Fucoxanthin Enhances Cisplatin-Induced Cytotoxicity via NFκB-Mediated Pathway and Downregulates DNA Repair Gene Expression in Human Hepatoma HepG2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
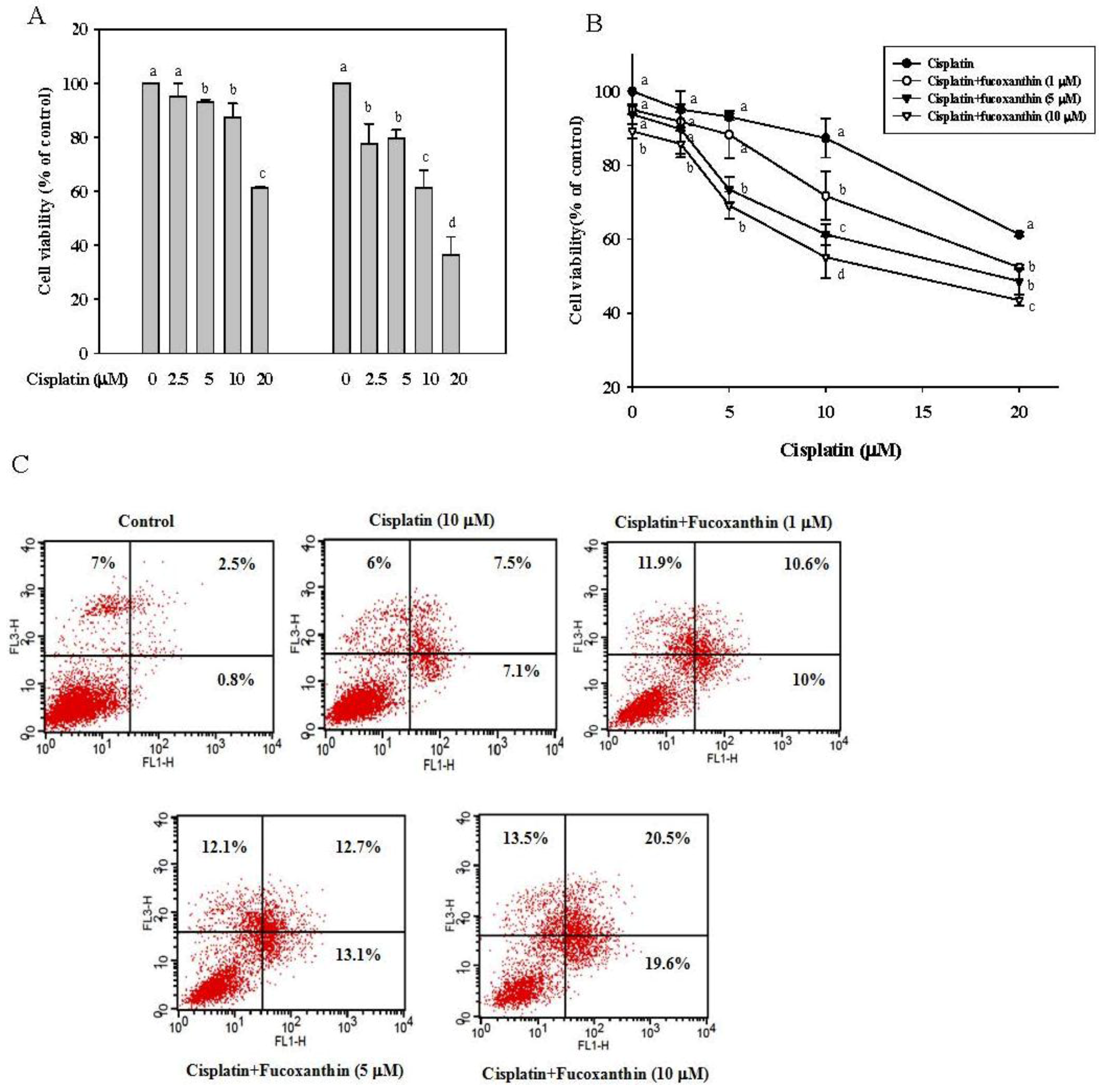
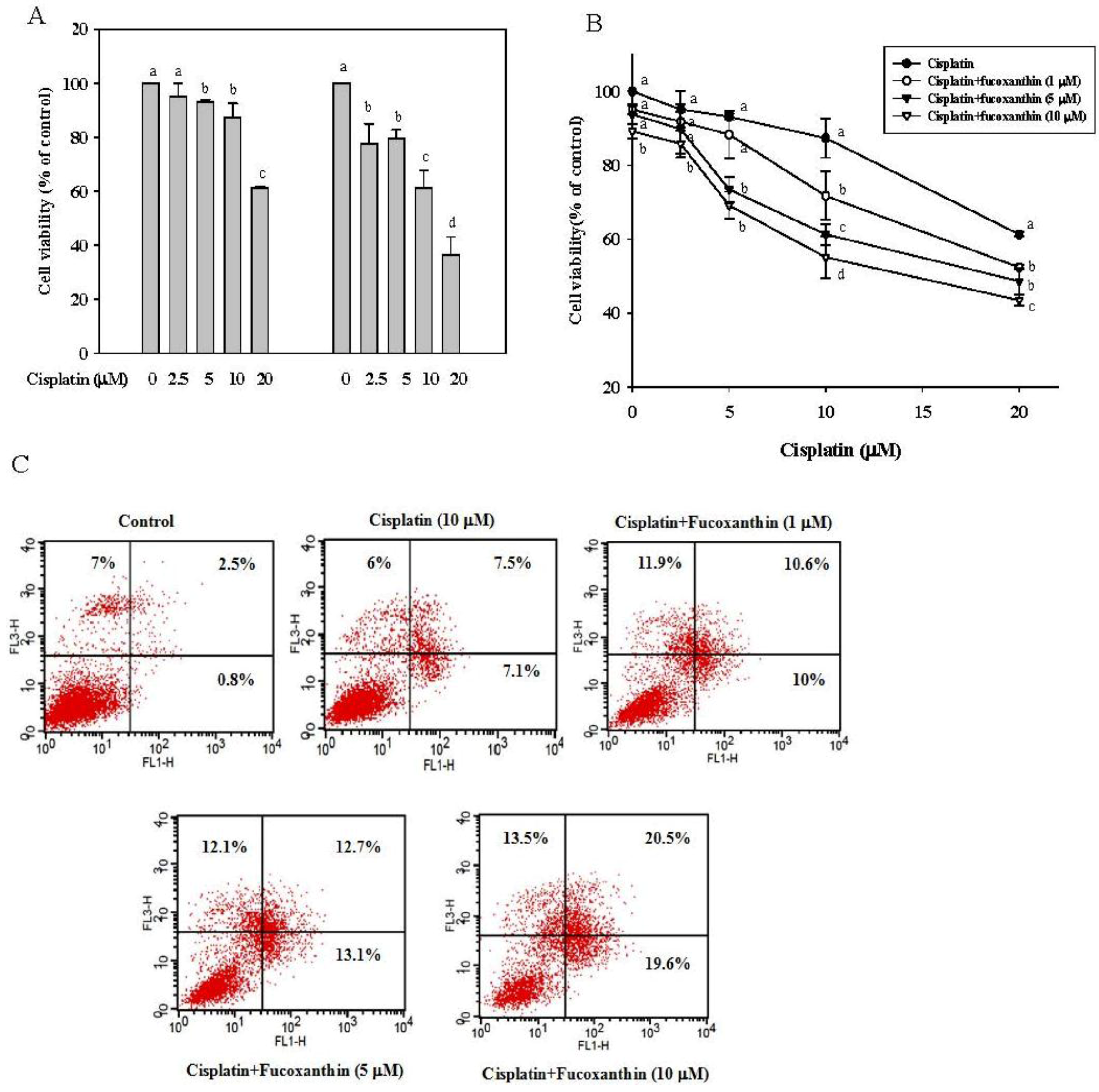
2.1. Fucoxanthin Increases the Sensitivity of Cisplatin in HepG2 Cells
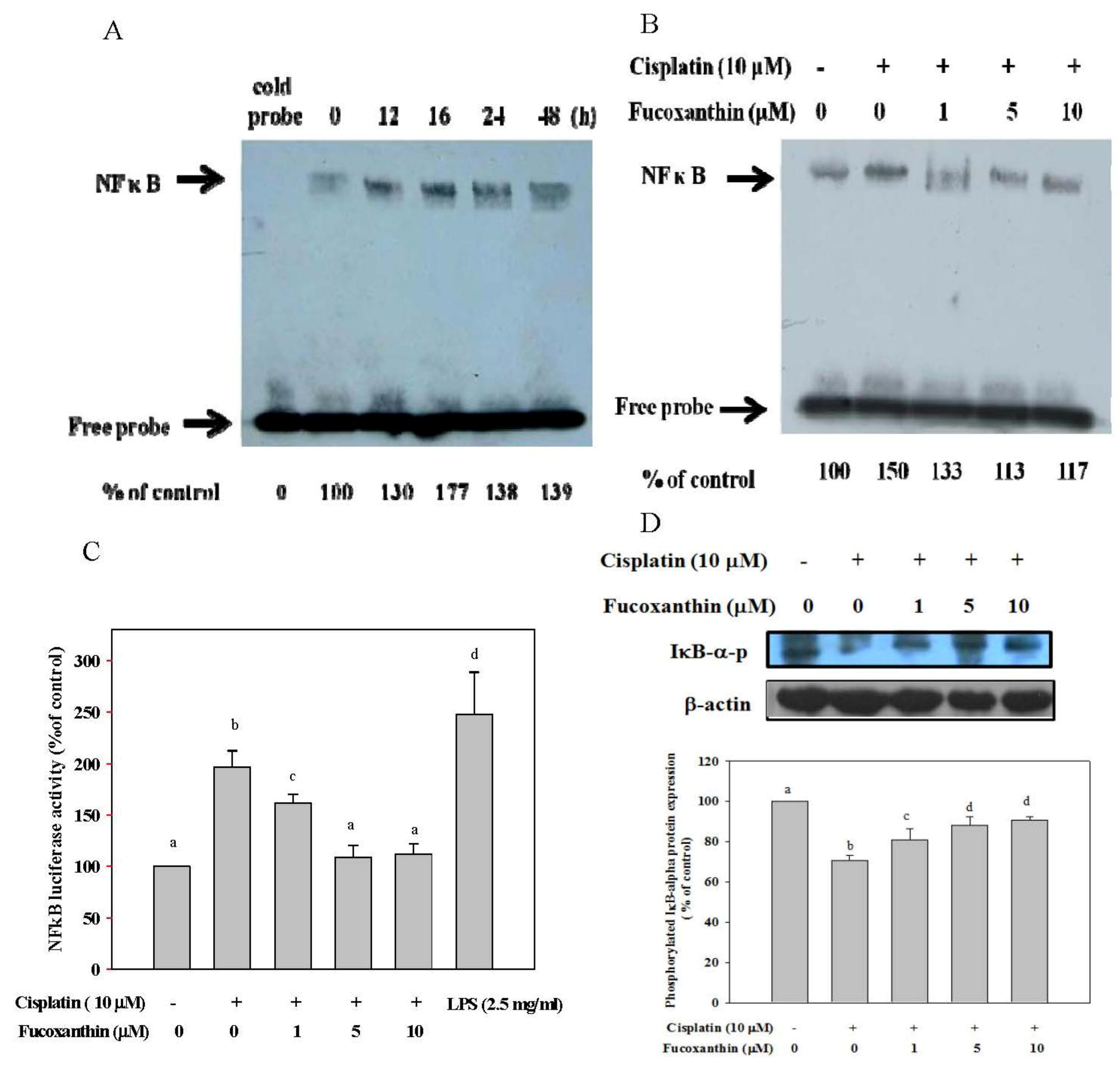
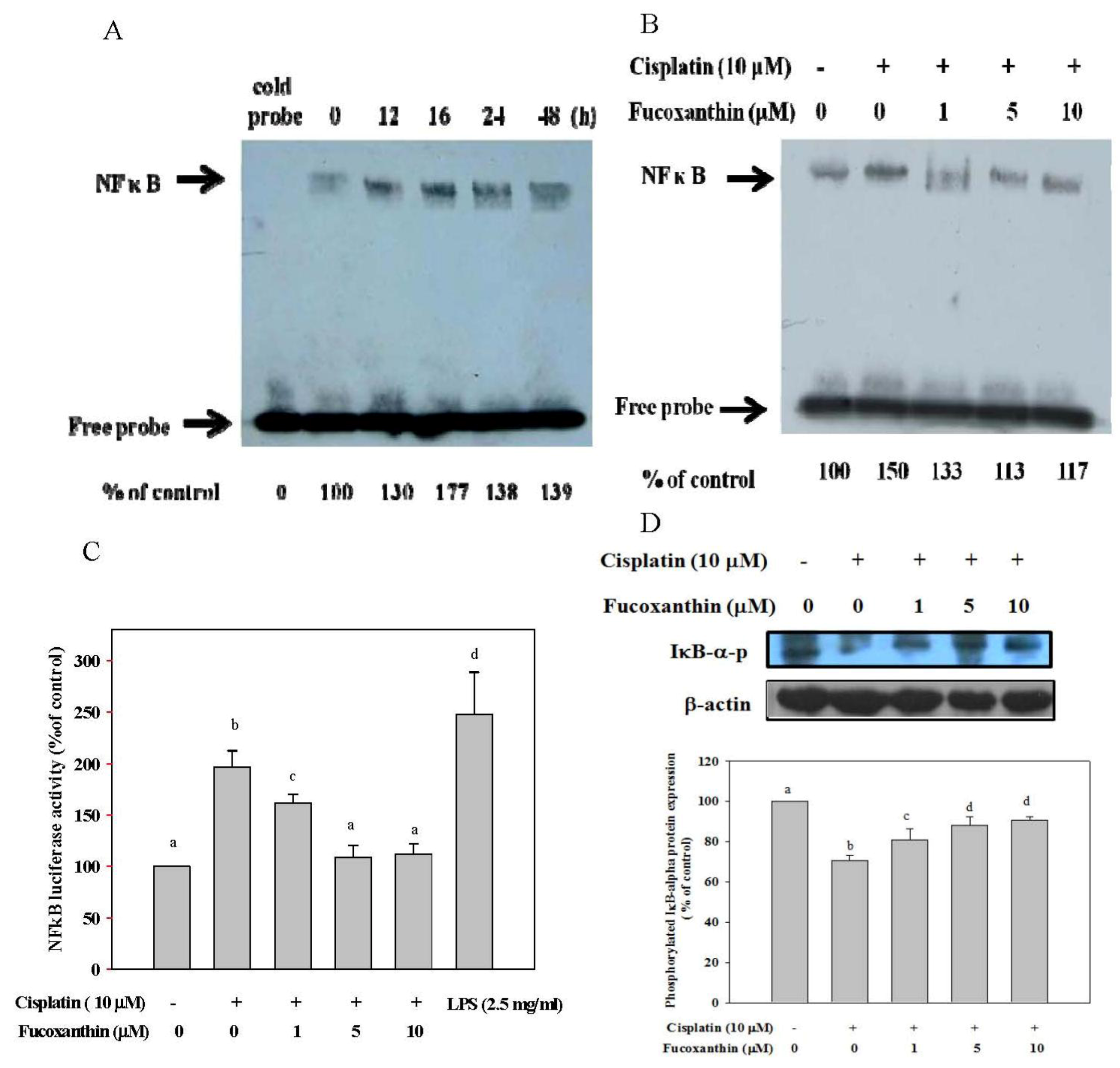
2.2. Fucoxanthin Attenuates the NFκB Expression Induced by Cisplatin and Restores the Phosphorylation of IκB-α Inhibited by Cisplatin
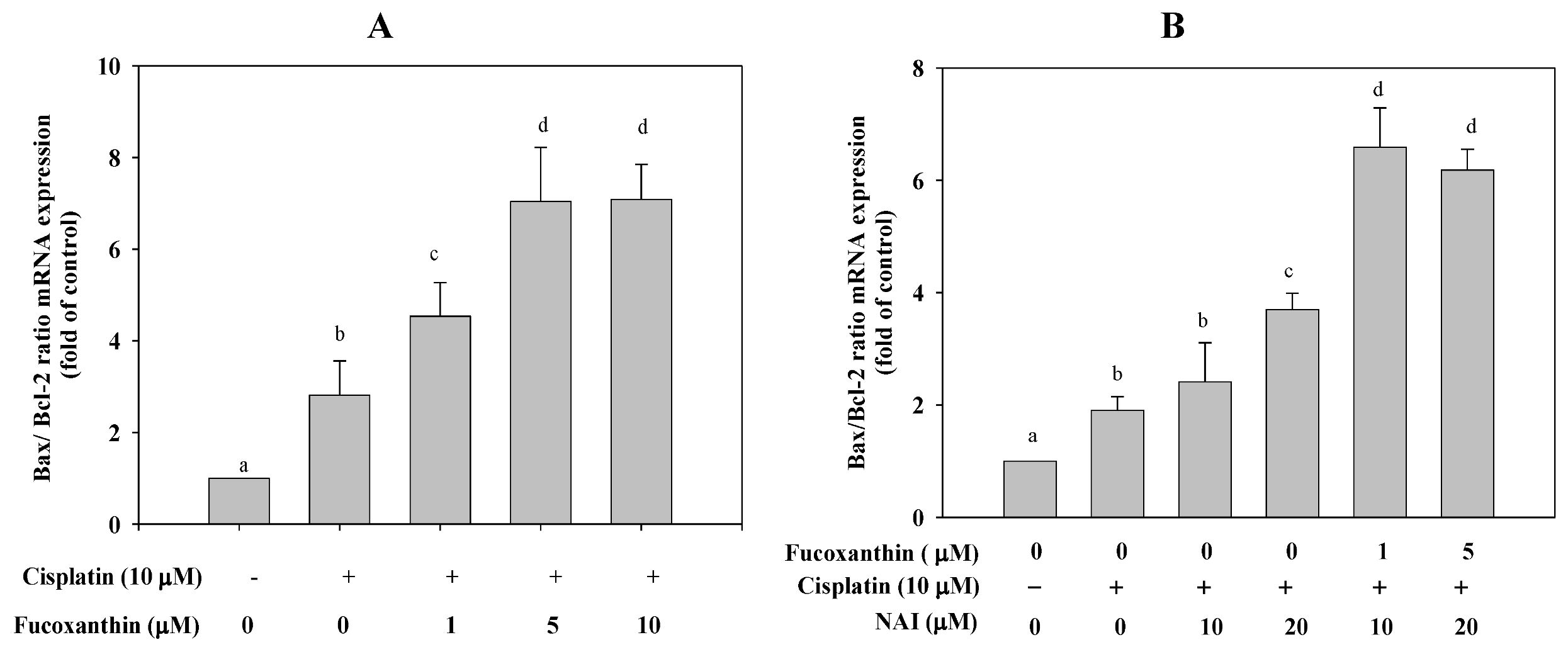
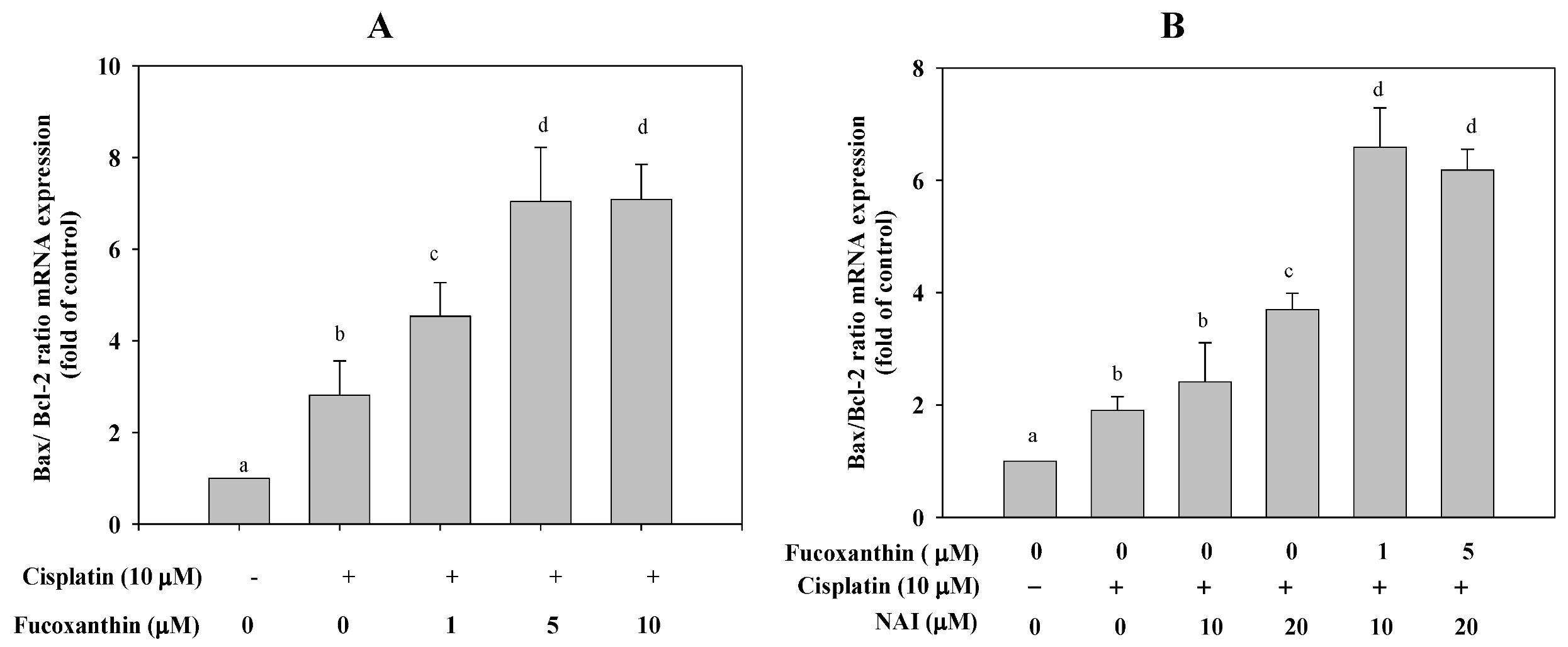
2.3. Fucoxanthin Combined with Cisplatin Increases the Ratio of Bax/Bcl-2 mRNA Expression in HepG2 Cells
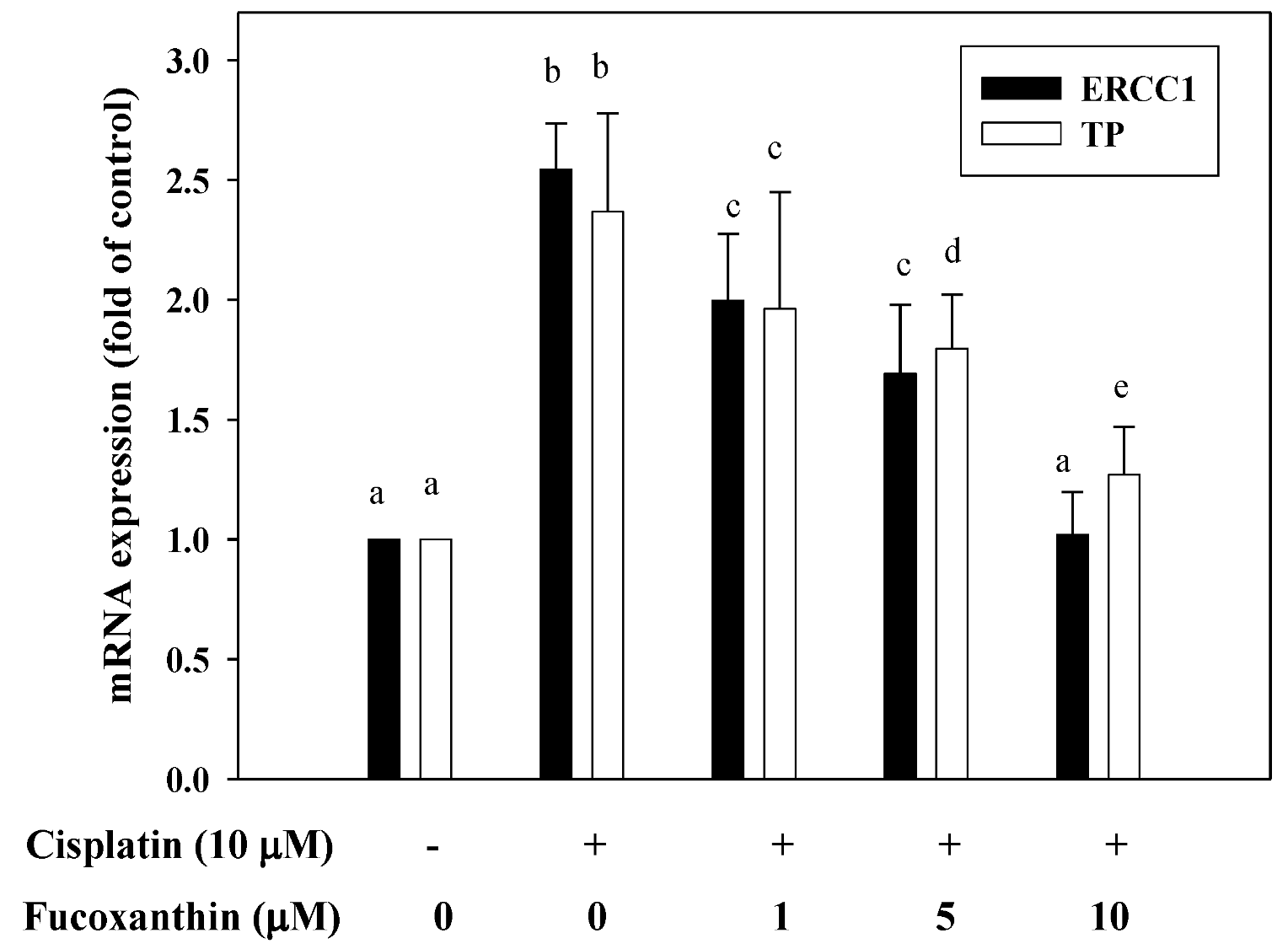
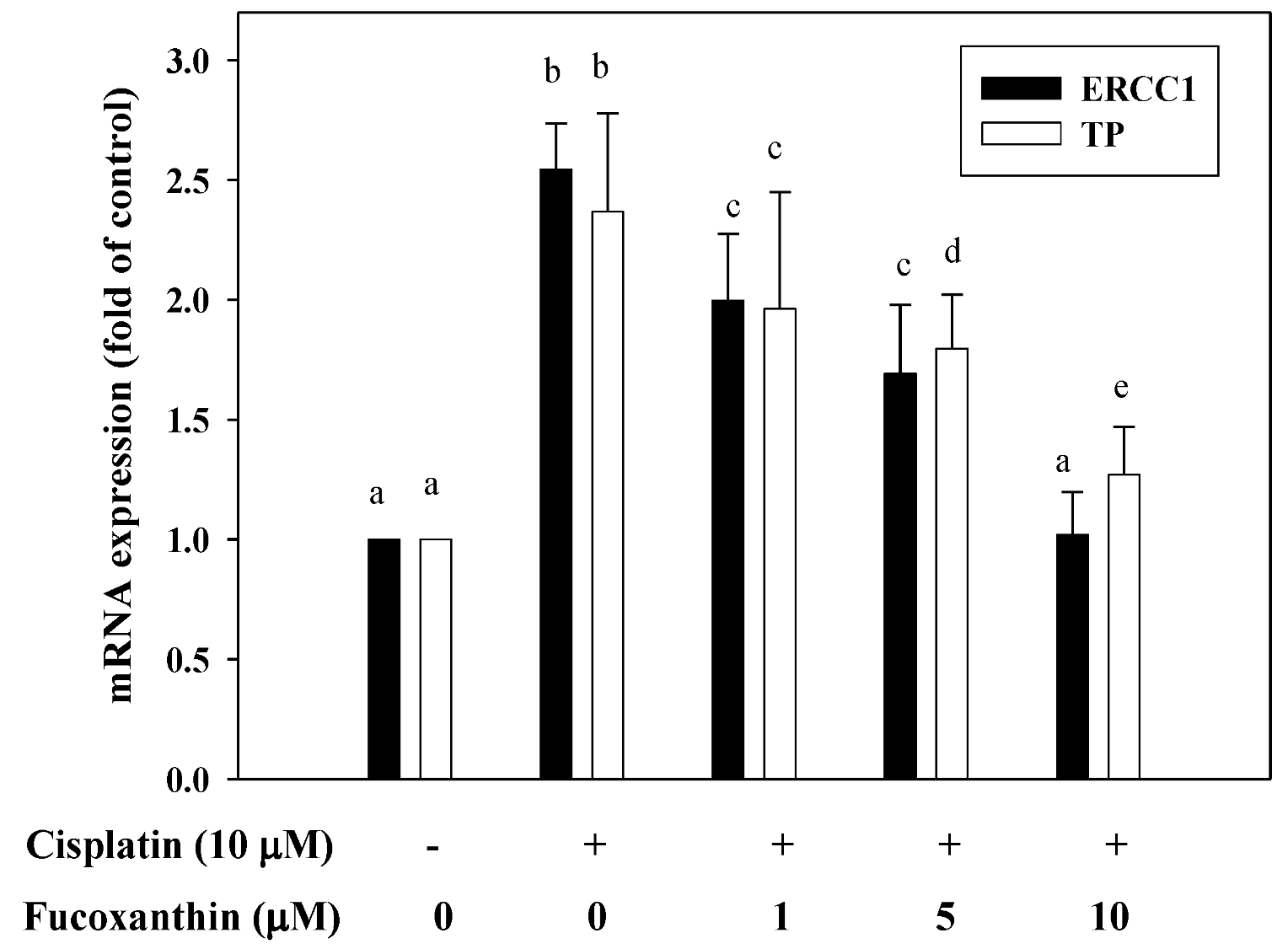
2.4. Fucoxanthin Attenuates mRNA Expression of ERCC1 and TP Induced by Cisplatin
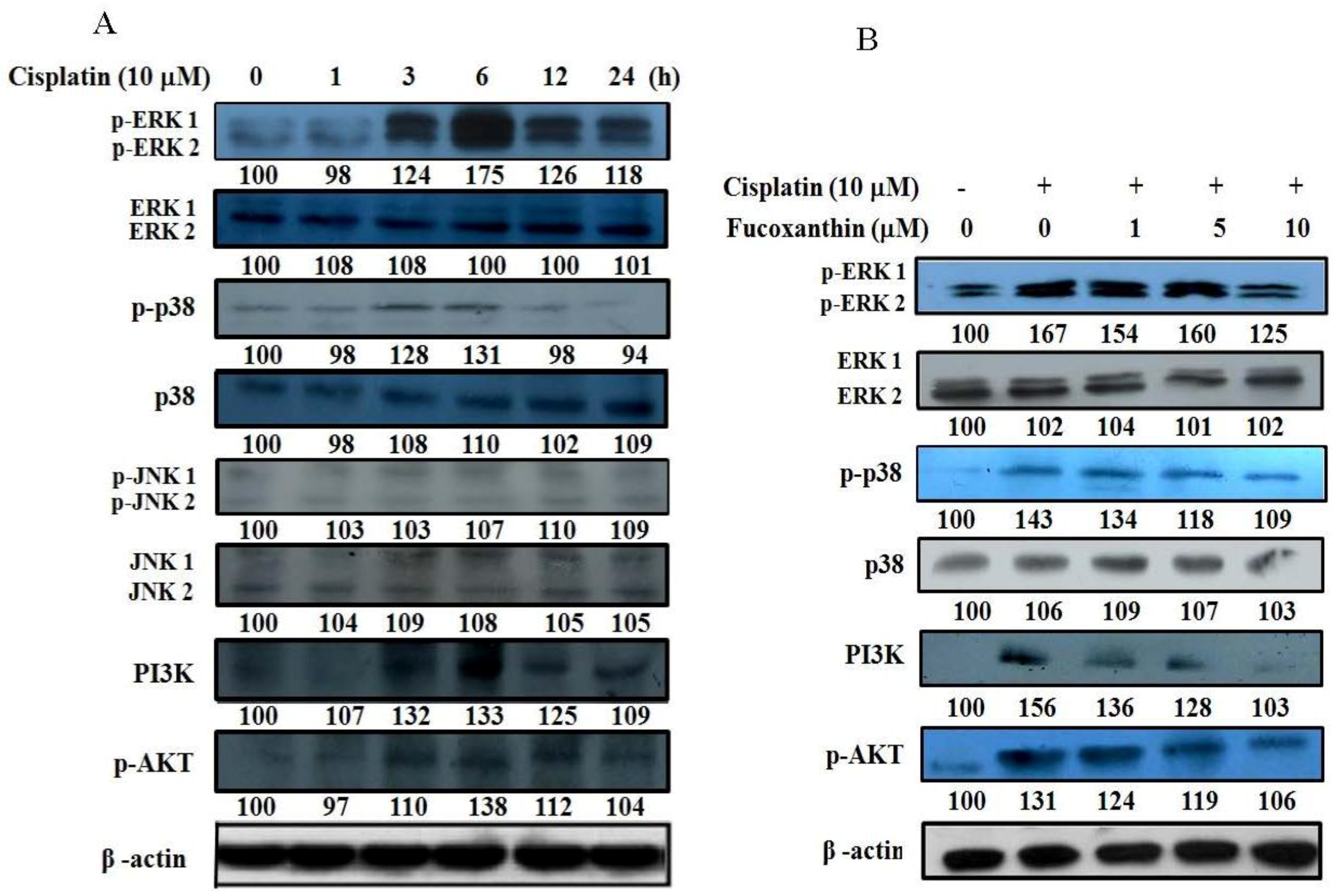
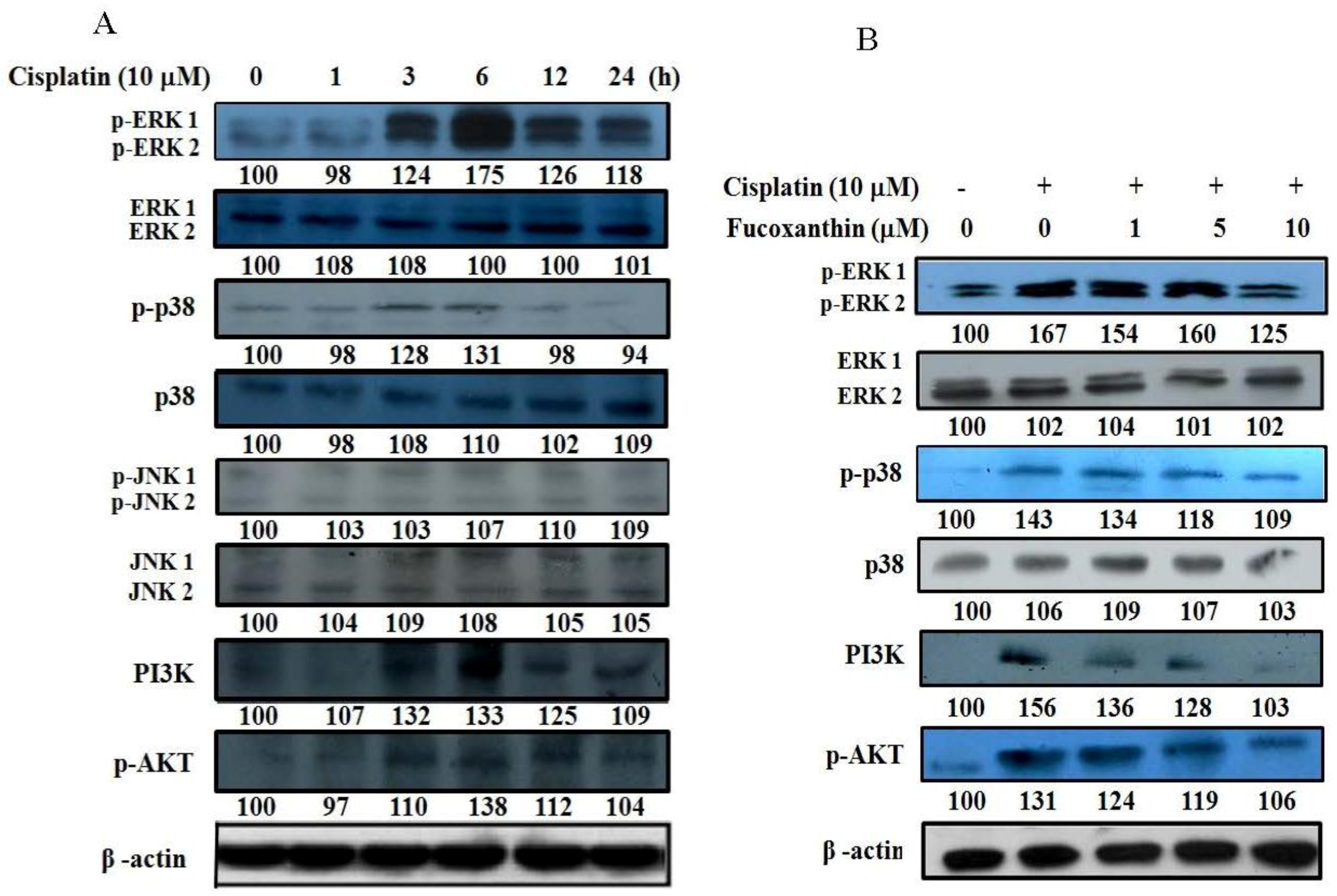
2.5. Fucoxanthin Attenuates the Phosphorylation of ERK1/2, p38, AKT and PI3K in HepG2 Cells
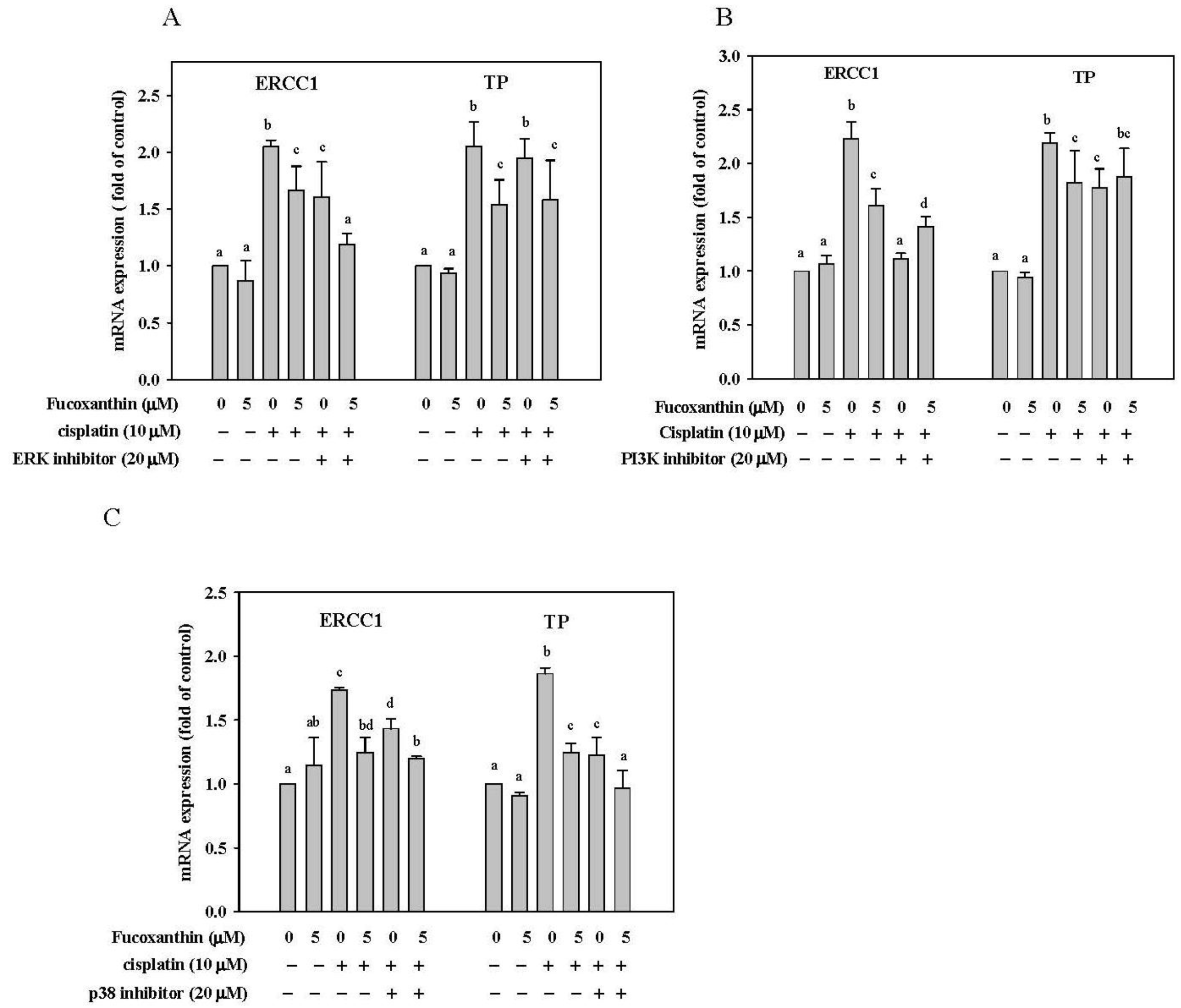
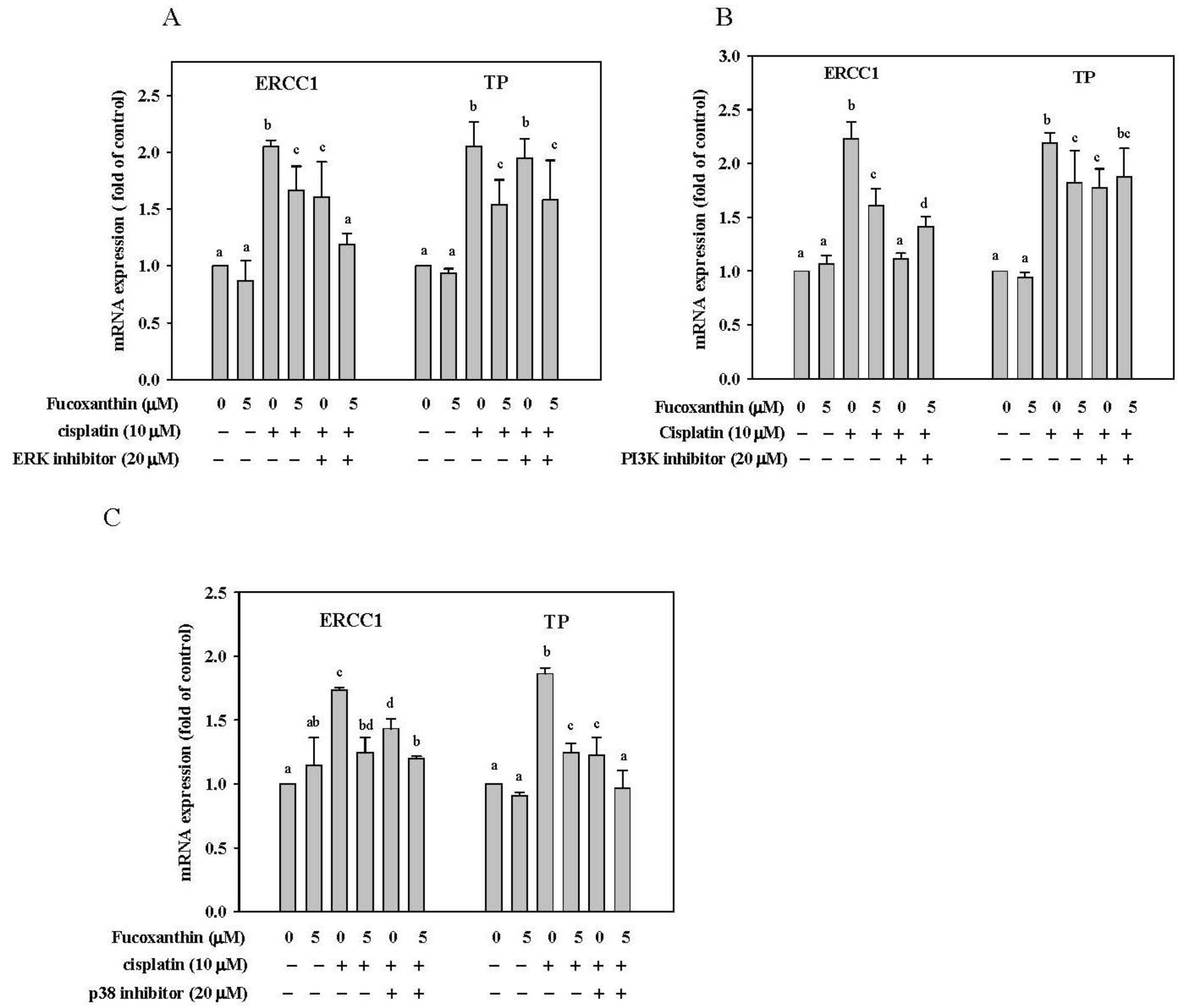
2.6. Effect of Fucoxanthin in Combination with ERK, p38 and PI3K Inhibitor on ERCC1 and TP mRNA Expression in HepG2 Cells
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Cultures
4.3. Assessment of Cell Viability
4.4. Real-Time Polymerase Chain Reaction
4.5. Western Blotting
4.6. Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
4.7. Transfection and Luciferase Reporter Gene Assays
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Supplementary Files
References
- Di Bisceglie, A.M. Epidemiology and clinical presentation of hepatocellular carcinoma. J. Vasc. Interv. Radiol. 2002, 13, S169–S171. [Google Scholar] [CrossRef]
- Montalto, G.; Cervello, M.; Giannitrapani, L.; Dantona, F.; Terranova, A.; Castagnetta, L.A. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann. N. Y. Acad. Sci. 2002, 963, 13–20. [Google Scholar]
- Chau, G.Y.; Lui, W.Y.; Tsay, S.H.; Chao, Y.; King, K.L.; Wu, C.W. Postresectional adjuvant intraportal chemotherapy in patients with hepatocellular carcinoma: A case-control study. Ann. Surg. Oncol. 2006, 13, 1329–1337. [Google Scholar] [CrossRef]
- Tong, S.W.; Yang, Y.X.; Hu, H.D.; An, X.; Ye, F.; Hu, P.; Ren, H.; Li, S.L.; Zhang, D.Z. Proteomic investigation of 5-fluorouracil resistance in a human hepatocellular carcinoma cell line. J. Cell. Biochem. 2012, 113, 1671–1680. [Google Scholar]
- Dietel, M. Molecular mechanisms and possibilities of overcoming drug resistance in gastrointestinal tumors. Recent Results Cancer Res. 1996, 142, 89–101. [Google Scholar] [CrossRef]
- Go, R.S.; Adjei, A.A. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J. Clin. Oncol. 1999, 17, 409–422. [Google Scholar]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef]
- Zorbas, H.; Keppler, B.K. Cisplatin damage: Are DNA repair proteins saviors or traitors to the cell? ChemBioChem 2005, 6, 1157–1166. [Google Scholar] [CrossRef]
- Van de Vaart, P.J.; van der Vange, N.; Zoetmulder, F.A.; van Goethem, A.R.; van Tellingen, O.; ten Bokkel Huinink, W.W.; Beijnen, J.H.; Bartelink, H.; Begg, A.C. Intraperitoneal cisplatin with regional hyperthermia in advanced ovarian cancer: Pharmacokinetics and cisplatin-DNA adduct formation in patients and ovarian cancer cell lines. Eur. J. Cancer 1998, 34, 148–154. [Google Scholar]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Shahzad, M.M.; Lopez-Berestein, G.; Sood, A.K. Novel strategies for reversing platinum resistance. Drug Resist. Updat. 2009, 12, 148–152. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- Shirota, Y.; Stoehlmacher, J.; Brabender, J.; Xiong, Y.P.; Uetake, H.; Danenberg, K.D.; Groshen, S.; Tsao-Wei, D.D.; Danenberg, P.V.; Lenz, H.J. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J. Clin. Oncol. 2001, 19, 4298–4304. [Google Scholar]
- Lord, R.V.; Brabender, J.; Gandara, D.; Alberola, V.; Camps, C.; Domine, M.; Cardenal, F.; Sanchez, J.M.; Gumerlock, P.H.; Taron, M.; et al. Low ERCC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin. Cancer Res. 2002, 8, 2286–2291. [Google Scholar]
- Zhou, W.; Gurubhagavatula, S.; Liu, G.; Park, S.; Neuberg, D.S.; Wain, J.C.; Lynch, T.J.; Su, L.; Christiani, D.C. Excision repair cross-complementation group 1 polymorphism predicts overall survival in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res. 2004, 10, 4939–4943. [Google Scholar]
- Selvakumaran, M.; Pisarcik, D.A.; Bao, R.; Yeung, A.T.; Hamilton, T.C. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003, 63, 1311–1316. [Google Scholar]
- Gossage, L.; Madhusudan, S. Current status of excision repair cross complementing-group 1 (ERCC1) in cancer. Cancer Treat. Rev. 2007, 33, 565–577. [Google Scholar] [CrossRef]
- Bijnsdorp, I.V.; Azijli, K.; Jansen, E.E.; Wamelink, M.M.; Jakobs, C.; Struys, E.A.; Fukushima, M.; Kruyt, F.A.; Peters, G.J. Accumulation of thymidine-derived sugars in thymidine phosphorylase overexpressing cells. Biochem. Pharmacol. 2010, 80, 786–792. [Google Scholar] [CrossRef]
- Nakayama, Y.; Inoue, Y.; Nagashima, N.; Katsuki, T.; Matsumoto, K.; Kadowaki, K.; Shibao, K.; Tsurudome, Y.; Hirata, K.; Sako, T.; et al. Expression levels of thymidine phosphorylase (TP) and dihydropyrimidine dehydrogenase (DPD) in patients with gastrointestinal cancer. Anticancer Res. 2005, 25, 3755–3761. [Google Scholar]
- Mori, S.; Takao, S.; Ikeda, R.; Noma, H.; Mataki, Y.; Wang, X.; Akiyama, S.; Aikou, T. Thymidine phosphorylase suppresses Fas-induced apoptotic signal transduction independent of its enzymatic activity. Biochem. Biophys. Res. Commun. 2002, 295, 300–305. [Google Scholar] [CrossRef]
- Ikeda, R.; Furukawa, T.; Mitsuo, R.; Noguchi, T.; Kitazono, M.; Okumura, H.; Sumizawa, T.; Haraguchi, M.; Che, X.F.; Uchimiya, H.; et al. Thymidine phosphorylase inhibits apoptosis induced by cisplatin. Biochem. Biophys. Res. Commun. 2003, 301, 358–363. [Google Scholar] [CrossRef]
- Jeung, H.C.; Che, X.F.; Haraguchi, M.; Furukawa, T.; Zheng, C.L.; Sumizawa, T.; Rha, S.Y.; Roh, J.K.; Akiyama, S. Thymidine phosphorylase suppresses apoptosis induced by microtubule-interfering agents. Biochem. Pharmacol. 2005, 70, 13–21. [Google Scholar]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Griffioen, A.W. A new role for NF-κB in angiogenesis inhibition. Cell. Death Differ. 2007, 14, 1393–1397. [Google Scholar] [CrossRef]
- Andela, V.B.; Gordon, A.H.; Zotalis, G.; Rosier, R.N.; Goater, J.J.; Lewis, G.D.; Schwarz, E.M.; Puzas, J.E.; O’Keefe, R.J. NFκB: A pivotal transcription factor in prostate cancer metastasis to bone. Clin. Orthop. Relat. Res. 2003, 415 (Suppl.), S75–S85. [Google Scholar]
- Tomita, M.; Kawakami, H.; Uchihara, J.N.; Okudaira, T.; Masuda, M.; Takasu, N.; Matsuda, T.; Ohta, T.; Tanaka, Y.; Ohshiro, K.; et al. Curcumin (diferuloylmethane) inhibits constitutive active NF-κB, leading to suppression of cell growth of human T-cell leukemia virus type I-infected T-cell lines and primary adult T-cell leukemia cells. Int. J. Cancer 2006, 118, 765–772. [Google Scholar] [CrossRef]
- Sharma, H.W.; Narayanan, R. The NF-κB transcription factor in oncogenesis. Anticancer Res. 1996, 16, 589–596. [Google Scholar]
- Nakanishi, C.; Toi, M. Nuclear factor-κB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef]
- Shou, Y.; Li, N.; Li, L.; Borowitz, J.L.; Isom, G.E. NF-κB-mediated up-regulation of Bcl-XS and Bax contributes to cytochrome c release in cyanide-induced apoptosis. J. Neurochem. 2002, 81, 842–852. [Google Scholar] [CrossRef]
- Sarkar, F.H.; Li, Y. Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Res. 2006, 66, 3347–3350. [Google Scholar] [CrossRef]
- Yeh, P.Y.; Chuang, S.E.; Yeh, K.H.; Song, Y.C.; Cheng, A.L. Involvement of nuclear transcription factor-κB in low-dose doxorubicin-induced drug resistance of cervical carcinoma cells. Biochem. Pharmacol. 2003, 66, 25–33. [Google Scholar]
- Dembitsky, V.M.; Maoka, T. Allenic and cumulenic lipids. Prog. Lipid Res. 2007, 46, 328–375. [Google Scholar] [CrossRef]
- Sachindra, N.M.; Sato, E.; Maeda, H.; Hosokawa, M.; Niwano, Y.; Kohno, M.; Miyashita, K. Radical scavenging and singlet oxygen quenching activity of marine carotenoid fucoxanthin and its metabolites. J. Agric. Food Chem. 2007, 55, 8516–8522. [Google Scholar] [CrossRef]
- Heo, S.J.; Jeon, Y.J. Protective effect of fucoxanthin isolated from Sargassum siliquastrum on UV-B induced cell damage. J. Photochem. Photobiol. B 2009, 95, 101–107. [Google Scholar] [CrossRef]
- Liu, C.L.; Liang, A.L.; Hu, M.L. Protective effects of fucoxanthin against ferric nitrilotriacetate-induced oxidative stress in murine hepatic BNL CL.2 cells. Toxicol. In Vitro 2011, 25, 1314–1319. [Google Scholar] [CrossRef]
- Liu, C.L.; Chiu, Y.T.; Hu, M.L. Fucoxanthin enhances HO-1 and NQO1 expression in murine hepatic BNL CL.2 cells through activation of the Nrf2/ARE system partially by its pro-oxidant activity. J. Agric. Food Chem. 2011, 59, 11344–11351. [Google Scholar]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Takahashi, N.; Kawada, T.; Miyashita, K. Fucoxanthin and its metabolite, fucoxanthinol, suppress adipocyte differentiation in 3T3-L1 cells. Int. J. Mol. Med. 2006, 18, 147–152. [Google Scholar]
- Jeon, S.M.; Kim, H.J.; Woo, M.N.; Lee, M.K.; Shin, Y.C.; Park, Y.B.; Choi, M.S. Fucoxanthin-rich seaweed extract suppresses body weight gain and improves lipid metabolism in high-fat-fed C57BL/6J mice. Biotechnol. J. 2010, 5, 961–969. [Google Scholar] [CrossRef]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Miyashita, K. Dietary combination of fucoxanthin and fish oil attenuates the weight gain of white adipose tissue and decreases blood glucose in obese/diabetic KK-Ay mice. J. Agric. Food Chem. 2007, 55, 7701–7706. [Google Scholar] [CrossRef]
- Nishino, H.; Tokuda, H.; Murakoshi, M.; Satomi, Y.; Masuda, M.; Onozuka, M.; Yamaguchi, S.; Takayasu, J.; Tsuruta, J.; Okuda, M.; et al. Cancer prevention by natural carotenoids. Biofactors 2000, 13, 89–94. [Google Scholar] [CrossRef]
- Kim, K.N.; Heo, S.J.; Yoon, W.J.; Kang, S.M.; Ahn, G.; Yi, T.H.; Jeon, Y.J. Fucoxanthin inhibits the inflammatory response by suppressing the activation of NF-κB and MAPKs in lipopolysaccharide-induced RAW 264.7 macrophages. Eur. J. Pharmacol. 2010, 649, 369–375. [Google Scholar] [CrossRef]
- Zaragoza, M.C.; Lopez, D.; Sáiz, M.P.; Poquet, M.; Perez, J.; Puig-Parellada, P.; Marmol, F.; Simonetti, P.; Gardana, C.; Lerat, Y.; et al. Toxicity and antioxidant activity in vitro and in vivo of two Fucus vesiculosus extracts. J. Agric. Food Chem. 2008, 56, 7773–7780. [Google Scholar]
- Yu, R.X.; Hu, X.M.; Xu, S.Q.; Jiang, Z.J.; Yang, W. Effects of fucoxanthin on proliferation and apoptosis in human gastric adenocarcinoma MGC-803 cells via JAK/STAT signal pathway. Eur. J. Pharmacol. 2011, 657, 10–19. [Google Scholar] [CrossRef]
- Satomi, Y.; Nishino, H. Implication of mitogen-activated protein kinase in the induction of G1 cell cycle arrest and gadd45 expression by the carotenoid fucoxanthin in human cancer cells. Biochim. Biophys. Acta 2009, 1790, 260–266. [Google Scholar] [CrossRef]
- Das, S.K.; Hashimoto, T.; Kanazawa, K. Growth inhibition of human hepatic carcinoma HepG2 cells by fucoxanthin is associated with down-regulation of cyclin D. Biochim. Biophys. Acta 2008, 1780, 743–749. [Google Scholar] [CrossRef]
- Yoshiko, S.; Hoyoku, N. Fucoxanthin, a natural carotenoid, induces G1 arrest and GADD45 gene expression in human cancer cells. In Vivo 2007, 21, 305–309. [Google Scholar]
- Liu, C.L.; Lim, Y.P.; Hu, M.L. Fucoxanthin attenuates rifampin-induced cytochrome P450 3A4 (CYP3A4) and multiple drug resistance 1 (MDR1) gene expression through pregnane X receptor (PXR)-mediated pathways in human hepatoma HepG2 and colon adenocarcinoma LS174T cells. Mar. Drugs 2012, 10, 242–257. [Google Scholar] [CrossRef] [Green Version]
- Viatour, P.; Bentires-Alj, M.; Chariot, A.; Deregowski, V.; de Leval, L.; Merville, M.P.; Bours, V. NF-κB2/p100 induces Bcl-2 expression. Leukemia 2003, 17, 1349–1356. [Google Scholar] [CrossRef]
- Minn, A.J.; Rudin, C.M.; Boise, L.H.; Thompson, C.B. Expression of bcl-xL can confer a multidrug resistance phenotype. Blood 1995, 86, 1903–1910. [Google Scholar]
- Altaha, R.; Liang, X.; Yu, J.J.; Reed, E. Excision repair cross complementing-group 1: Gene expression and platinum resistance. Int. J. Mol. Med. 2004, 14, 959–970. [Google Scholar]
- Ko, J.C.; Su, Y.J.; Lin, S.T.; Jhan, J.Y.; Ciou, S.C.; Cheng, C.M.; Chiu, Y.F.; Kuo, Y.H.; Tsai, M.S.; Lin, Y.W. Emodin enhances cisplatin-induced cytotoxicity via down-regulation of ERCC1 and inactivation of ERK1/2. Lung Cancer 2010, 69, 155–164. [Google Scholar] [CrossRef]
- Tsai, M.S.; Weng, S.H.; Kuo, Y.H.; Chiu, Y.F.; Lin, Y.W. Synergistic effect of curcumin and cisplatin via down-regulation of thymidine phosphorylase and excision repair cross-complementary 1 (ERCC1). Mol. Pharmacol. 2011, 80, 136–146. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Basu, A.; Tu, H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cδ. Biochem. Biophys. Res. Commun. 2005, 334, 1068–1073. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Mabuchi, S.; Ohmichi, M.; Nishio, Y.; Hayasaka, T.; Kimura, A.; Ohta, T.; Saito, M.; Kawagoe, J.; Takahashi, K.; Yada-Hashimoto, N.; et al. Inhibition of NFκB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J. Biol. Chem. 2004, 279, 23477–23485. [Google Scholar]
- Ohta, T.; Ohmichi, M.; Hayasaka, T.; Mabuchi, S.; Saitoh, M.; Kawagoe, J.; Takahashi, K.; Igarashi, H.; Du, B.; Doshida, M.; et al. Inhibition of phosphatidylinositol 3-kinase increases efficacy of cisplatin in in vivo ovarian cancer models. Endocrinology 2006, 147, 1761–1769. [Google Scholar]
- Andrieux, L.O.; Fautrel, A.; Bessard, A.; Guillouzo, A.; Baffet, G.; Langouet, S. GATA-1 is essential in EGF-mediated induction of nucleotide excision repair activity and ERCC1 expression through ERK2 in human hepatoma cells. Cancer Res. 2007, 67, 2114–2123. [Google Scholar]
- Bessard, A.; Coutant, A.; Rescan, C.; Ezan, F.; Fremin, C.; Courselaud, B.; Ilyin, G.; Baffet, G. An MLCK-dependent window in late G1 controls S phase entry of proliferating rodent hepatocytes via ERK-p70S6K pathway. Hepatology 2006, 44, 152–163. [Google Scholar] [CrossRef]
- Chen, C.C.; Chen, L.C.; Liang, Y.; Tsang, N.M.; Chang, Y.S. Epstein-Barr virus latent membrane protein 1 induces the chemotherapeutic target, thymidine phosphorylase, via NF-κB and p38 MAPK pathways. Cell. Signal. 2010, 22, 1132–1142. [Google Scholar] [CrossRef]
- Liu, C.L.; Huang, Y.S.; Hosokawa, M.; Miyashita, K.; Hu, M.L. Inhibition of proliferation of a hepatoma cell line by fucoxanthin in relation to cell cycle arrest and enhanced gap junctional intercellular communication. Chem. Biol. Interact. 2009, 182, 165–172. [Google Scholar] [CrossRef]
- Lin, C.Y.; Huang, C.S.; Hu, M.L. The use of fetal bovine serum as delivery vehicle to improve the uptake and stability of lycopene in cell culture studies. Br. J. Nutr. 2007, 98, 226–232. [Google Scholar] [CrossRef]
- Lim, Y.P.; Kuo, S.C.; Lai, M.L.; Huang, J.D. Inhibition of CYP3A4 expression by ketoconazole is mediated by the disruption of pregnane X receptor, steroid receptor coactivator-1, and hepatocyte nuclear factor 4alpha interaction. Pharmacogenetics Genomics 2009, 19, 11–24. [Google Scholar] [CrossRef]
- Chen, Y.C.; Kuo, T.C.; Lin-Shiau, S.Y.; Lin, J.K. Induction of HSP70 gene expression by modulation of Ca(+2) ion and cellular p53 protein by curcumin in colorectal carcinoma cells. Mol. Carcinog. 1996, 17, 224–234. [Google Scholar] [CrossRef]
- Shukla, S.; Maclennan, G.T.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Constitutive activation of P I3 K-Akt and NF-κB during prostate cancer progression in autochthonous transgenic mouse model. Prostate 2005, 64, 224–239. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, C.-L.; Lim, Y.-P.; Hu, M.-L. Fucoxanthin Enhances Cisplatin-Induced Cytotoxicity via NFκB-Mediated Pathway and Downregulates DNA Repair Gene Expression in Human Hepatoma HepG2 Cells. Mar. Drugs 2013, 11, 50-66. https://doi.org/10.3390/md11010050
Liu C-L, Lim Y-P, Hu M-L. Fucoxanthin Enhances Cisplatin-Induced Cytotoxicity via NFκB-Mediated Pathway and Downregulates DNA Repair Gene Expression in Human Hepatoma HepG2 Cells. Marine Drugs. 2013; 11(1):50-66. https://doi.org/10.3390/md11010050
Chicago/Turabian StyleLiu, Cheng-Ling, Yun-Ping Lim, and Miao-Lin Hu. 2013. "Fucoxanthin Enhances Cisplatin-Induced Cytotoxicity via NFκB-Mediated Pathway and Downregulates DNA Repair Gene Expression in Human Hepatoma HepG2 Cells" Marine Drugs 11, no. 1: 50-66. https://doi.org/10.3390/md11010050
APA StyleLiu, C.-L., Lim, Y.-P., & Hu, M.-L. (2013). Fucoxanthin Enhances Cisplatin-Induced Cytotoxicity via NFκB-Mediated Pathway and Downregulates DNA Repair Gene Expression in Human Hepatoma HepG2 Cells. Marine Drugs, 11(1), 50-66. https://doi.org/10.3390/md11010050