Leucettamols, Bifunctionalized Marine Sphingoids, Act as Modulators of TRPA1 and TRPM8 Channels





Abstract
:
1. Introduction

2. Results and Discussion
2.1. Chemistry
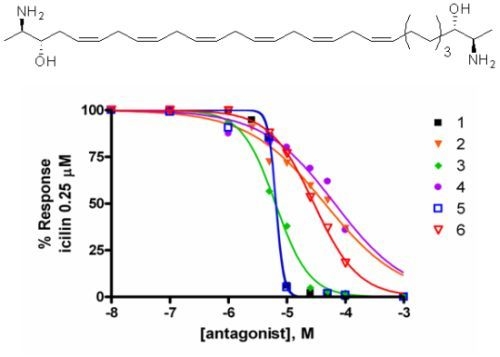
2.2. Activity at CB Receptors and TRP Channels

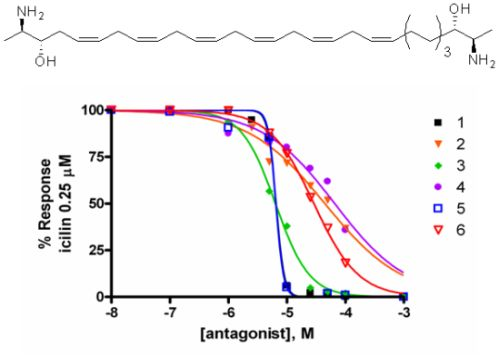{kind=link}
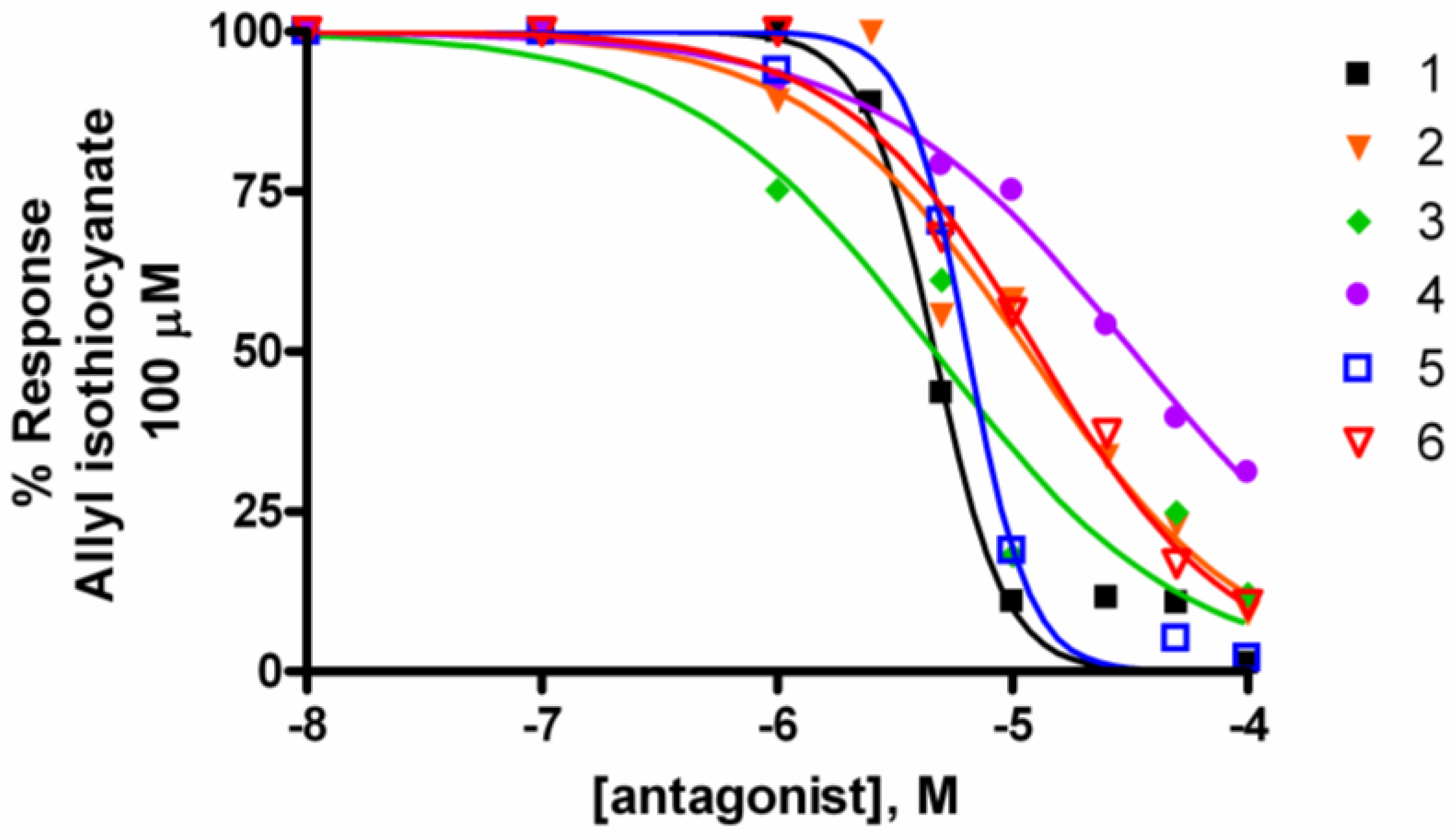{kind=link}
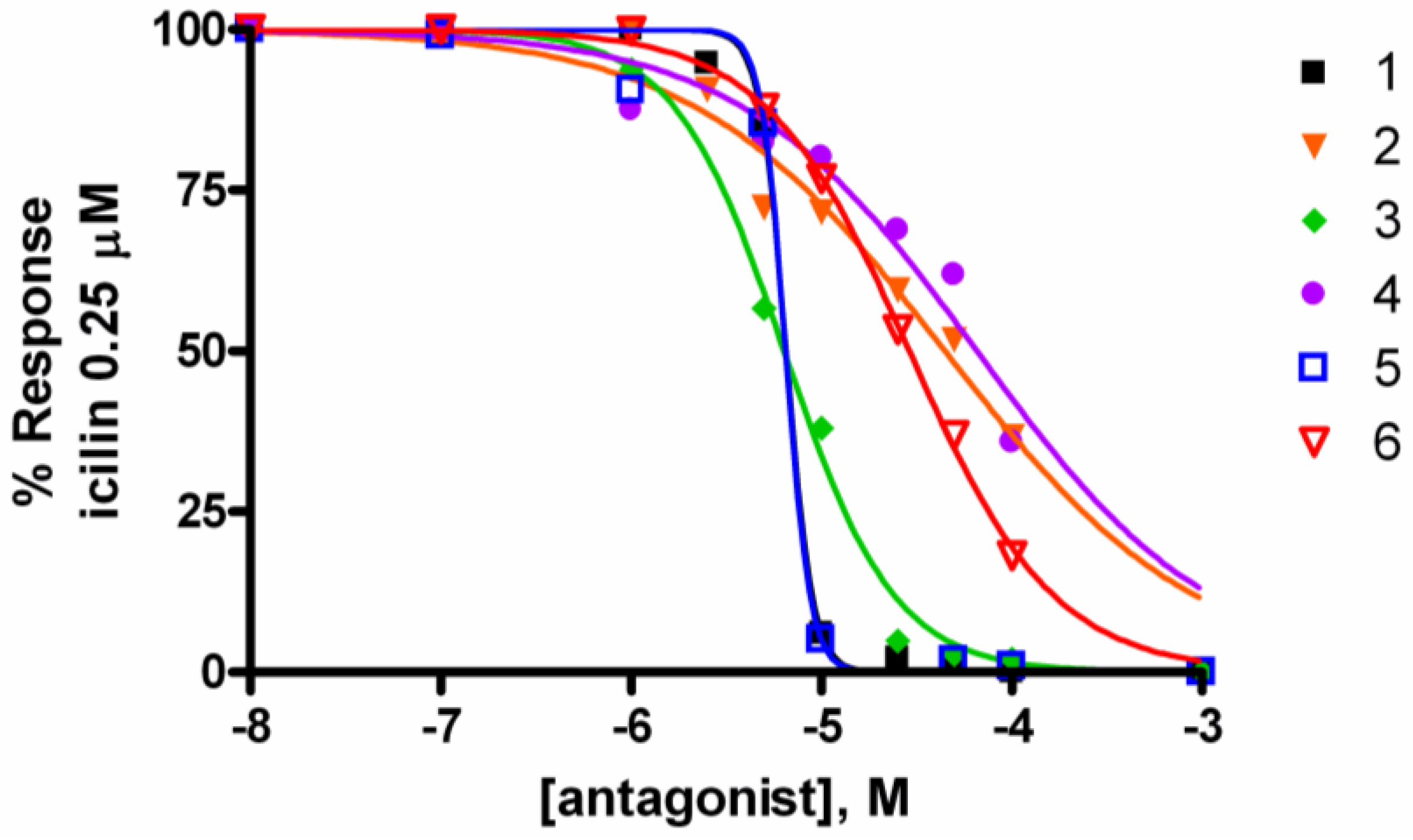{kind=link}
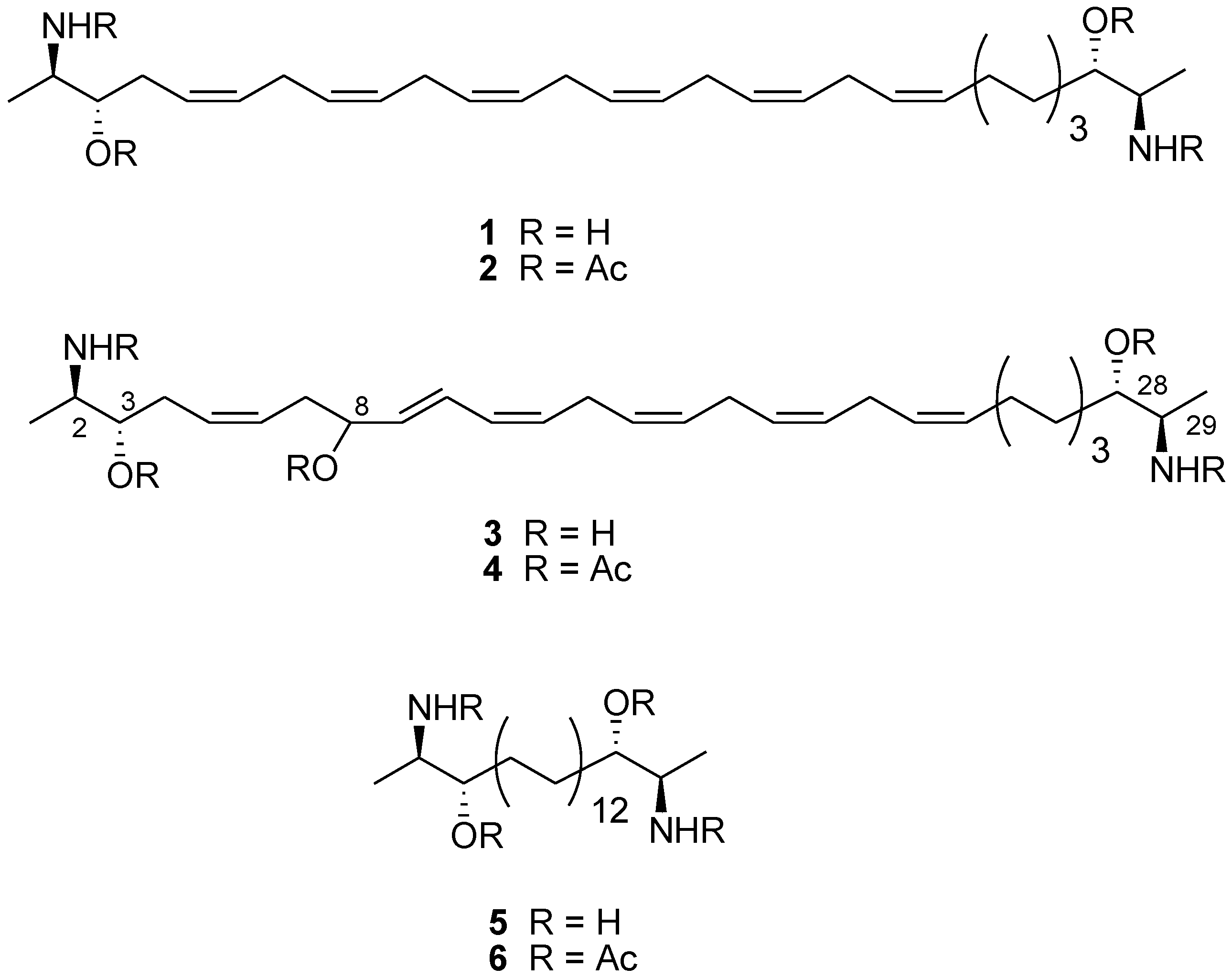{kind=link}
| Potency EC50 TRPA1 (μM) | Efficacy TRPA1 (% AITC 100 μM) | IC50 Inh. TRPA1 μM (AITC 100 μM) | IC50 Inh. TRPM8 μM (icilin 0.25 μM) | |
|---|---|---|---|---|
| Leucettamol A (1) | 3.7 ± 1.7 | 101.9 ± 12.4 | 4.7 ± 0.2 | 6.5 ± 0.3 |
| 2 | 9.4 ± 2.2 | 139.2 ± 7.5 | 11.6 ± 2.3 | 44.6 ± 10.1 |
| Leucettamol B (3) | 5.9 ± 1.9 | 103.3 ± 10.6 | 4.7 ± 0.9 | 6.4 ± 1.0 |
| 4 | 3.5 ± 2.5 | 109.6 ± 21.0 | 32.6 ± 4.8 | 65.7 ± 8.6 |
| 5 | 2.6 ± 0.1 | 69.2 ± 0.1 | 6.5 ± 0.4 | 6.5 ± 0.3 |
| 6 | 9.7 ± 3.3 | 100.0 ± 9.7 | 12.7 ± 0.8 | 29.0 ± 0.6 |
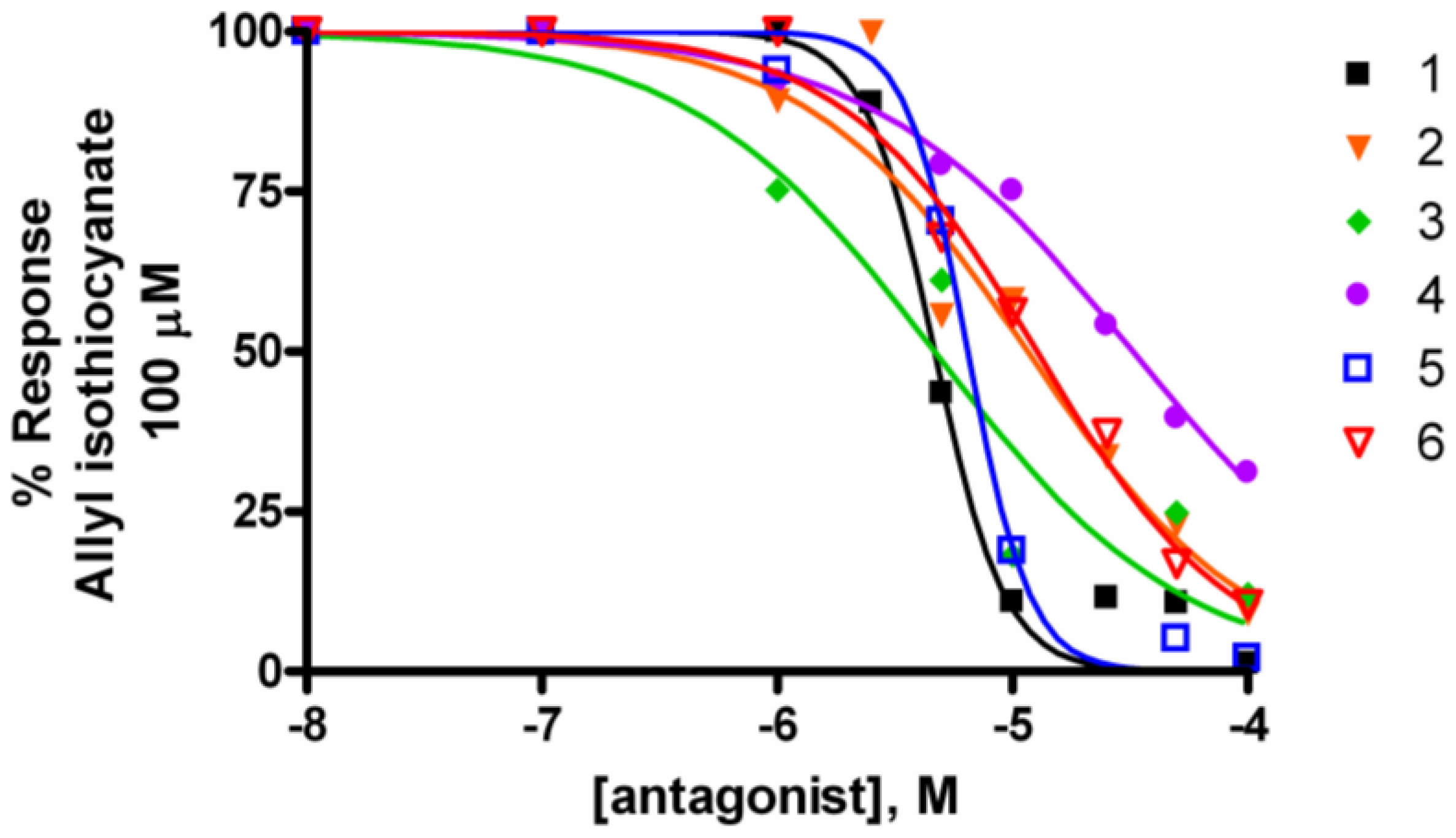

3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Material, Extraction and Isolation
3.3. Acetylation of Leucettamols
3.4. Reduction of Leucettamol A and Acetylation of Compound 5
3.5. In Vitro Assays with TRP Receptors
3.6. CB1 and CB2 Receptor Binding Assays
4. Conclusions
Acknowledgments
References
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V.; Bindels, R.J.; Bruford, E.A.; Caterina, M.J.; Clapham, D.E.; Harteneck, C.; Heller, S.; Julius, D.; et al. A unified nomenclature for the superfamily of TRP cation channels. Mol. Cell 2002, 9, 229–231. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP channels, a remarkably functional family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef]
- Zhang, Y.; Hoon, M.A.; Chandrashekar, J.; Mueller, K.L.; Cook, B.; Wu, D.Q.; Zuker, C.S.; Ryba, N.J.P. Coding of sweet, bitter, and umami tastes: Different receptor cells sharing similar signaling pathway. Cell 2003, 112, 293–301. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor, a heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef]
- Appendino, G.; Minassi, A.; Pagani, A.; Ech-Chadad, A. The role of natural products in the ligand deorphanization of TRP channels. Curr. Pharm. Des. 2008, 14, 2–17. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Preti, D.; Materazzi, S.; Geppetti, P. Transient receptor potential ankyrin 1 (TRPA1) channel as emerging target for novel analgesics and anti-inflammatory agents. J. Med. Chem. 2010, 53, 5085–5107. [Google Scholar] [CrossRef]
- Knowlton, W.M.; Bifolck-Fischer, A.; Bautista, D.M.; McKemy, D.D. TRPM8, but not TRPA1, is required for neural and behavioral responses to acute noxious cold temperatures and cold-mimetics in vivo. Pain 2010, 150, 340–350. [Google Scholar] [CrossRef]
- Brignell, J.L.; Chapman, V.; Kendall, D.A. Comparison of icilin- and cold-evoked responses of spinal neurones, and their modulation of mechanical activity, in a model of neuropathic pain. Brain Res. 2008, 1215, 87–96. [Google Scholar] [CrossRef]
- Harrington, A.M.; Hughes, P.A.; Martin, C.M.; Yang, J.; Castro, J.; Isaacs, N.J.; Blackshaw, L.A.; Brierley, S.M. A novel role for TRPM8 in visceral afferent function. Pain 2011, 152, 1459–1468. [Google Scholar] [CrossRef]
- Zhang, L.; Barritt, G.J. TRPM8 in prostate cancer cells: A potential diagnostic and prognostic marker with a secretory function? Endocr. Relat. Cancer 2006, 13, 27–38. [Google Scholar] [CrossRef]
- Zhang, L.; Barritt, G.J. Evidence that TRPM8 is an androgen-dependent Ca2+ channel required for the survival of prostate cancer cells. Cancer Res. 2004, 64, 8365–8373. [Google Scholar] [CrossRef]
- Meves, H. Arachidonic acid and ion channels: An update. Br. J. Pharmacol. 2008, 155, 4–16. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar]
- Vetter, I.; Lewis, R.J. Natural product ligands of TRP channels. Adv. Exp. Med. Biol. 2011, 704, 41–85. [Google Scholar] [CrossRef]
- Bandell, M.; Story, G.M.; Hwang, S.W.; Viswanath, V.; Eid, S.R.; Petrus, M.J.; Earley, T.J.; Patapoutian, A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004, 41, 849–857. [Google Scholar] [CrossRef]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stucky, C.L.; Jordt, S.E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar]
- Costa, B.; Giagnoni, G.; Franke, C.; Trovato, A.E.; Colleoni, M. Vanilloid TRPV1 receptor mediates the anti-hyperalgesic effect of the non-psychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br. J. Pharmacol. 2004, 143, 247–250. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T.; Nilius, B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424, 434–438. [Google Scholar]
- Bassoli, A.; Borgonovo, G.; Caimi, S.; Scaglioni, L.; Morini, G.; Moriello, A.S.; Di Marzo, V.; de Petrocellis, L. Taste-guided identification of high potency TRPA1 agonists from Perilla frutescens. Bioorg. Med. Chem. 2009, 17, 1636–1639. [Google Scholar]
- Appendino, G.; Ligresti, A.; Minassi, A.; Cascio, M.G.; Allarà, M.; Taglialatela-Scafati, O.; Pertwee, R.G.; de Petrocellis, L.; Di Marzo, V. Conformationally constrained fatty acid ethanolamides as cannabinoid and vanilloid receptor probes. J. Med. Chem. 2009, 52, 3001–3009. [Google Scholar]
- Avonto, C.; Taglialatela-Scafati, O.; Pollastro, F.; Minassi, A.; Di Marzo, V.; de Petrocellis, L.; Appendino, G. An NMR spectroscopic method to identify and classify thiol-trapping agents: Revival of Michael acceptors for drug discovery? Angew. Chem. Int. Ed. 2011, 50, 467–471. [Google Scholar]
- Nassini, R.; Materazzi, S.; Vriens, J.; Prenen, J.; Benemei, S.; de Siena, G.; La Marca, G.; Andrè, E.; Preti, D.; Avonto, C.; et al. The “headache tree”, via umbellulone and TRPA1, activates the trigeminovascular system. Brain 2012, 135, 376–390. [Google Scholar] [CrossRef]
- Chianese, G.; Fattorusso, E.; Taglialatela-Scafati, O.; Bavestrello, G.; Calcinai, B.; Dien, H.A.; Ligresti, A.; Di Marzo, V. Desulfohaplosamate, a new phosphate-containing steroid from Dasychalina sp., is a selective cannabinoid CB2 receptor ligand. Steroids 2011, 76, 998–1002. [Google Scholar] [CrossRef]
- Fattorusso, C.; Persico, M.; Calcinai, B.; Cerrano, C.; Parapini, S.; Taramelli, D.; Novellino, E.; Romano, A.; Scala, F.; Fattorusso, E.; et al. Manadoperoxides A-D from the Indonesian sponge Plakortis cfr. Simplex: Further insights on the structure-activity relationships of simple 1,2-dioxane antimalarials. J. Nat. Prod. 2010, 73, 1138–1145. [Google Scholar] [CrossRef]
- Fattorusso, E.; Romano, A.; Taglialatela-Scafati, O.; Irace, C.; Maffettone, C.; Bavestrello, G.; Cerrano, C. Oxygenated cembranoids of the decaryiol type from the Indonesian soft coral Lobophytum sp. Tetrahedron 2009, 65, 2898–2904. [Google Scholar] [CrossRef]
- Fattorusso, E.; Luciano, P.; Putra, M.Y.; Taglialatela-Scafati, O.; Ianaro, A.; Panza, E.; Bavestrello, G.; Cerrano, C. Chloroscabrolides, chlorinated norcembranoids from the Indonesian soft coral Sinularia sp. Tetrahedron 2011, 67, 7983–7988. [Google Scholar] [CrossRef]
- Kong, F.H.; Faulkner, D.J. Leucettamines A and B, two antimicrobial lipids from the calcareous sponge Leucetta microraphis. J. Org. Chem. 1993, 58, 970–971. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Takeuchi, T.; Rotinsulu, H.; Mangindaan, R.E.P.; van Soest, R.W.M.; Ukai, K.; Kobayashi, H.; Namikoshi, M.; Ohta, T.; Yokosawa, H. Leucettamol A: A new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg. Med. Chem. Lett. 2008, 18, 6319–6320. [Google Scholar]
- Dalisay, D.S.; Tsukamoto, S.; Molinski, T.F. Absolute configuration of the α,ω-bifunctionalized sphingolipid leucettamol A from Leucetta microrhaphis by deconvoluted exciton coupled CD. J. Nat. Prod. 2009, 72, 353–359. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Starowicz, K.; Moriello, A.S.; Vivese, M.; Orlando, P.; Di Marzo, V. Regulation of transient receptor potential channels of melastatin type 8 (TRPM8): Effect of cAMP, cannabinoid CB(1) receptors and endovanilloids. Exp. Cell Res. 2007, 313, 1911–1920. [Google Scholar] [CrossRef]
- Ortar, G.; de Petrocellis, L.; Morera, L.; Moriello, A.S.; Orlando, P.; Morera, E.; Nalli, M.; Di Marzo, V. (−)-Menthylamine derivatives as potent and selective antagonists of transient receptor potential melastatin type-8 (TRPM8) channels. Bioorg. Med. Chem. Lett. 2010, 20, 2729–2732. [Google Scholar]
- Tamayo, N.A.; Bo, Y.; Gore, V.; Ma, V.; Nishimura, N.; Tang, P.; Deng, H.; Klionsky, L.; Lehto, S.G.; Wang, W.; et al. Fused piperidines as a novel class of potent and orally available transient receptor potential melastatin type 8 (TRPM8) antagonists. J. Med. Chem. 2012, 55, 1593–1611. [Google Scholar] [CrossRef]
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Poerzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 2011, 286, 39328–39335. [Google Scholar]
- Chubanov, V.; Mederos y Schnitzler, M.; Meisner, M.; Schafer, S.; Abstiens, K.; Hofmann, T.; Gudermann, T. Natural and synthetic modulators of SK (Kca2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br. J. Pharmacol. 2012, 166, 1357–1376. [Google Scholar] [CrossRef]
- Talareva, K.; Yasumatsu, K.; Yoshida, R.; Margolskee, R.F.; Voets, T.; Ninomiya, Y.; Nilius, B. The taste transduction channel: TRPM5 is a locus for bitter-sweet taste interactions. FASEB J. 2008, 22, 1343–1355. [Google Scholar]
- Cuypers, E.; Yanagihara, A.; Karlsson, E.; Tytgat, J. Jellyfish and other cnidarian envenomations cause pain by affecting TRPV1 channels. FEBS Lett. 2006, 580, 5728–5732. [Google Scholar] [CrossRef]
- Cuypers, E.; Yanagihara, A.; Rainier, J.D.; Tytgat, J. TRPV1 as a key determinant in ciguatera and neurotoxic shellfish poisoning. Biochem. Biophys. Res. Commun. 2007, 361, 214–217. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Bisogno, T.; Davis, J.B.; Pertwee, R.G.; Di Marzo, V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: Inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000, 483, 52–56. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Vellani, V.; Schiano-Moriello, A.; Marini, P.; Magherini, P.C.; Orlando, P.; Di Marzo, V. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J. Pharmacol. Exp. Ther. 2008, 325, 1007–1015. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Denisenko, V.A.; Stonik, V.A.; Milgrom, Y.N.; Rashkes, Y.W. Rhizochalin, a novel secondary metabolite of mixed biosynthesis from the sponge Rhizochalina incrustata. Tetrahedron Lett. 1989, 30, 6581–6584. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Santalova, E.A.; Pokanevich, E.V.; Molinski, T.F.; Stonik, V.A. Rhizochalin A, a novel two-headed sphingolipid from the sponge Rhizochalina incrustata. J. Nat. Prod. 2005, 68, 255–257. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Dmitrenok, P.S.; Zakharenko, A.M.; Denisenko, V.A.; Guzii, A.G.; Li, R.; Skepper, C.K.; Molinski, T.F.; Stonik, V.A. Rhizochalins C and D from the sponge Rhizochalina incrustata: A rare threo-sphingolipid and a facile method for determination of the carbonyl position in alpha,omega-bifunctionalized ketosphingolipids. J. Nat. Prod. 2007, 70, 1991–1998. [Google Scholar] [CrossRef]
- Molinski, T.F.; Makarieva, T.N.; Stonik, V.A. (−)-Rhizochalin is a dimeric enantiomorphic (2R)-sphingolipid: Absolute configuration of pseudo-C2v-symmetric bis-2-amino-3-alkanols by CD. Angew. Chem. Int. Ed. 2000, 39, 4076–4079. [Google Scholar] [CrossRef]
- Nicholas, G.M.; Hong, T.W.; Molinski, T.F.; Lerch, M.L.; Cancilla, M.T.; Lebrilla, C.B. Oceanapiside, an antifungal bis-α,ω-amino alcohol glycoside from the marine sponge Oceanapia phillipensis. J. Nat. Prod. 1999, 62, 1678–1681. [Google Scholar] [CrossRef]
- Zhou, B.N.; Mattern, M.P.; Johnson, R.K.; Kingston, D.G.I. Structure and stereochemistry of a novel bioactive sphingolipid from a Calyx sp. Tetrahedron 2001, 57, 9549–9554. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chianese, G.; Fattorusso, E.; Putra, M.Y.; Calcinai, B.; Bavestrello, G.; Moriello, A.S.; Petrocellis, L.D.; Marzo, V.D.; Taglialatela-Scafati, O. Leucettamols, Bifunctionalized Marine Sphingoids, Act as Modulators of TRPA1 and TRPM8 Channels. Mar. Drugs 2012, 10, 2435-2447. https://doi.org/10.3390/md10112435
Chianese G, Fattorusso E, Putra MY, Calcinai B, Bavestrello G, Moriello AS, Petrocellis LD, Marzo VD, Taglialatela-Scafati O. Leucettamols, Bifunctionalized Marine Sphingoids, Act as Modulators of TRPA1 and TRPM8 Channels. Marine Drugs. 2012; 10(11):2435-2447. https://doi.org/10.3390/md10112435
Chicago/Turabian StyleChianese, Giuseppina, Ernesto Fattorusso, Masteria Yunovilsa Putra, Barbara Calcinai, Giorgio Bavestrello, Aniello Schiano Moriello, Luciano De Petrocellis, Vincenzo Di Marzo, and Orazio Taglialatela-Scafati. 2012. "Leucettamols, Bifunctionalized Marine Sphingoids, Act as Modulators of TRPA1 and TRPM8 Channels" Marine Drugs 10, no. 11: 2435-2447. https://doi.org/10.3390/md10112435
APA StyleChianese, G., Fattorusso, E., Putra, M. Y., Calcinai, B., Bavestrello, G., Moriello, A. S., Petrocellis, L. D., Marzo, V. D., & Taglialatela-Scafati, O. (2012). Leucettamols, Bifunctionalized Marine Sphingoids, Act as Modulators of TRPA1 and TRPM8 Channels. Marine Drugs, 10(11), 2435-2447. https://doi.org/10.3390/md10112435