1. Introduction
Chemotherapy remains an important and irreplaceable modality of cancer treatment that utilizes chemical drugs to kill, or inhibit, the growth of cancer cells. To date, hundreds of compounds have been used in cancer chemotherapy [
1]. Employment of chemotherapy in patients with cancer, however, often fails to achieve clinical benefit due to severe adverse effects. Furthermore, development of resistant to chemotherapeutic drugs, particularly through multidrug resistance (MDR), represents a major impediment to successful chemotherapy [
2]. Tumor cells are found to adopt multiple mechanisms to resist drugs, including decreased uptake of drugs and/or enhanced efflux of drugs, altered metabolism of drugs, alterations in drug targets, activation of the detoxification system, enhanced DNA repair ability and inhibition of apoptosis [
2]. Hence, an increasing demand for the availability of new lead compounds drives an intense search for new bioactive natural products. Currently, it is of particular note that pharmacologically-active compounds derived from marine organisms represent a promising source for developing new investigational anti-cancer drugs, as they produce a wide variety of metabolites that are structurally unique and biologically active [
3].
Breast cancer remains the most commonly diagnosed type of cancer among women and a major health problem worldwide [
1]. Despite many advances in the treatment of breast cancer, resistance to anti-breast cancer agents represents a major challenge to the development of successful treatment. Mechanistically, resistance to chemotoxic agents in breast cancer patients is most often associated with the action of ATP-binding cassette (ABC) drug transporters, which include ABC transporter-subfamily B member 1 (ABCB1, or P-glycoprotein/P-gp), subfamily C member 1 (ABCC1, or multidrug resistance-associated protein 1/MRP1) and subfamily G member 2 (ABCG2, or breast cancer resistance protein/BCRP) [
2,
4].
Several new studies have shown that growth factor receptor-mediated signal transduction, such as the HER2/phosphoinositide-3-kinases(PI3K)/Akt signaling pathway, has been implicated in conferring resistance to conventional chemotherapy against breast cancer [
5,
6,
7,
8]. Moreover, the PI3K/Akt pathway has been implicated in the regulation of a wide variety of cellular processes including survival, proliferation, growth and metabolism [
9]. Genetic deregulation of PI3K activity has been found in a wide variety of tumor types, including somatic oncogenic gain-of-function mutations and amplification of genes encoding key components along the pathway [
10,
11]. Several new studies suggest that HER2/PI3K/Akt signaling may confer resistance to a panel of chemotherapeutic agents with different mechanisms of actions: adriamycin (an anthracycline) [
6,
7,
8], mitoxantrone (an anthracycline) [
8], 5-fluorourocil (an antimetabolite) [
7,
8], etoposide (a DNA-damaging agent) [
7,
8], camptothecin (a topoisomerase I inhibitor) [
7]
etc. Such resistance may be relevant to the effects of MDR in breast cancer. It has been documented that activation of Akt by HER2/PI-3K plays an important role in conferring broad-spectrum chemoresistance in breast cancer cells. Thus, Akt is believed to be a novel molecular target for therapies that would improve the outcome of anti-breast cancer chemotherapy in patients with breast cancer [
12].
Anthracyclines are the most commonly used anti-cancer treatment. Anthracycline drugs, such as Epirubicin and Mitoxantrone, are still in the first-line treatment against many types of cancer, including acute leukemia, Hodgkin’s lymphoma and breast cancer [
13,
14]. Clinical application of anthracyclines, however, is limited by their tendency to induce multidrug resistance [
14]. It is therefore important to identify new anthracenedione derivatives with improved pharmacological and toxicological profiles. In our previous study, SZ-685C [
15], a secondary metabolite purified from the mangrove endophytic fungus No. 1403, collected from the South China Sea, was found to exhibit structural similarity to anthracyclines, the anti-cancer drug widely used in the clinic. We demonstrated that SZ-685C induces apoptosis through Akt signaling, which consequently led to antitumor effects both
in vitro and
in vivo, suggesting that SZ-685C may be a potentially promising Akt inhibitor and anti-cancer drug candidate [
15].
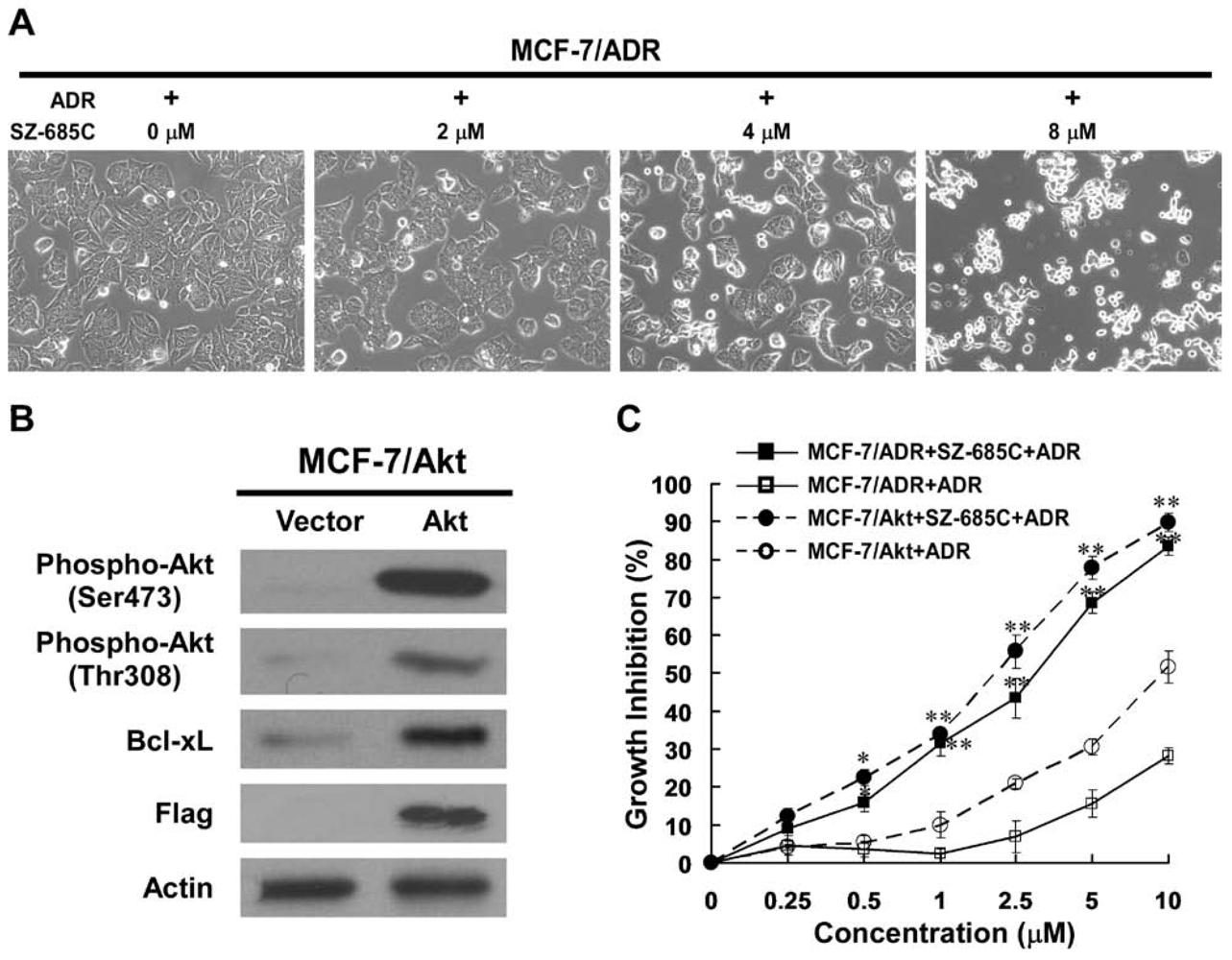
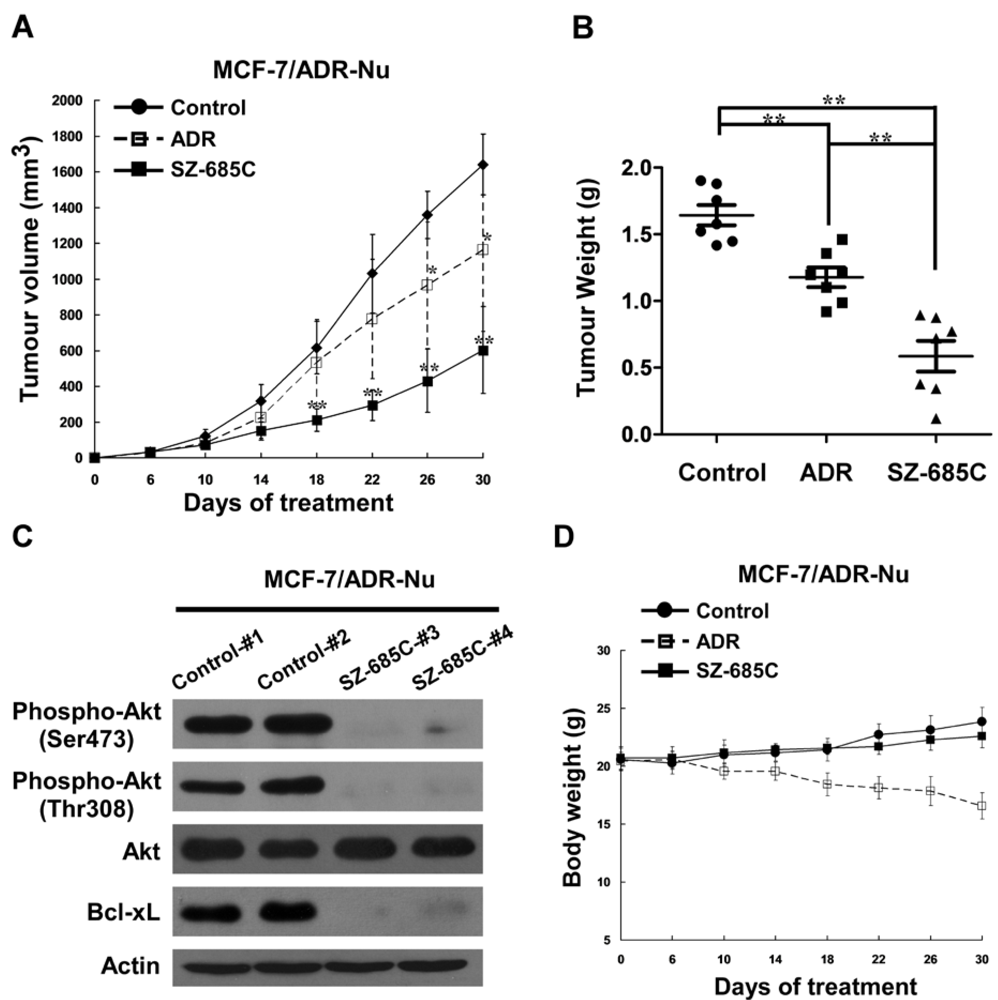
The current study attempts to address whether SZ-685C can reverse chemoresistance by suppressing the Akt signaling in human breast cancer cells. Our results demonstrated that SZ-685C suppressed the proliferation of ADR-resistant MCF-7/ADR and MCF-7/Akt breast cancer cells as well as the growth of MCF-7/ADR xenografts, suggesting a potentially promising approach to the treatment of ADR-resistant breast cancer.
3. Discussion
Anthracyclines, such as ADR, are a class of anti-cancer chemotherapeutic drugs commonly used in the clinic to treat various types of human cancer, including breast cancer. Clinical application of anthracyclines, however, is frequently impeded by their severe adverse side effects, e.g., cardiotoxicity [
18,
19], and myelosuppression [
20], as well as the tendency to induce multidrug resistance [
21]. There is, therefore, an urgent need to identify new anthracenedione derivatives with improved pharmacological and toxicological profiles. In our previous study that aimed at screening novel anticancer lead compounds from a metabolite library derived from marine microorganisms isolated from the South China Sea, SZ-685C was selected for its potent anticancer activity
in vitro as well as
in vivo [
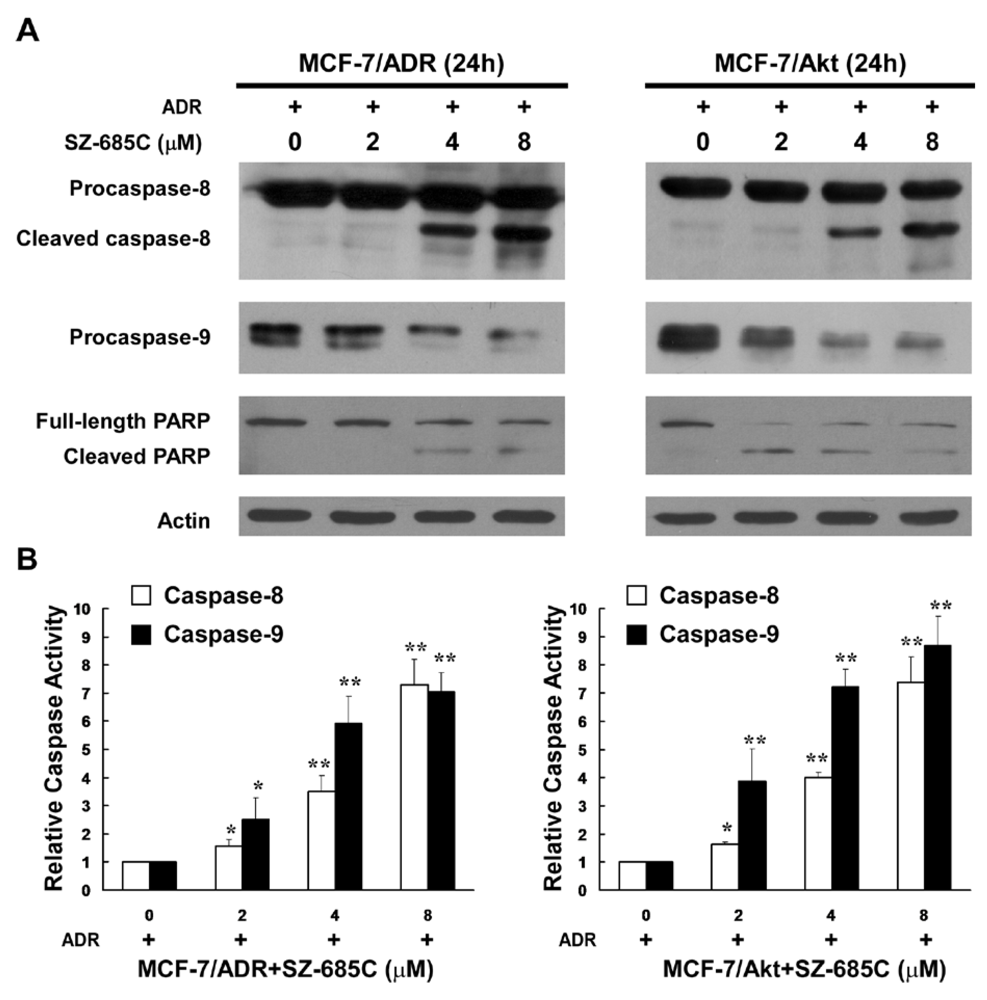
15]. We have demonstrated that SZ-685C selectively kills cancer cells via activating both caspase-8- and caspase-9-mediated apoptotic mechanisms by, at least in part, suppressing the phosphorylation of Akt. In our current study, we provide evidence that SZ-685C is able to kill breast cancer cells resistant to the anti-cancer effect of ADR, and thereby might represent a new, potentially promising approach to the treatment of anthrocycline-resistant breast cancer.
This study employed a cellular model MCF-7/ADR that was originally derived from the human breast cancer cell line MCF-7, and selected for resistance to ADR. It is of note that the parental cell line of the MCF-7/ADR and MCF-7/Akt models,
i.e., the MCF-7 cell line, is relatively well differentiated and estrogen-dependent, containing receptors for estrogen and progesterone [
22,
23]. Interestingly, these estrogen responses are lost in MCF-7/ADR cells. It has been reported that the MCF-7/ADR cells form tumors in the nude mice in the absence of exogenous estrogen administration, while the formation of tumors in nude mice by wild-type MCF-7 cells is estrogen-dependent [
24]. Furthermore, MDR presented by MCF-7/ADR cells is also associated with a loss of mitogenic response to estrogen and the development of cross-resistance to the anti-estrogen, 4-hydroxytamoxifen. These changes in the hormonal sensitivity and estrogen-independent tumorigenicity of the multidrug-resistant MCF-7/ADR cell line might be attributable to loss of estrogen receptor and a concomitant increase in the level of other receptors, such as those for epidermal growth factor. The results obtained from our current study warrants further exploration of the possibility of SZ-685C to sensitize breast cancer cells to anti-estrogen drugs.
A second test model used in our study,
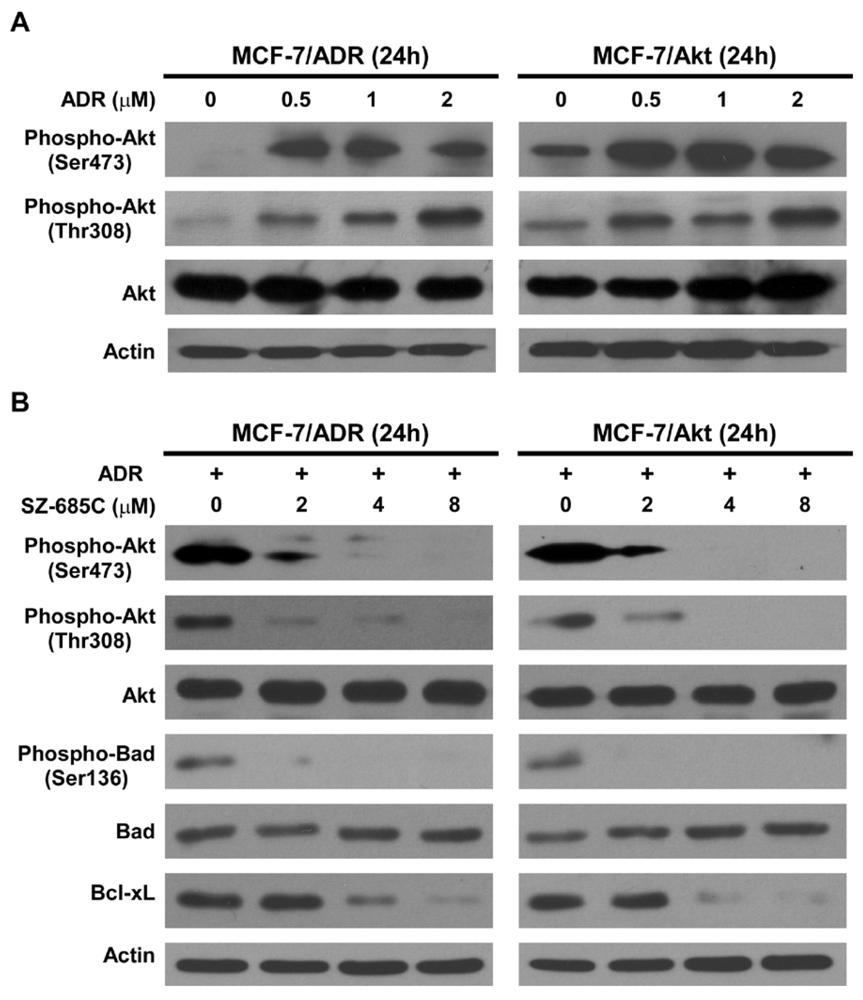
i.e., the MCF-7/Akt cell, is a derivative of the MCF-7 cells, engineered to express constitutively activated Akt. As a central mediator of PI3K signaling, Akt plays a key role in protecting cells from various apoptotic stimuli by phosphorylating diverse downstream targets, such as Forkhead family transcription factors, Bad, Bcl-xL, InB kinase, MDM2 and procaspase-9 [
9,
25]. Akt also interacts, directly or indirectly, with numerous other regulatory proteins, including cyclin D, p21
Cip1, glycogen synthase kinase (GSK)-3, and mTOR [
9,
25]. The PI3K/Akt pathway has thus been implicated in the regulation of a wide variety of cellular processes, including survival, proliferation, growth, and metabolism. Under physiological conditions, PI3Ks were localized to the plasma membrane upon activation, where it phosphorylates the substrate phosphatidylinositol-4, 5-diphosphate (PIP2), integral components of the plasma membrane, to phosphatidylinositol-3, 4, 5-triphosphate (PIP3) [
26]. PIP3 functions as a membrane-bound second messenger by recruiting a subset of pleckstrin homology (PH), domain-containing proteins, such as Akt and 3-phosphoinositide-dependent kinase 1 (PDK1), to the membrane, in which they become activated and initiate downstream signaling events [
26].
It was previously reported that MCF-7 cells expressing a constitutively active Akt, in which the phospholipid-interactive PH domain of Akt was replaced by a farnesylation sequence for constitutive membrane anchorage, showed a broad-spectrum chemoresistance on breast cancer cells [
6,
7]. Here, we also generated MCF-7/Akt stable cell lines by using the same approach, in which Akt protein was cleavable by the farnesyl-esterases that were expressed in mammalian cells, thereby allowing its cytoplasmic relocalization. Our results obtained from the current study indicate that the Akt kinase in MCF-7/Akt was constitutively active, as expected, and was sufficient to mediate resistance to ADR. The results are in line with the observation that the modified MCF-7 cells expressing constitutively activated Akt or a high level of HER2 were resistant to a panel of chemotherapeutic agents [
6,
7,
8]. These results strongly support the hypothesis that deregulated over-activation of Akt plays an important role in resistance to chemotherapy. SZ-685C, a novel anthracycline compound, as evidenced in this study, is able to potently suppress the phosphorylation of Akt and subsequently induce apoptosis in ADR-resistant breast cancer cells in which Akt kinase is over-activated. The involvement of Akt in human cancer oncogenesis and chemoresistance indicates that Akt is an important target for screening anti-cancer drugs against various types of cancer cells including those resistant to conventional chemotherapy.
4. Experimental Section
4.1. Chemicals
Unless otherwise stated, laboratory chemicals, bacterial culture reagents, antibiotics and disposable labware were purchased from the Sangon Biotech (Shanghai, China), Sigma-Aldrich (St. Louis, MO, USA), or BBI (Gaithersburg, MD, USA). SZ-685C was prepared and purified from mangrove endophytic fungus No. 1403 as previously reported [
15], and its structure was identified by interpretation of spectral data (MS,
1H NMR,
13C NMR, 2D NMR) and X-ray single crystal diffractive technique. The compound was dissolved in 0.5% dimethylsulfoxide (DMSO) at a concentration of 1 mM as stock solution, and diluted according to experimental requirements when used. ADR was purchased from Merck (Merck KGaA, Darmstadt, Germany).
4.2. Cell Culture
MCF-7/ADR (Human breast cancer), K562/ADR (Human erythromyeloid leukemia), and HL-60/ADR cells (Human promyelocytic leukemia) were obtained from Keygen Biotech (China). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco-Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 2 mM L-glutamine, 100 μg/mL streptomycin and 100 Units/mL penicillin (Gibco-Invitrogen, Carlsbad, CA), 1 μM ADR (to maintain ADR resistance phenotype, upon treatment with or without SZ-685C). All of the cells used in this study were maintained at 37 °C in a humidified atmosphere, 5% CO2 incubator.
4.3. Establishment of Stable Cell Lines Expressing Constitutively Activated Akt
Flag-tagged constitutively activated (PI3K-independent) Akt expression vector was constructed by replacing the phospholipid-interactive PH domain of Akt1 with a farnesylation sequence for directing constitutive membrane anchoring, termed Flag-DPH-Akt1-farn vector, as previously described [
7]. The final PCR product encoded Akt1 that lacks the PH domain (aminoacids 1-106) with an
N-terminal fusion of the Flag epitope (DYKDDDDK) and a
C-terminal fusion of a poly-basic tail and a farnesylation sequence (KKKKKKSKTKCVIM). The PCR fragment was subcloned into the pMSCV-Puro vector (Invitrogen, Carlsbad, CA, USA) in the restriction sites Bgl II and EcoR I. The final Akt sequence was verified by nucleotide sequencing analysis. Recombinant retroviruses were prepared by transfecting 2 μg of each of the pMSCV-Flag-DPH-Akt1-farn plasmid DNAs and pMSCV-Puro vector into the packaging cells 293FT, together with 2 μg of the pIK plasmid. These viruses were used to infect MCF-7 cells, and stably transduced cell lines were selected in DMEM medium supplemented with puromycin (5 μg/mL). To verify the expression of the Flag-DPH-Akt1-farn proteins, cell lysates were collected and examined using Western blotting analysis.
4.4. Cell Viability Analysis
Cells (1 × 10
4 cells/well) in growth medium were seeded in 96-well flat-bottom plates (in triplicates) for 24 h in the absence or presence of various concentrations of SZ-685C and 1 μM ADR for additional 48 h. Cell viability was measured by using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2
H-tetrazolium) assay to monitor cell proliferation, according to the manufacturer’s recommendations. Briefly, 20 μL MTS solution (CellTiter 96Aqueous One Solution reagent, Promega, Madison, WI, USA) were added to each well and incubated for an additional 4 h at 37 °C. The absorbance was measured at 490 nm using a microplate reader (Bio-Tek Synergy 2, Winooski, VT, USA). The control group was defined as cells that were treated with 1 μM ADR and not exposed to SZ-685C. Cell growth inhibition was determined using the following formula according to a previously published method: growth inhibition (%) = (1 − O.D. of treated cells/O.D. of control cells) × 100% [
15]. The half maximal inhibitory concentration (IC
50) was calculated with Bliss’s software and the data were analyzed by SPSS [
15]. The resistance factor (RF) was calculated as the ratio of the IC
50 for the resistant cell line to the IC
50 for the sensitive cell line. All experiments were performed three times from which mean values were calculated.
4.5. Annexin V-FITC/Propidium Iodide (PI) Staining Assay
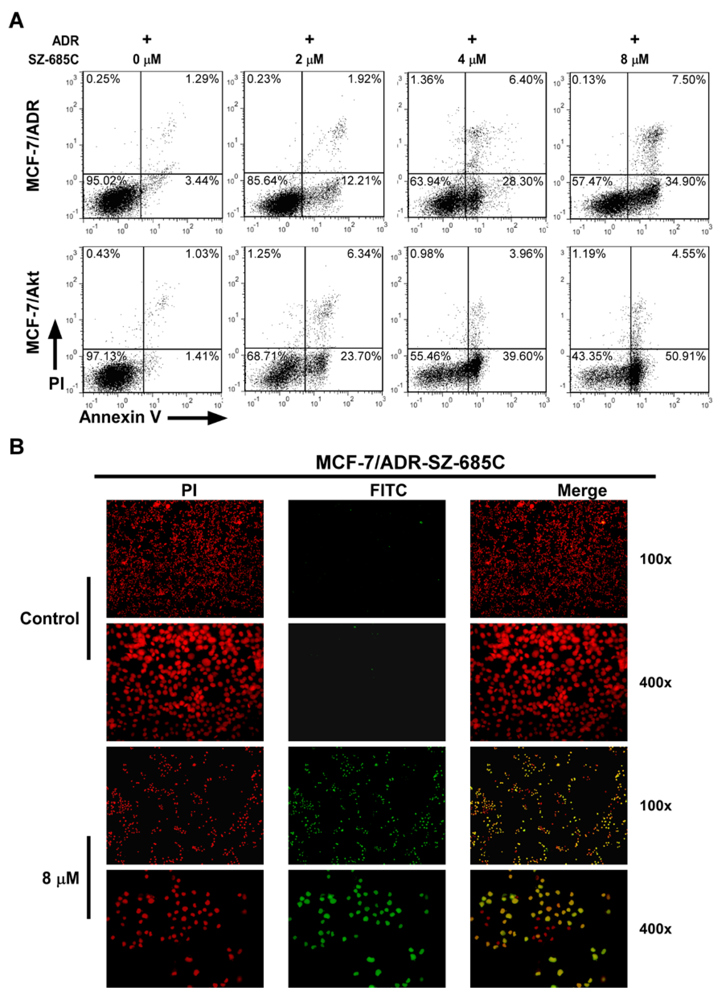
The Annexin V/Propidium iodide (PI) dual staining assay was employed to detect viable and early apoptotic cells. Cells (2 × 10
5 cells/well) were seeded in 60 mm plates and allowed to settle for 24 h before treatment with various concentrations (0, 2, 4, 8 μM) of SZ-685C and 1 μM ADR for an additional 12 h. The cells were trypsinized after washing for three times with PBS, and incubated in 500 μL binding buffer containing annexin V-FITC and PI in the dark for 10 min at room temperature, according to the manufacturer’s instructions (Keygen Biotech, China), followed by measurement with flow cytometric (FCM) analysis (FCAScan; Becton-Dickinson Immunocytochemistry Systems, San Jose, CA, USA). The percentage of apoptotic cells was defined by their distribution in a fluorescence dot plot using WinMdi 2.8 software [
27].
4.7. Caspase Activity Assay
Activity of Caspase-8 and Caspase-9 was measured using a caspase colorimetric assay kit (Keygen Biotech, China), according the manufacturer’s protocol. Briefly, following treatment of SZ-685C at different concentrations and 1 μM ADR for 48 h, cells were harvested and washed with PBS for two times, then resuspended in chilled 1 × lysis buffer. Cells lysates were centrifuged for 1 min at 10,000 × g after incubation on ice for 60 min. The supernatant was collected in a fresh tube, and the total protein concentration was determined by the Bradford protein assay kit (Keygen Biotech, China), according to the manufacturer’s protocol. Subsequently, 150 μg of each sample was diluted with 50 μL lysis buffer and added to 50 μL of 2 × reaction buffer containing 10 mM DTT in a 96-well plate. Then, 5 μL of a colorigenic substrate, IETD-pNA or LEHD-pNA, was added to each well, and the plate was incubated at 37 °C in the dark for 4 h. Absorbance at 405 nm was determined using a microplate reader (Bio-Tek Synergy 2, Winooski, VT).
4.8. Western Blotting Analysis
After treatment with SZ-685C or ADR at different concentrations for 48 h, floating cells and adherent cells were collected and lysed in 1× sample buffer containing 50 mM Tris-HCl (pH 7.4), 1 mM PMSF, 10% glycerol, 6% SDS, 5% mercaptoethanol and 0.1% bromophenol blue before sonication. The protein concentration of the extracts was determined by the Bicinchoninic Acid Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL), according to the manufacturer’s instructions with BSA as the standard. The total cell lysate (40 μg of protein) was subjected to SDS-PAGE, and then transferred to PVDF membranes. After blocking with blocking buffer (Tris-buffered saline, TBS, containing 5% non-fat milk) for 1 h at room temperature, the membranes were incubated overnight at 4 °C with specific primary antibodies, namely: mouse anti-human caspase-8, mouse anti-human caspase-9 (BD Biosciences, San Jose, CA, USA); rabbit anti-human PARP, rabbit anti-human phospho-Akt (Ser473), rabbit anti-human phospho-Akt (Thr 308), rabbit anti-human Akt, Phospho-Bad (Ser136) antibody, Bad antibody, Bcl-xL Antibody (Cell Signaling, Beverly, MA, USA), and monoclonal anti-Actin antibody (Sigma-Aldrich, St. Louis, MO, USA). Further incubation with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies, depending on the primary antibody used, was performed for 1 h at room temperature. Immuno-reactive bands were detected using enhanced chemiluminescence kit (Thermo Fisher Scientific, Rockford, IL, USA) with Kodak film. Actin was used as a loading control for quantity normalization.
4.9. Xenografted Tumor Model and Anti-Tumor Effect of SZ-685C in Vivo
Female BALB/c-nu mice (18-20 g) were purchased from the Shanghai Laboratory Animal Center, Chinese Academy Sciences (Shanghai SLAC Laboratory Animal Co. LTD.), and were housed in barrier facilities on a 12 h light/dark cycle. Three days before implanting MCF-7/ADR cells, mice received a 0.8 mg β-estradiol pellet (Innovative Research, Toledo, OH, USA) subcutaneously in their front-back area. On day zero, MCF-7/ADR cells (1 × 107 cells in 0.1 mL/mouse) were inoculated subcutaneously in the right mammary pad. Mice were ear-tagged and randomly divided into two treatment groups and a control group in a blinded fashion before treatment. On day six, formed tumors were measured, and the two treatment groups of animals received an intraperitoneal (i.p.) dosage of 200 μL SZ-685C (50 mg/kg body weight) and ADR (8 mg/kg body weight), respectively, every four days, while the vehicle-control animals received daily i.p. doses of 200 μL 0.5% DMSO solution per mouse. Tumors were measured every three days in a blinded manner by measuring perpendicular diameters with a digital caliper, and tumor volumes (mm3) were calculated using the following formula: volume = width × width × length × π/6. Data were presented as the means ± standard deviation (SD) of seven mice in each group. At the endpoint of the experiment, all the animals were euthanized, and the tumors were dissected and weighed. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University.
4.10. Statistical Analysis
The data given in the text are expressed as means ± SD. For MTS assay, statistical analyses were undertaken using the Student’s t test. Statistical significances of the differences in tumor volumes between treatment and control groups were determined by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




