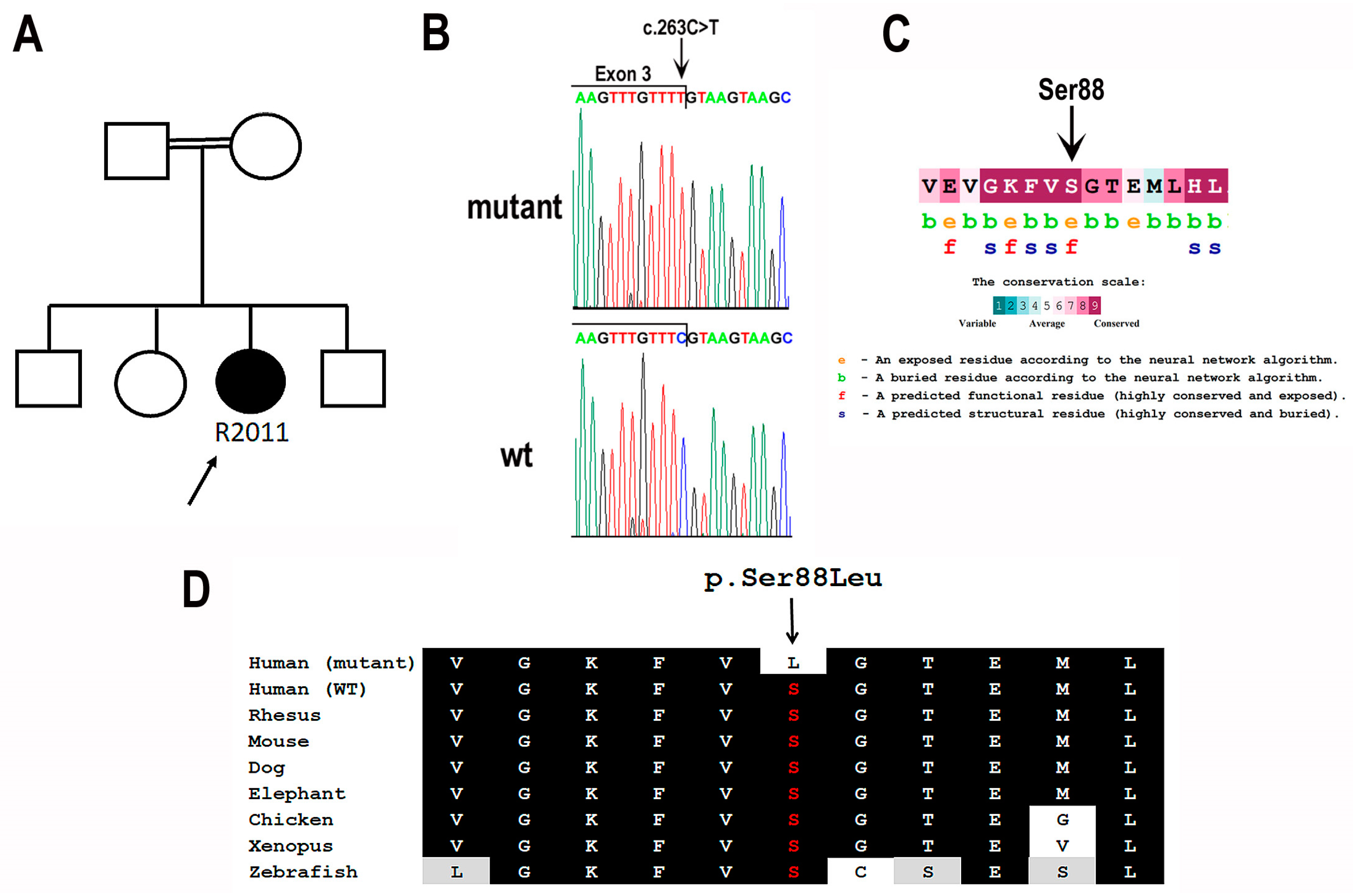
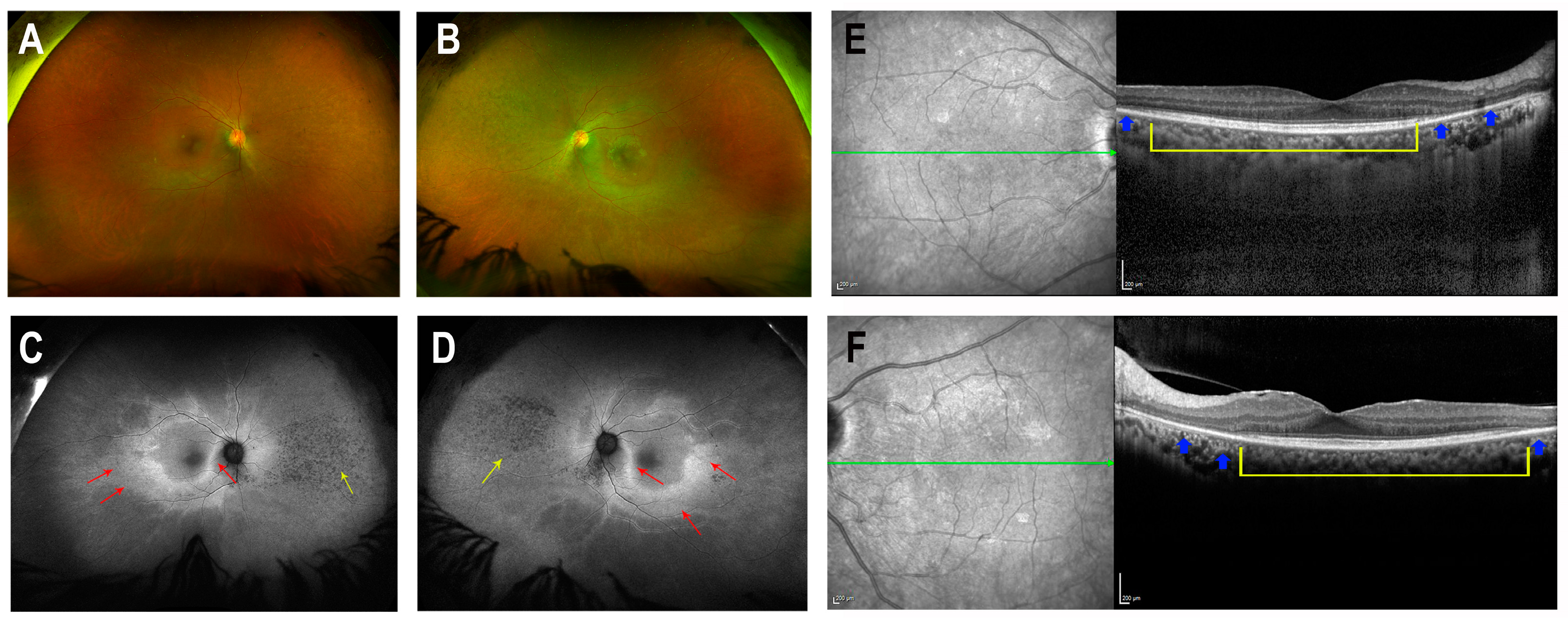
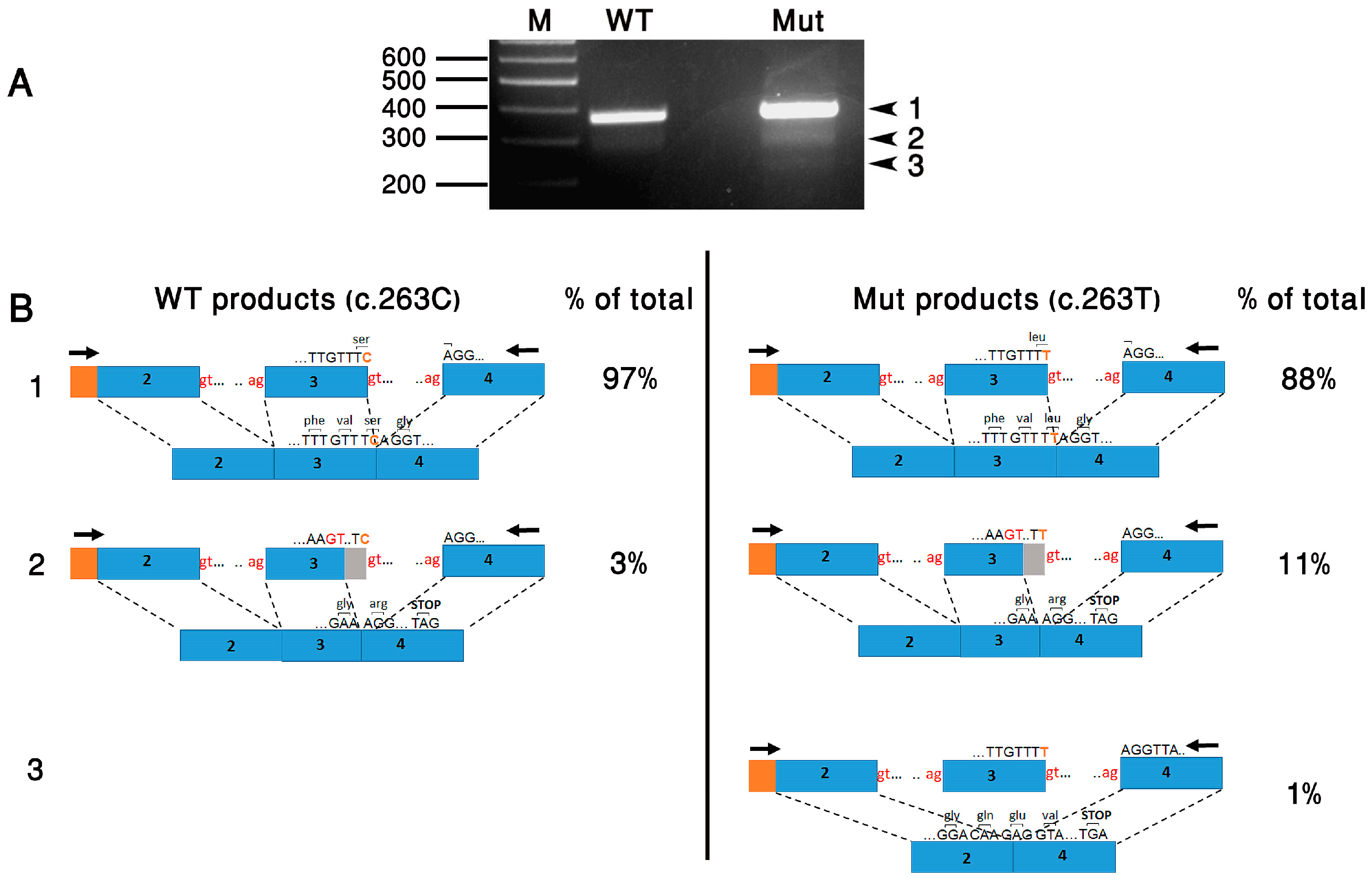
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef]
- Fahim, A.T.; Daiger, S.P.; Weleber, R.G. Nonsyndromic Retinitis Pigmentosa Overview. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, J.H.L., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef]
- Forsyth, R.; Gunay-Aygun, M. Bardet-Biedl Syndrome Overview. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, J.H.L., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Focsa, I.O.; Budisteanu, M.; Balgradean, M. Clinical and genetic heterogeneity of primary ciliopathies (Review). Int. J. Mol. Med. 2021, 48, 176. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Petriman, N.A.; Lorentzen, E. Moving proteins along in the cilium. eLife 2020, 9, e55254. [Google Scholar] [CrossRef]
- Hsu, Y.; Garrison, J.E.; Kim, G.; Schmitz, A.R.; Searby, C.C.; Zhang, Q.; Datta, P.; Nishimura, D.Y.; Seo, S.; Sheffield, V.C. BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet. 2017, 13, e1007057. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.; Garrison, J.E.; Seo, S.; Sheffield, V.C. The absence of BBSome function decreases synaptogenesis and causes ectopic synapse formation in the retina. Sci. Rep. 2020, 10, 8321. [Google Scholar] [CrossRef]
- Stoetzel, C.; Muller, J.; Laurier, V.; Davis, E.E.; Zaghloul, N.A.; Vicaire, S.; Jacquelin, C.; Plewniak, F.; Leitch, C.C.; Sarda, P.; et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am. J. Hum. Genet. 2007, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.Y.; Swiderski, R.E.; Searby, C.C.; Berg, E.M.; Ferguson, A.L.; Hennekam, R.; Merin, S.; Weleber, R.G.; Biesecker, L.G.; Stone, E.M.; et al. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am. J. Hum. Genet. 2005, 77, 1021–1033. [Google Scholar] [CrossRef]
- Veleri, S.; Bishop, K.; Dalle Nogare, D.E.; English, M.A.; Foskett, T.J.; Chitnis, A.; Sood, R.; Liu, P.; Swaroop, A. Knockdown of Bardet-Biedl syndrome gene BBS9/PTHB1 leads to cilia defects. PLoS ONE 2012, 7, e34389. [Google Scholar] [CrossRef]
- Ece Solmaz, A.; Onay, H.; Atik, T.; Aykut, A.; Cerrah Gunes, M.; Ozalp Yuregir, O.; Bas, V.N.; Hazan, F.; Kirbiyik, O.; Ozkinay, F. Targeted multi-gene panel testing for the diagnosis of Bardet Biedl syndrome: Identification of nine novel mutations across BBS1, BBS2, BBS4, BBS7, BBS9, BBS10 genes. Eur. J. Med. Genet. 2015, 58, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Hjortshoj, T.D.; Gronskov, K.; Philp, A.R.; Nishimura, D.Y.; Riise, R.; Sheffield, V.C.; Rosenberg, T.; Brondum-Nielsen, K. Bardet-Biedl syndrome in Denmark—Report of 13 novel sequence variations in six genes. Hum. Mutat. 2010, 31, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Jeziorny, K.; Antosik, K.; Jakiel, P.; Mlynarski, W.; Borowiec, M.; Zmyslowska, A. Next-Generation Sequencing in the Diagnosis of Patients with Bardet-Biedl Syndrome—New Variants and Relationship with Hyperglycemia and Insulin Resistance. Genes 2020, 11, 1283. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.R.; Nazir, A.; Imtiaz, S.; Paracha, S.A.; Waryah, Y.M.; Ujjan, I.D.; Anwar, I.; Iqbal, A.; Santoni, F.A.; Shah, I.; et al. Delineating the Spectrum of Genetic Variants Associated with Bardet-Biedl Syndrome in Consanguineous Pakistani Pedigrees. Genes 2023, 14, 404. [Google Scholar] [CrossRef] [PubMed]
- Sathya Priya, C.; Sen, P.; Umashankar, V.; Gupta, N.; Kabra, M.; Kumaramanickavel, G.; Stoetzel, C.; Dollfus, H.; Sripriya, S. Mutation spectrum in BBS genes guided by homozygosity mapping in an Indian cohort. Clin. Genet. 2015, 87, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Abu-Safieh, L.; Al-Anazi, S.; Al-Abdi, L.; Hashem, M.; Alkuraya, H.; Alamr, M.; Sirelkhatim, M.O.; Al-Hassnan, Z.; Alkuraya, B.; Mohamed, J.Y.; et al. In search of triallelism in Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2012, 20, 420–427. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Koenekoop, R.K.; Senechal, A.; De Baere, E.B.; de Ravel, T.; Banfi, S.; Kohl, S.; Ayuso, C.; Sharon, D.; Hoyng, C.B.; et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch. Ophthalmol. 2012, 130, 1425–1432. [Google Scholar] [CrossRef]
- Shevach, E.; Ali, M.; Mizrahi-Meissonnier, L.; McKibbin, M.; El-Asrag, M.; Watson, C.M.; Inglehearn, C.F.; Ben-Yosef, T.; Blumenfeld, A.; Jalas, C.; et al. Association between missense mutations in the BBS2 gene and nonsyndromic retinitis pigmentosa. JAMA Ophthalmol. 2015, 133, 312–318. [Google Scholar] [CrossRef]
- Abu Safieh, L.; Aldahmesh, M.A.; Shamseldin, H.; Hashem, M.; Shaheen, R.; Alkuraya, H.; Al Hazzaa, S.A.; Al-Rajhi, A.; Alkuraya, F.S. Clinical and molecular characterisation of Bardet-Biedl syndrome in consanguineous populations: The power of homozygosity mapping. J. Med. Genet. 2010, 47, 236–241. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Safieh, L.A.; Alkuraya, H.; Al-Rajhi, A.; Shamseldin, H.; Hashem, M.; Alzahrani, F.; Khan, A.O.; Alqahtani, F.; Rahbeeni, Z.; et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol. Vis. 2009, 15, 2464–2469. [Google Scholar]
- Goyal, S.; Jager, M.; Robinson, P.N.; Vanita, V. Confirmation of TTC8 as a disease gene for nonsyndromic autosomal recessive retinitis pigmentosa (RP51). Clin. Genet. 2016, 89, 454–460. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Iqbal, M.; Wang, Y.; Masuda, T.; Chen, Y.; Bowne, S.; Sullivan, L.S.; Waseem, N.H.; Bhattacharya, S.; Daiger, S.P.; et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010, 86, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Bujakowska, K.M.; Zhang, Q.; Siemiatkowska, A.M.; Liu, Q.; Place, E.; Falk, M.J.; Consugar, M.; Lancelot, M.E.; Antonio, A.; Lonjou, C.; et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet. 2015, 24, 230–242. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Neveling, K.; Kohl, S.; Banin, E.; Rotenstreich, Y.; Sharon, D.; Falik-Zaccai, T.C.; Hipp, S.; Roepman, R.; Wissinger, B.; et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am. J. Hum. Genet. 2012, 90, 102–109. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Yoshitake, K.; Akahori, M.; Ikeo, K.; Gekka, T.; Tsuneoka, H.; Iwata, T. Novel C8orf37 Mutations in Patients with Early-onset Retinal Dystrophy, Macular Atrophy, Cataracts, and High Myopia. Ophthalmic Genet. 2016, 37, 68–75. [Google Scholar] [PubMed]
- Ravesh, Z.; El Asrag, M.E.; Weisschuh, N.; McKibbin, M.; Reuter, P.; Watson, C.M.; Baumann, B.; Poulter, J.A.; Sajid, S.; Panagiotou, E.S.; et al. Novel C8orf37 mutations cause retinitis pigmentosa in consanguineous families of Pakistani origin. Mol. Vis. 2015, 21, 236–243. [Google Scholar] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Grimberg, J.; Nawoschik, S.; Belluscio, L.; McKee, R.; Turck, A.; Eisenberg, A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989, 17, 8390. [Google Scholar] [CrossRef]
- Panneman, D.M.; Hitti-Malin, R.J.; Holtes, L.K.; de Bruijn, S.E.; Reurink, J.; Boonen, E.G.M.; Khan, M.I.; Ali, M.; Andreasson, S.; De Baere, E.; et al. Cost-effective sequence analysis of 113 genes in 1192 probands with retinitis pigmentosa and Leber congenital amaurosis. Front. Cell Dev. Biol. 2023, 11, 1112270. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar] [CrossRef]
- Riise, R.; Andreasson, S.; Borgastrom, M.K.; Wright, A.F.; Tommerup, N.; Rosenberg, T.; Tornqvist, K. Intrafamilial variation of the phenotype in Bardet-Biedl syndrome. Br. J. Ophthalmol. 1997, 81, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Manara, E.; Paolacci, S.; D’Esposito, F.; Abeshi, A.; Ziccardi, L.; Falsini, B.; Colombo, L.; Iarossi, G.; Pilotta, A.; Boccone, L.; et al. Mutation profile of BBS genes in patients with Bardet-Biedl syndrome: An Italian study. Ital. J. Pediatr. 2019, 45, 72. [Google Scholar] [CrossRef] [PubMed]
- Perea-Romero, I.; Solarat, C.; Blanco-Kelly, F.; Sanchez-Navarro, I.; Bea-Mascato, B.; Martin-Salazar, E.; Lorda-Sanchez, I.; Swafiri, S.T.; Avila-Fernandez, A.; Martin-Merida, I.; et al. Allelic overload and its clinical modifier effect in Bardet-Biedl syndrome. NPJ Genom. Med. 2022, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Yildiz Bolukbasi, E.; Mumtaz, S.; Afzal, M.; Woehlbier, U.; Malik, S.; Tolun, A. Homozygous mutation in CEP19, a gene mutated in morbid obesity, in Bardet-Biedl syndrome with predominant postaxial polydactyly. J. Med. Genet. 2018, 55, 189–197. [Google Scholar] [CrossRef]
- Denniston, A.K.; Beales, P.L.; Tomlins, P.J.; Good, P.; Langford, M.; Foggensteiner, L.; Williams, D.; Tsaloumas, M.D. Evaluation of visual function and needs in adult patients with bardet-biedl syndrome. Retina 2014, 34, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Maltese, P.E.; Colombo, L.; Martella, S.; Rossetti, L.; El Shamieh, S.; Sinibaldi, L.; Passarelli, C.; Coppe, A.M.; Buzzonetti, L.; Falsini, B.; et al. Genetics of Inherited Retinal Diseases in Understudied Ethnic Groups in Italian Hospitals. Front. Genet. 2022, 13, 914345. [Google Scholar] [CrossRef]
- Kousi, M.; Soylemez, O.; Ozanturk, A.; Mourtzi, N.; Akle, S.; Jungreis, I.; Muller, J.; Cassa, C.A.; Brand, H.; Mokry, J.A.; et al. Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy. Nat. Genet. 2020, 52, 1145–1150. [Google Scholar] [CrossRef]
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 2004, 13, R65–R71. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Right Eye (µV/ms) | Left Eye (µV/ms) | Normal Reference Limit (µV-Minimum/ms-Maximum) | ||
|---|---|---|---|---|
| DA 0.01 | a wave (amp/implicit time) | −1.519/36.5 | −9.616/45 | 1.0/47 |
| b wave (amp/implicit time) | 14.3/94.5 | 9.754/97.5 | 70/108.6 | |
| DA 3.0 | a wave (amp/implicit time) | −73.98/17.5 | −48.26/17 | −94/20.3 |
| b wave (amp/implicit time) | 36.72/57.5 | 37.48/43.5 | 174/56.8 | |
| DA 10.0 | a wave (amp/implicit time) | −91.94/15.5 | −69.22/15.5 | −113/15.42 |
| b wave (amp/implicit time) | 51.5/53.5 | 45.83/35 | 200/54.3 | |
| LA 3.0 | a wave (amp/implicit time) | −10.63/12.5 | −7/16 | −15.2/16.7 |
| b wave (amp/implicit time) | 30/32.5 | 40.52/32.5 | 56.6/34 | |
| LA FLICKER 30 Hz | Amplitude/ time (msec) | 32.21/30.5 | 18.31/29 | 57.2/30.3 |
| Trough (msec) | 15 | 15 | 15.3 |
| Prediction Tool | Prediction (Score) | Scale |
|---|---|---|
| Mutation Taster | Deleterious (1) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| Missense prediction tools | ||
| Revel | Deleterious (0.76) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| Polyphen2 | Deleterious (1) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| DANN | Deleterious (1) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| MetaLR | Deleterious (0.71) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| BayesDel | Deleterious (0.13) | (−1.29334)–0.75731, Deleterious: >(−0.0570105) |
| SIFT | Uncertain (0.001) | 0–1, Deleterious: <0.001 |
| FATHMM | Uncertain (−1.74) | (−16.13)–10.64, Deleterious: <(−4.14) |
| PrimateAI | Uncertain (0.74) | 0–1, Deleterious: >0.803 |
| AlphaMissense | Uncertain (0.477) | 0–1, Deleterious: >0.761 |
| Splice-site prediction tools | ||
| dbscSNV Ada | Deleterious (0.99) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| dbscSNV RF | Deleterious (0.74) | 0–1, higher scores reflecting a greater likelihood that the variant is deleterious |
| SpliceAI | Uncertain (0.12) | 0–1, Deleterious: >0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deitch, I.; Itskov, S.; Panneman, D.; Abu Shtaya, A.; Saban, T.; Goldberg, Y.; Ehrenberg, M.; Cremers, F.P.M.; Roosing, S.; Ben-Yosef, T. Autosomal Recessive Rod–Cone Dystrophy with Mild Extra-Ocular Manifestations Due to a Splice-Affecting Variant in BBS9. Curr. Issues Mol. Biol. 2024, 46, 2566-2575. https://doi.org/10.3390/cimb46030163
Deitch I, Itskov S, Panneman D, Abu Shtaya A, Saban T, Goldberg Y, Ehrenberg M, Cremers FPM, Roosing S, Ben-Yosef T. Autosomal Recessive Rod–Cone Dystrophy with Mild Extra-Ocular Manifestations Due to a Splice-Affecting Variant in BBS9. Current Issues in Molecular Biology. 2024; 46(3):2566-2575. https://doi.org/10.3390/cimb46030163
Chicago/Turabian StyleDeitch, Iris, Sofia Itskov, Daan Panneman, Aasem Abu Shtaya, Tal Saban, Yael Goldberg, Miriam Ehrenberg, Frans P. M. Cremers, Susanne Roosing, and Tamar Ben-Yosef. 2024. "Autosomal Recessive Rod–Cone Dystrophy with Mild Extra-Ocular Manifestations Due to a Splice-Affecting Variant in BBS9" Current Issues in Molecular Biology 46, no. 3: 2566-2575. https://doi.org/10.3390/cimb46030163