2. Results and Discussion
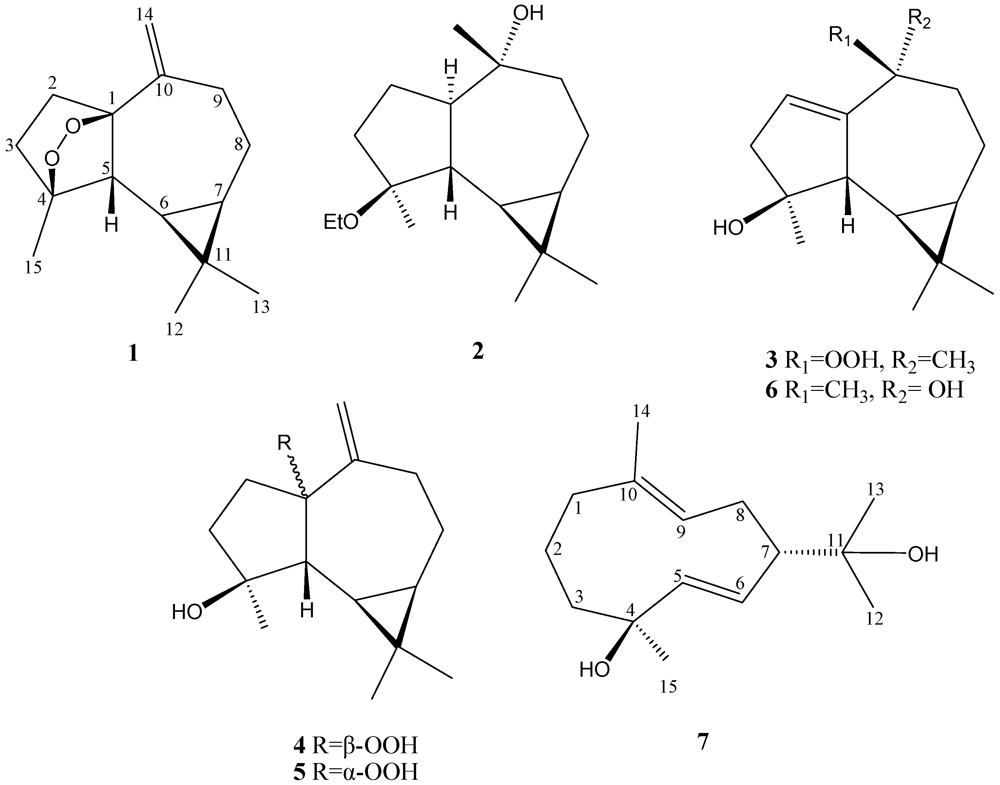
Lochmolin A (
1) was isolated as a colorless oil. Its molecular formula, C
15H
22O
2, was established by HREIMS (
m/z 234.1620, [M]
+), implying five degrees of unsaturation. The
13C NMR spectral data of
1 (
Table 1), showed the presence of 15 carbon atoms, including three methyls (δ
C 28.5, 24.8, and 16.2) and two quaternary sp
3oxycarbons (δ
C 89.3 and 83.1), as assigned by the DEPT spectrum, suggesting the oxygenated sesquiterpenoid nature of
1. The NMR signals (
Table 1 and
Table 2) observed at δ
C 112.7 (CH
2) and 151.5 (C), δ
H 5.02 and 4.89 (each 1H, s) showed the presence of one 1,1-disubstituted double bond. Thus, the tetracyclic structure of
1 was revealed. In the
1H-
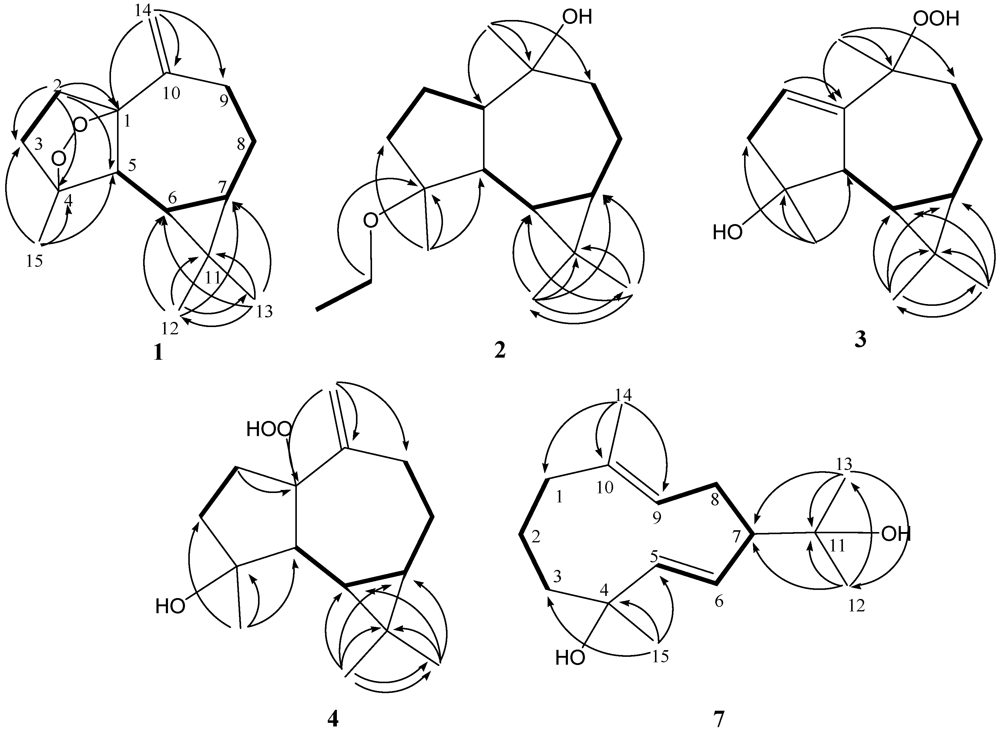
1H COSY spectrum it was possible to identify two different structural units, which were assembled with the assistance of an HMBC experiment. Key HMBC correlations of H
2-2 to C-1, C-3, C-4 and C-5; H
3-12 to C-6, C-7, C-11 and C-13; H
3-13 to C-6, C-7, C-11 and C-12; H
2-14 to C-1, C-9 and C-10; H
3-15 to C-3, C-4 and C-5 permitted the establishment of the aromadendrane-type skeleton of
1 (
Figure 1). Furthermore, the two additional oxygen atoms could be used to form an endoperoxide bridge in the cyclopentane moiety of the molecule from the downfield chemical shifts of the sp
3 carbons C-1 (δ 89.3, C) and C-4 (δ 83.1, C). The presence of a cyclopropane was further confirmed by the upfield chemical shifts of H-6 (δ 0.08) and H-7 (δ 0.54).
Table 1.
13C NMR spectroscopic data for compounds 1–7.
Table 1.
13C NMR spectroscopic data for compounds 1–7.
| | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|
| 1 | 89.3 | 47.3 | 150.1 | 101.1 | 96.8 | 155.2 | 41.1 |
| 2 | 36.0 | 23.4 | 125.2 | 29.6 | 29.1 | 117.3 | 23.4 |
| 3 | 38.9 | 37.8 | 45.3 | 39.0 | 39.6 | 45.1 | 40.8 |
| 4 | 83.1 | 79.3 | 82.2 | 81.7 | 81.0 | 82.2 | 73.0 |
| 5 | 56.2 | 49.9 | 52.6 | 55.2 | 57.3 | 53.8 | 143.1 |
| 6 | 27.4 | 25.1 | 27.4 | 26.6 | 21.4 | 27.2 | 128.4 |
| 7 | 23.0 | 26.2 | 27.0 | 24.2 | 27.6 | 27.4 | 57.9 |
| 8 | 20.9 | 20.1 | 18.8 | 20.6 | 25.2 | 20.2 | 24.7 |
| 9 | 31.4 | 40.3 | 38.3 | 33.1 | 34.1 | 43.4 | 129.5 |
| 10 | 151.5 | 80.4 | 83.0 | 147.1 | 150.7 | 73.9 | 131.9 |
| 11 | 18.4 | 19.0 | 20.3 | 18.5 | 19.9 | 19.0 | 71.9 |
| 12 | 28.5 | 28.9 | 28.5 | 28.4 | 28.6 | 28.3 | 26.8 |
| 13 | 16.2 | 16.4 | 16.0 | 16.0 | 15.7 | 16.0 | 26.9 |
| 14 | 112.7 | 25.7 | 23.0 | 116.2 | 113.3 | 27.3 | 16.6 |
| 15 | 24.8 | 18.8 | 22.9 | 24.3 | 27.6 | 22.4 | 23.4 |
| OEt | | 55.2 | | | | | |
| | 16.3 | | | | | |
Table 2.
1H NMR spectral data for compounds 1–4.
Table 2.
1H NMR spectral data for compounds 1–4.
| | 1 | 2 | 3 | 4 |
|---|
| 1 | | 1.05 m | | |
| 2 | 2.01 m2.45 m | 1.70 m1.80 m | 5.66 d (3.0) | 2.19 m2.46 m |
| 3 | 1.95 m2.05 m | 1.62 m1.72 m | 2.27 dd (17.0, 3.0)2.61 d (17.0) | 1.91 m1.97 m |
| 5 | 1.77 d (12.0)
a | 2.38 dd (11.0, 5.5) | 2.31 d (11.0) | 1.68 d (11.5) |
| 6 | 0.08 dd (12.0, 10.0) | 0.64 m | 0.27 dd (11.0, 9.5) | 0.19 dd (11.5, 9.0) |
| 7 | 0.54 dd (17.0, 10.0) | 0.67 m | 0.53 m | 0.58 m |
| 8 | 1.48 m1.87 m | 0.89 m1.86 m | 1.46 m1.65 m | 1.40 m1.80 m |
| 9 | 2.30 m2.65 m | 1.48 m1.68 m | 1.64 m1.92 dd (14.5, 5.5) | 2.35 m2.63 m |
| 12 | 1.00 s | 1.05 s | 1.02 s | 1.02 s |
| 13 | 1.06 s | 0.97 s | 1.12 s | 1.04 s |
| 14 | 4.89 s5.02 s | 1.24 s | 1.49 s | 5.08 s5.14 s |
| 15 | 1.30 s | 1.05 s | 1.40 s | 1.27 s |
| 16 | | 3.40 m3.44 m | | |
| 17 | | 1.12 t (7.0) | | |
| 10-OOH | | | 7.64 s | |
Figure 1.
Selected 1H-1H COSY (▬) and HMBC (→) correlations of 1–4 and 7.
Figure 1.
Selected 1H-1H COSY (▬) and HMBC (→) correlations of 1–4 and 7.
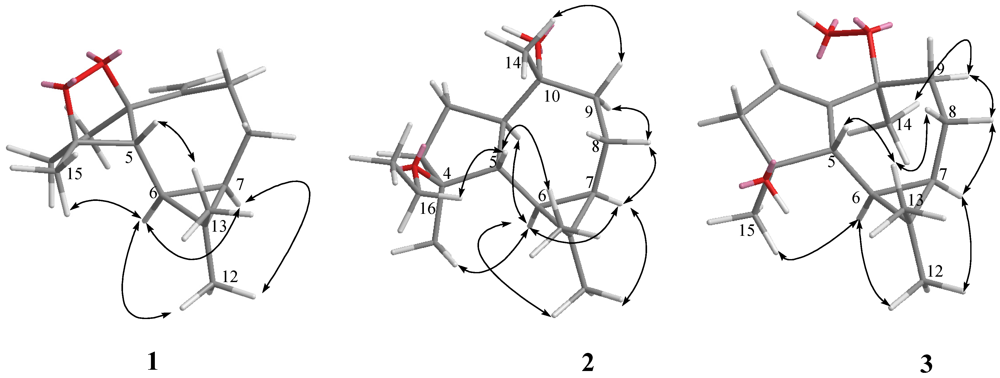
The relative configurations of five chiral centers at C-1, C-4, C-5, C-6, and C-7 in
1 were elucidated by NOE analysis (
Figure 2). It was found that H-6 showed NOE correlations with H-7, H
3-12, and H
3-15; H-7 (δ 0.54) showed NOE correlations with H
3-12; and H-5 (δ 1.77) showed NOE correlations with H
3-13. Thus H-6, H-7, and H
3-15 were assumed to be positioned on the α face, and H-5 was assumed to be positioned on the β face. On the basis of these results, lochmolin A (
1) was found to possess the (1
R*,4
S*,5
R*,6
R*,7
R*) configuration.
Figure 2.
Key NOESY correlations for 1–3.
Figure 2.
Key NOESY correlations for 1–3.
Lochmolin B (
2) was obtained as a colorless oil. HRESIMS showed the molecular formula C
17H
30O
2, requiring three degrees of unsaturation. The IR spectrum suggested the presence of hydroxy group (3437 cm
−1). The 3H triplet appearing at δ 1.12 (
J = 7.0 Hz) in the
1H NMR spectrum and the methylene carbon signal at δ 55.2 in the
13C NMR spectrum were ascribable to an ethoxy group. Comparison of the NMR data (
Table 1) of
2 with those of
1 also showed the aromadendrane skeleton of
2. In the 2D NMR spectra, including
1H-
1H COSY and HMBC (
Figure 1), three segregate consecutive proton spin systems, H-1 to H
2-3, H-5 to H
2-9, and CH
2 to CH
3 of an ethoxy group, were found in the
1H-
1H COSY spectrum. The detailed analysis of HMBC correlations further established the planar structure of
2. The relative structure of
2 was elucidated by the analysis of NOE correlations, as shown in
Figure 2. It was found that H-6 (δ 0.64, m) showed NOE interactions with H-1 (δ 1.05, m), H-7 (δ 0.67, m), H
3-12 (δ 1.05, s) and H
3-15 (δ 1.05, s), and H-7 (δ 0.67, m) showed NOE correlations with H-8α (δ 1.86, m) and H
3-12 (δ 1.05, s); therefore, assuming the α-orientation of H-1, all of H-6, H-7, H
3-12 and H
3-15 should also be positioned on the α face. One of the methylene protons at C-9 (δ 1.68, m) exhibited NOE correlations with H-8α (δ 1.86, m) and was characterized as H-9α, while the other (δ 1.48, m) was assigned as H-9β. NOE correlations observed between H-9β and H
3-14, and H-5 with both H
3-13 (δ 0.97, s) and protons of OCH
2, reflected the β-orientation of H-5 and H
3-14. On the basis of the above findings (
Figure 2), the relative structure of lochmolin B (
2) was determined.
The HRESIMS spectrum of lochmolin C (
3) exhibited a molecular ion peak at
m/z 275.1622 ([M + Na]
+), consistent with the molecular formula C
15H
24O
3 and implying four degrees of unsaturation. The IR absorption of
3 also revealed the presence of hydroxy group (3437 cm
−1). Comparison of the NMR data (
Table 1 and
Table 2) of
3 with those of
2 showed the appearance of an additional trisubstituted double bond in
3. The NMR chemical shifts for C-4 and C-10 of
3 (δ 82.2 and δ 83.0, respectively), were found to be shifted downfield in comparison with the analogous data of
2 (δ 79.3 and δ 80.4), suggesting that the 4-OEt and 10-OH of
2 might be replaced by the 4-OH and 10-OOH (δ 7.64, s) in
3. This could be confirmed from the carbon shifts of both hydroxylated quaternary carbons C-4 (δ 82.2) and C-10 (δ 73.9) of
6 (latter discussed) which showed the identical chemical shift of C-4 of compound
3. By analysis of 2D NMR spectra (HMQC,
1H-
1H COSY, and HMBC), compound
3 was shown to possess the same molecular framework as that of
2. Investigation of the NOESY spectrum of
3 (
Figure 2) revealed the NOE interactions of H-7 (δ 0.53, m) with H-8α (δ 1.65, m), H-8α with H-9α (δ 1.92, dd,
J = 14.5, 5.5 Hz), and H-9α with H
3-14 (δ 1.49, s), suggesting the α-orientation of H
3-14. Further analysis of other NOE interactions revealed that
3 possessed the same relative configurations at C-4, C-5, C-6, and C-7 as those of
2.
Lochmolin D (
4) was also isolated as a colorless oil with a molecular formula of C
15H
24O
3. The ESIMS and NMR spectroscopic data of
4 (
Table 1) showed the presence of a hydroxy and hydroperoxy moiety [δ 81.7 (C), and 101.1 (C)]. Comparison of the NMR data of
4 with those
1 revealed that the two differences between both compounds were the replacement of the endoperoxide bridge moiety at C-1 and C-4 in
1 by the hydroxy at C-4 and the hydroperoxy at C-1 in
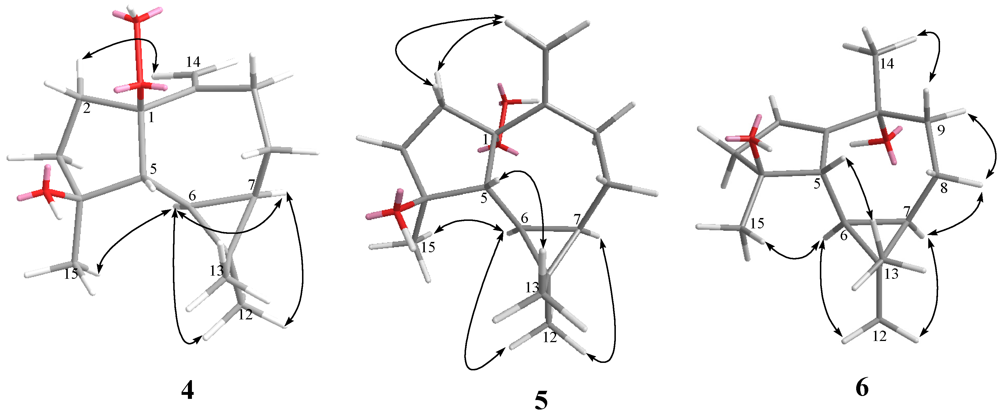
4. The relative structure of
4 was elucidated by the analysis of NOE correlations, as shown in
Figure 3. It was found that H-6 (δ 0.19, dd,
J = 11.5, 9.0 Hz) showed NOE interactions with H-7 (δ 0.58, m), H
3-12 (δ 1.02, s), and H
3-15 (δ 1.27, s), but not with H-5 (δ 1.68, d,
J = 9.0 Hz); therefore, assuming an α-orientation of H-6, H-7 and H
3-15 should also be positioned on the α face, and H-5 should be placed on the β face. One of the sp
3methylene proton at C-2 (δ 2.19, m) exhibited NOE correlations with one of the sp
2methylene proton at C-14 (δ 5.08, s), suggesting the β-orientation of 1-OOH by inspecting the molecular model of
4. If the 1-OOH was placed on the α face as in the case of
5 (latter discussed), both protons at C-2 were found to exhibit NOE correlations with one of the sp
2 proton at C-14 by molecular modeling study. On the basis of the above findings, the relative structure of
4 was determined.
Figure 3.
Key NOESY correlations for 4–6.
Figure 3.
Key NOESY correlations for 4–6.
HRESIMS and NMR spectroscopic data (
Table 1 and
Table 3) revealed that lochmolin E (
5) has the same molecular formula, C
15H
24O
3, as that of
4. By analysis of 2D NMR spectra, including
1H-
1H COSY, HMQC, and HMBC, compound
5 was shown to possess the same molecular framework as that of
4. Comparison of the NMR data of
5 with those of
4 revealed that the only difference between the compounds was the replacement of the β-hydroperoxy group at C-1 in
4 by the α-hydroperoxy group in
5. From the NOESY spectrum, it was found both protons of H
2-2 (each 1H, δ 2.19 and 2.24, m) showed NOE interactions with one of the sp
2methylene proton at C-14 (δ 4.97, s), suggesting the α-orientation of 1-OOH by investigation of the molecular model (
Figure 3). Further analysis of other NOE interactions revealed that
5 possesses the same relative configurations at C-4, C-5, C-6, and C-7, as those of
4. Therefore,
5was found to be the C-1 epimer of
4.
Lochmolin F (
6) was obtained as a colorless oil and exhibited an ion peak at
m/z 236.1774 ([M]
+) by HREIMS, appropriate for the molecular formula C
15H
24O
2. Comparison of the NMR data of
6 with those of
3 revealed that the only difference between both compounds was the replacement of a hydroperoxy group at C-10 in
3 by the hydroxy moiety in
6. This was evidenced from the upfield chemical shifts induced by a hydroxy group at C-10 (δc 73.9) and H
3-14 (δ
H 1.32) in
6 relative to those of
3. The relative configuration of
6 was determined by analysis of key NOE correlations (
Figure 3).
The metabolite lochmolin G (
7) was also obtained as a colorless oil. Its HRESIMS spectroscopic data (
m/z 261.1828) suggested the molecular formula C
15H
26O
2, requiring three degrees of unsaturation. IR absorption was observed at 3303 cm
−1, suggesting the presence of hydroxy group in
7. In the
13C NMR and DEPT spectra (
Table 1), signals of four methyls, four sp
3methylenes, one sp
3methine, three sp
2methines, two sp
3 quaternary carbons, and one sp
2 quaternary carbons were observed. The
13C NMR data of
7 (
Table 1) revealed the presence of one trisubstituted and one 1,2-disubstituted carbon-carbon double bond [δc 131.9 (C) and 129.5 (CH); 143.1 (CH) and 128.4 (CH)]. Two hydroxylated carbons (δc 73.0 and 71.9) were also assigned from the
13C NMR spectrum. The remaining one degree of unsaturation identified
7 as a cyclic compound. The planar structure of metabolite
7 was elucidated by analysis of
1H-
1H COSY and HMBC correlations (
Figure 1). Key HMBC correlations from H
3-12 to C-7, C-11, and C-13; H
3-13 to C-7, C-11, and C-12; H
3-14 to C-1, C-9, and C-10; H
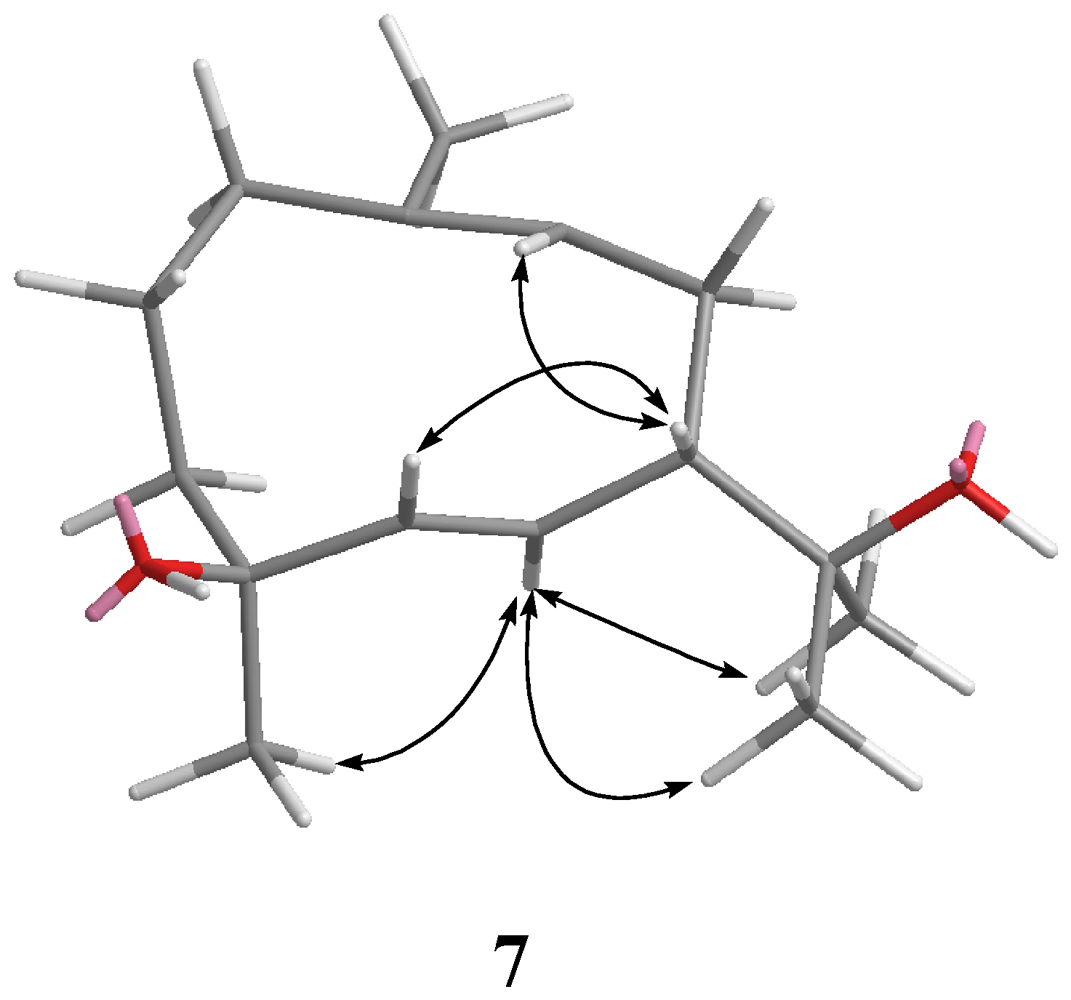
3-15 to C-3, C-4, and C-5 permitted the establishment of the germacrane skeleton. In the NOESY spectrum of
7 (
Figure 4), observation of the NOE correlations between H-6 and H
3-12, H
3-13 and H
3-15, and between H-5 and H-7, suggested that H
3-15 is α-oriented, and H-7 is β-oriented. The
E geometries were assigned for the 5,6- and 9,10- double bonds on the basis of the upfield chemical shift of C-14 (δ 16.6) and the large coupling constant between H-5 and H-6 (
J = 16.0 Hz). Therefore, the relative structure of
7 was established.
Table 3.
1H NMR spectral data for compounds 5–7.
Table 3.
1H NMR spectral data for compounds 5–7.
| | 5 | 6 | 7 |
|---|
| 1 | | | 2.24 m |
| | | | 2.29 m |
| 2 | 2.19 m | 5.54 dd (3.0, 1.5)
a | 1.61 m |
| | 2.24 m | | |
| 3 | 1.78 m | 2.25 dd (16.5, 3.0) | 1.65 m |
| | 1.98 m | 2.55 d (16.5) | 1.70 m |
| 5 | 1.75 d (12.0) | 1.96 d (10.0) | 5.44 d (16.0) |
| 6 | 0.50 dd (12.0, 9.5) | 0.29 dd (10.0, 9.5) | 5.13 dd (16.0, 10.0) |
| 7 | 0.81 m | 0.57 m | 2.23 m |
| 8 | 0.99 m | 0.98 m | 2.07 m |
| | 2.08 m | 1.90 m | 2.27 m |
| 9 | 2.32 dd (13.0, 6.5) | 1.59 m | 4.90 brd (11.5) |
| | 2.44 t (13.0) | 1.89 m | |
| 12 | 1.07 s | 1.03 s | 1.11 s |
| 13 | 1.01 s | 1.08 s | 1.16 s |
| 14 | 4.97 s | 1.32 s | 1.53 s |
| | 4.99 s | | |
| 15 | 1.33 s | 1.37 s | 1.37 s |
| 1-OOH | 7.06 s | | |
Figure 4.
Key NOESY correlations for 7.
Figure 4.
Key NOESY correlations for 7.
Cytotoxicity of compounds 1–7 against the proliferation of a limited panel of cancer cell lines, including human cervical epitheloid (HeLa), liver (SK-Hep1), and melanin (B-16) carcinoma cells, was evaluated. The results showed all of compounds were not cytotoxic toward these three cancer cell lines. The anti-inflammatory activities of 1–7 against the accumulation of pro-inflammatory iNOS and COX-2 proteins in RAW264.7 macrophage cells were evaluated by Western blot analysis. It was found that 1–7 could not reduce the accumulation of iNOS protein induced by LPS. At a concentration of 1 μM, only compound 1 could reduce the level of LPS-induced COX-2 to 36.6 ± 3.8%. At a concentration of 10 μM, compounds 1, 3, and 4 reduced the accumulation of LPS-induced COX-2 to 8.7 ± 4.5%, 61.0 ± 6.0%, and 83.4 ± 6.4%, respectively. At a concentration of 100 μM, 1–4 could further reduce the levels of induced COX-2 to 1.7 ± 1.3%, 17.6 ± 2.2%, 32.8 ± 3.2%, and 71.3 ± 7.2%, respectively, in comparison with those of control cells stimulated with LPS only. Thus, compound 1 might be considered to be a promising COX-2 inhibiting agent.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



