An Aeroplysinin-1 Specific Nitrile Hydratase Isolated from the Marine Sponge Aplysina cavernicola

Abstract
:1. Introduction

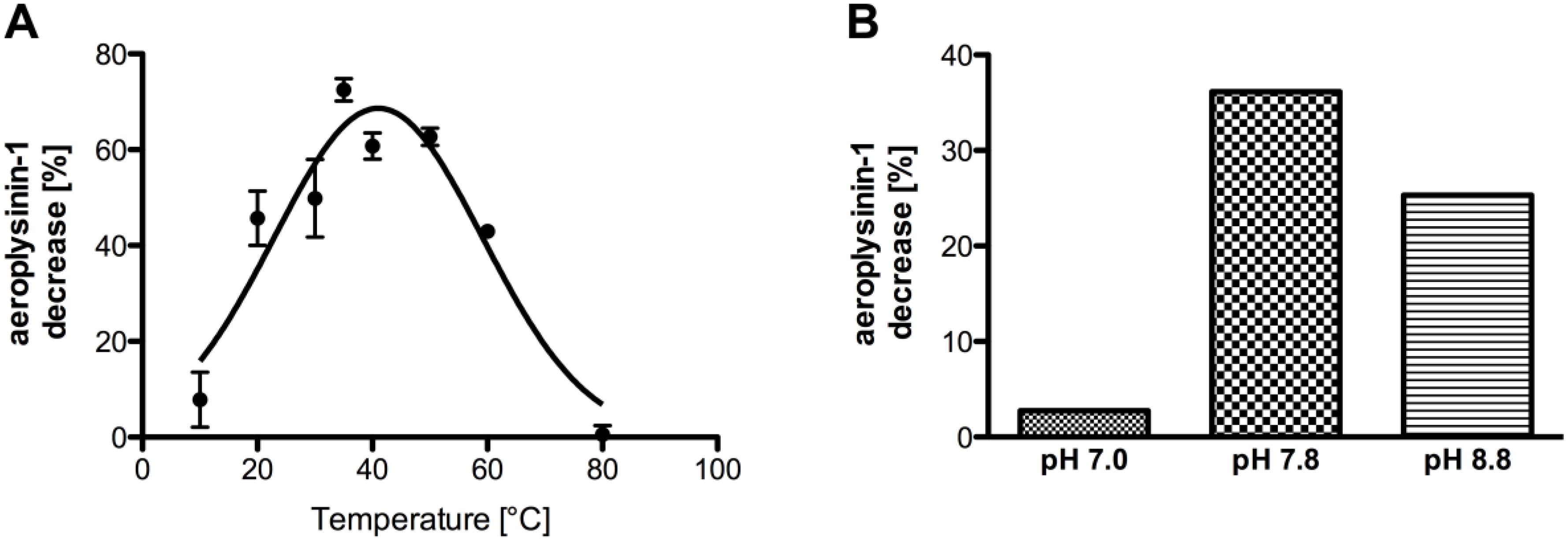
2. Results
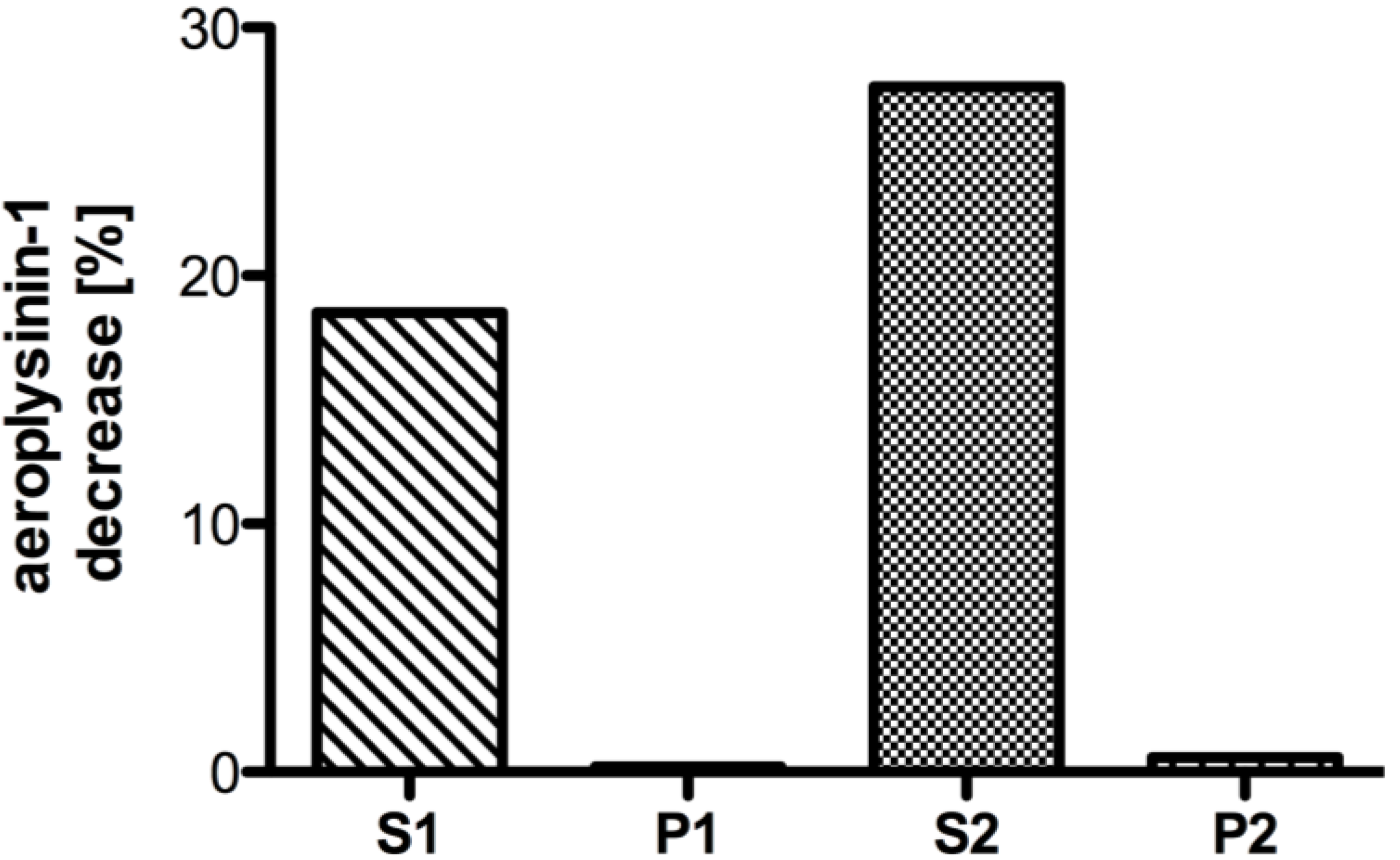
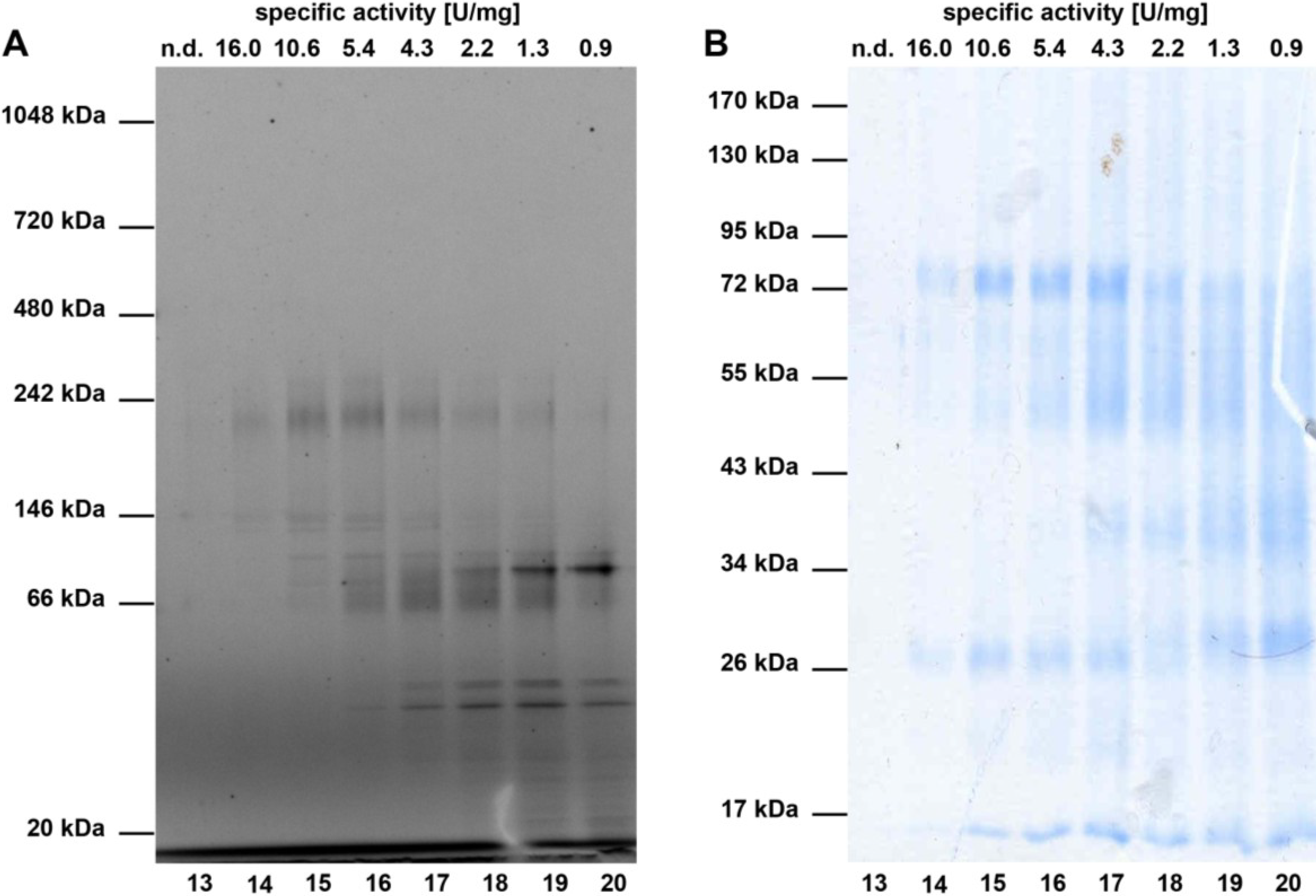
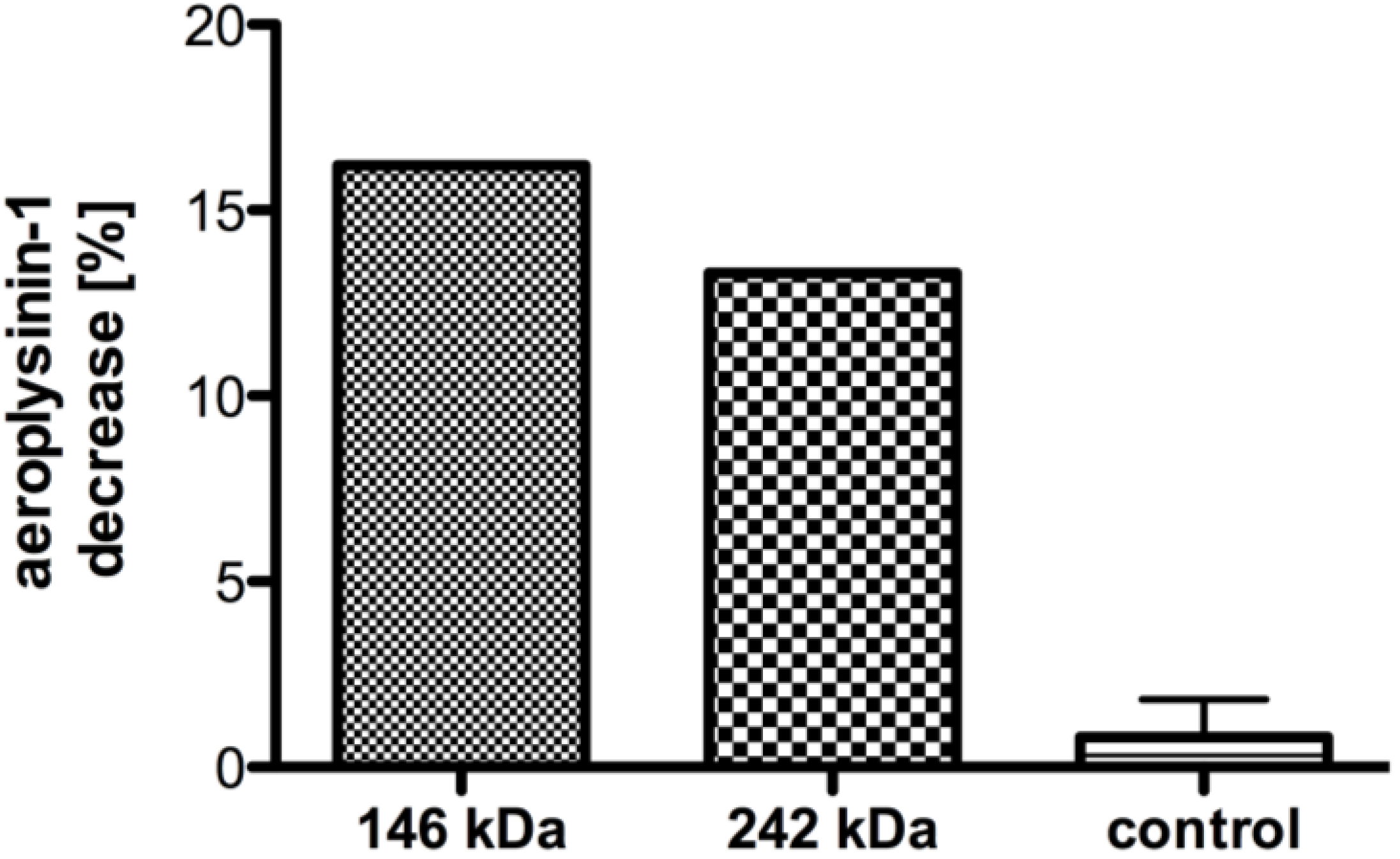
2.1. Partial Purification and Kinetics of the Nitrile Hydratases




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification step | Total protein (mg) | Total activity (U) | Specific activity (U/mg) | Yield (%) |
|---|---|---|---|---|
| Cell-free extract | 114.1 | 36.1 | 0.31 | 100 |
| Ammonium sulphate fractionation | 30.9 | 10.8 | 0.35 | 29.9 |
| HiTrap Q Sepharose XL | 2.4 | 1.08 | 0.45 | 2.9 |
| Superdex 200 10/300 | 0.05 | 0.81 | 16.2 | 1.3 |

| Molecular weight ([M + H]+) | Charge (z) | Mass-charge ratio (m/z) | Sequence |
|---|---|---|---|
| 914.46 | ++ | 457.73 | LSSEFGFK |
| 960.56 | ++ | 480.78 | FVTPLDLR |
| 1187.64 | ++ | 594.37 | WDETVVALVR |
| 1937.92 | +++ | 646.64 | (FN)FDLTHQQQLDYLR |
| 2269.15 | +++ | 757.05 | DLPASANDLPYFLLHAQLDR |

2.2. Influence of Different Metal Ions on the Enzymatic Activity

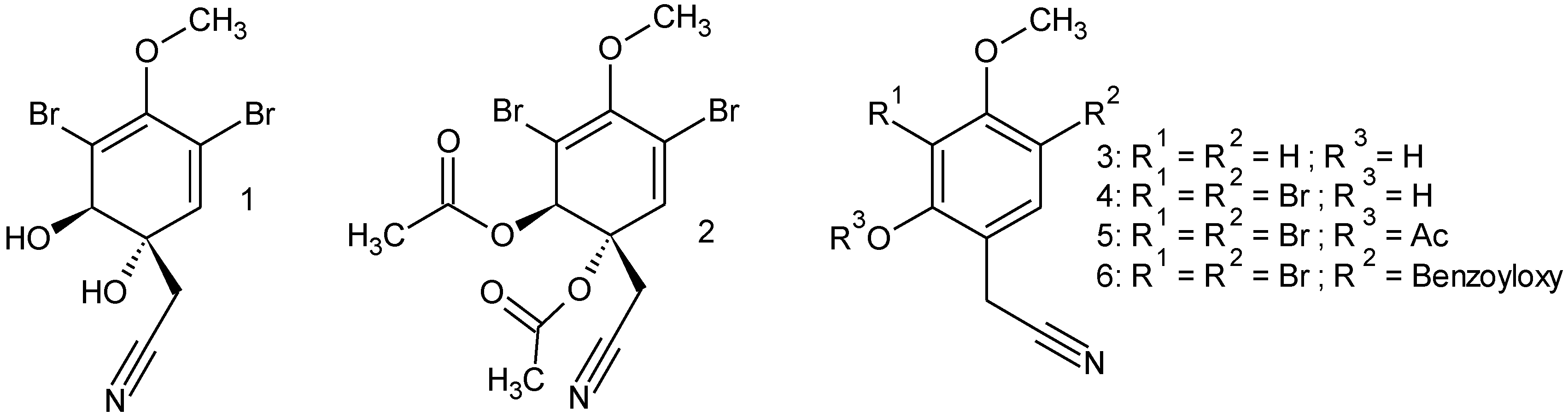
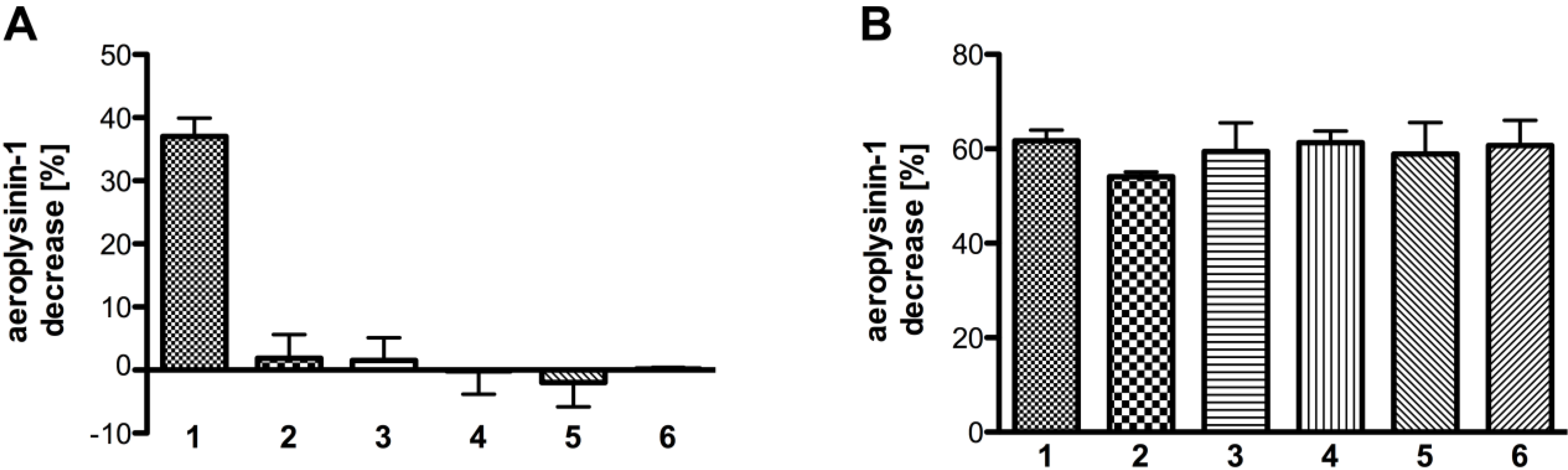
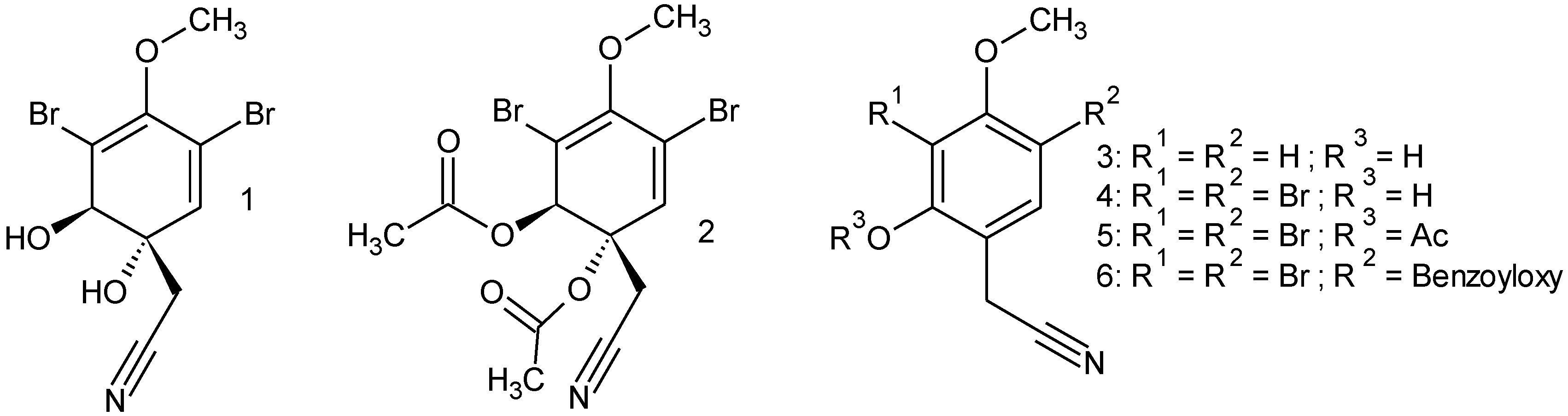
2.3. Substrate Specificity

3. Discussion
4. Experimental Section
4.1. Origin of Sponge Material
4.2. Protein Extraction
4.3. Ammonium Sulphate Fractionation
4.4. Ion Exchange Chromatography
4.5. Size Exclusion Chromatography
4.6. Protein Analysis
4.7. Activity Assay
4.8. Analysis of Metals in the Supernatant S2
4.9. Activity Assay with Excised Protein Bands from Blue Native-PAGE
4.10. Influence of Different Metal Ions on the Enzymatic Activity
4.11. Substrate Specificity
4.12. Inhibition Assay
4.13. Enzyme Kinetics
4.14. Restoring Enzymatic Activity
4.15. Synthesis
4.15.1. Diacetylaeroplysinin-1 (2)
4.15.2. 2-Hydroxy-4-methoxyphenylacetonitril (3)
4.15.3. 3,5-Dibromo-2-hydroxy-4-methoxyphenylacetonitril (4)
4.15.4. 3,5-Dibromo-2-acetyl-4-methoxyphenylacetonitril (5)
4.15.5. 3,5-Dibromo-2-benzoyloxy-4-methoxyphenylacetonitril (6)
4.16. Mass Spectrometric Analysis of Peptide Fragments
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Putz, A.; Proksch, P. Chemical defence in marine ecosystems. Annu. Plant Rev. 2010, 39, 162–213. [Google Scholar]
- Paul, V.J.; van Alstyne, K.L. Activation of chemical defenses in the tropical green algae Halimeda spp. J. Exp. Mar. Biol. Ecol. 1992, 160, 191–203. [Google Scholar] [CrossRef]
- Jung, V.; Pohnert, G. Rapid wound-activated transformation of the green algal defensive metabolite caulerpenyne. Tetrahedron 2001, 57, 7169–7172. [Google Scholar] [CrossRef]
- Teeyapant, R.; Proksch, P. Biotransformation of brominated compounds in the marine sponge Verongia aerophoba—evidence for an induced chemical defense? Naturwissenschaften 1993, 80, 369–370. [Google Scholar] [CrossRef]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Magno, S.; Carrano, L.; Pansini, M. Chemistry of Verongida sponges. VII. Bromo compounds from the Caribbean sponge Aplysina archeri. Tetrahedron 1996, 52, 9863–9868. [Google Scholar] [CrossRef]
- Thoms, C.; Ebel, R.; Hentschel, U.; Proksch, P. Sequestration of dietary alkaloids by the spongivorous marine mollusc Tylodina perversa. Z. Naturforsch. C 2003, 58, 426–432. [Google Scholar]
- Thoms, C.; Wolff, M.; Padmakumar, K.; Ebel, R.; Proksch, P. Chemical defense of Mediterranean sponges Aplysina cavernicola and Aplysina aerophoba. Z. Naturforsch. C J. Biosci. 2004, 59, 113–122. [Google Scholar]
- Ebel, R.; Brenzinger, M.; Kunze, A.; Gross, H.J.; Proksch, P. Wound activation of protoxins in marine sponge Aplysina aerophoba. J. Chem. Ecol. 1997, 23, 1451–1462. [Google Scholar] [CrossRef]
- Thoms, C.; Ebel, R.; Proksch, P. Activated chemical defense in Aplysina sponges revisited. J. Chem. Ecol. 2006, 32, 97–123. [Google Scholar] [CrossRef]
- Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Bioactive natural products from marine sponges and fungal endophytes. Phytochem. Rev. 2010, 9, 475–489. [Google Scholar] [CrossRef]
- Putz, A. Secondary metabolites from marine sponges, with focus on the chemical ecology and biochemical characterisation of the stress induced biotransformation of Aplysina alkaloids. Ph.D. Thesis, University Düsseldorf, Düsseldorf, Germany, 2009. [Google Scholar]
- Prasad, S.; Bhalla, T.C. Nitrile hydratases (NHases): At the interface of academia and industry. Biotechnol. Adv. 2010, 28, 725–741. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimizu, S. Metalloenzyme nitrile hydratase: Structure, regulation, and application to biotechnology. Nat. Biotechnol. 1998, 16, 733–736. [Google Scholar] [CrossRef]
- Cyanide in Biology; Vennesland, B.; Conn, E.E.; Knowles, C.J.; Westley, J.; Wissing, F. (Eds.) Academic Press: London, England, 1981; p. 548.
- Nagasawa, T.; Yamada, H. Microbial transformations of nitriles. Trends Biotechnol. 1989, 7, 153–158. [Google Scholar] [CrossRef]
- Hjort, C.M.; Godtfredsen, S.E.; Emborg, C. Isolation and characterization of a nitrile hydratase from a Rhodococcus sp. J. Chem. Technol. Biotechnol. 1990, 48, 217–226. [Google Scholar]
- Kobayashi, M.; Yanaka, N.; Nagasawa, T.; Yamada, H. Primary structure of an aliphatic nitrile-degrading enzyme, aliphatic nitrilase, from Rhodococcus rhodochrous K22 and expression of its gene and identification of its active site residue. Biochemistry 1992, 31, 9000–9007. [Google Scholar] [CrossRef]
- Endo, I.; Odaka, M.; Yohda, M. An enzyme controlled by light: The molecular mechanism of photoreactivity in nitrile hydratase. Trends Biotechnol. 1999, 17, 244–249. [Google Scholar] [CrossRef]
- Van, P.S.; Quignard, S.; Kubac, D.; Sorokin, D.Y.; van, R.F.; Sheldon, R.A. Nitrile hydratase CLEAs: The immobilization and stabilization of an industrially important enzyme. Green Chem. 2008, 10, 395–400. [Google Scholar] [CrossRef]
- Shaw, N.M.; Robins, K.T.; Kiener, A. Lonza: 20 years of biotransformations. Adv. Synth. Catal. 2003, 345, 425–435. [Google Scholar] [CrossRef]
- Marron, A.O.; Akam, M.; Walker, G. Nitrile hydratase genes are present in multiple eukaryotic supergroups. PLoS One 2012, 7, e32867. [Google Scholar]
- Kobayashi, M.; Shimizu, S. Nitrile hydrolases. Curr. Opin. Chem. Biol. 2000, 4, 95–102. [Google Scholar] [CrossRef]
- Dyballa, N.; Metzger, S. Fast and sensitive coomassie staining in quantitative proteomics. Methods Mol. Biol. 2012, 893, 47–59. [Google Scholar] [CrossRef]
- Kinter, M.; Sherman, N.E. Protein Sequencing and Identification Using Tandem Mass Spectrometry; Wiley-Interscience: New York, NY, USA, 2000. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Asano, Y.; Fujishiro, K.; Tani, Y.; Yamada, H. Microbial degradation of nitrile compounds. Part V. Aliphatic nitrile hydratase from Arthrobacter sp. J-1. Purification and characterization. Agric. Biol. Chem. 1982, 46, 1165–1174. [Google Scholar] [CrossRef]
- Rezende, R.P.; Dias, J.C.T.; Rosa, C.A.; Carazza, F.; Linardi, V.R. Utilization of nitriles by yeasts isolated from a Brazilian gold mine. J. Gen. Appl. Microbiol. 1999, 45, 185–192. [Google Scholar] [CrossRef]
- Rezende, R.P.; Dias, J.C.T.; Monteiro, A.S.; Carraza, F.; Linardi, V.R. The use of acetonitrile as the sole nitrogen and carbon source by Geotrichum sp. JR1. Braz. J. Microbiol. 2004, 35, 117–120. [Google Scholar] [CrossRef]
- Maier-Greiner, U.H.; Obermaier-Skrobranek, B.M.; Estermaier, L.M.; Kammerloher, W.; Freund, C.; Wulfing, C.; Burkert, U.I.; Matern, D.H.; Breuer, M.; Eulitz, M.; et al. Isolation and properties of a nitrile hydratase from the soil fungus Myrothecium verrucaria that is highly specific for the fertilizer cyanamide and cloning of its gene. Proc. Natl. Acad. Sci. USA 1991, 88, 4260–4264. [Google Scholar] [CrossRef]
- Foerstner, K.U.; Doerks, T.; Muller, J.; Raes, J.; Bork, P. A nitrile hydratase in the eukaryote Monosiga brevicollis. PLoS One 2008, 3, e3976. [Google Scholar]
- Walker, G.; Dorrell, R.G.; Schlacht, A.; Dacks, J.B. Eukaryotic systematics: A user’s guide for cell biologists and parasitologists. Parasitology 2011, 138, 1638–1663. [Google Scholar] [CrossRef]
- Vacelet, J. Electron-microscope study of association between bacteria and sponges of genus Verongia (Dictyoceratida). J. Microsc. Biol. Cell 1975, 23, 271–283. [Google Scholar]
- Vacelet, J.; Donadey, C. Electron-microscope study of association between some sponges and bacteria. J. Exp. Mar. Biol. Ecol. 1977, 30, 301–314. [Google Scholar] [CrossRef]
- Bayer, K.; Scheuermayer, M.; Fieseler, L.; Hentschel, U. Genomic mining for novel FADH2-dependent halogenases in marine sponge-associated microbial consortia. Mar. Biotechnol. 2013, 15, 63–72. [Google Scholar] [CrossRef]
- Payne, M.S.; Wu, S.; Fallon, R.D.; Tudor, G.; Stieglitz, B.; Turner, I.M., Jr.; Nelson, M.J. A Stereoselective cobalt-containing nitrile hydratase. Biochemistry 1997, 36, 5447–5454. [Google Scholar] [CrossRef]
- Nagasawa, T.; Takeuchi, K.; Yamada, H. Characterization of a new cobalt-containing nitrile hydratase purified from urea-induced cells of Rhodococcus rhodochrous J1. Eur. J. Biochem. 1991, 196, 581–589. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nagasawa, T.; Yamada, H. Enzymic synthesis of acrylamide: A success story not yet over. Trends Biotechnol. 1992, 10, 402–408. [Google Scholar] [CrossRef]
- Okamoto, S.; Eltis, L.D. Purification and characterization of a novel nitrile hydratase from Rhodococcus sp. RHA1. Mol. Microbiol. 2007, 65, 828–838. [Google Scholar] [CrossRef]
- Kim, S.H.; Oriel, P. Cloning and expression of the nitrile hydratase and amidase genes from Bacillus sp. BR449 into Escherichia coli. Enzym. Microb. Technol. 2000, 27, 492–501. [Google Scholar] [CrossRef]
- Nojiri, M.; Yohda, M.; Odaka, M.; Matsushita, Y.; Tsujimura, M.; Yoshida, T.; Dohmae, N.; Takio, K.; Endo, I. Functional expression of nitrile hydratase in Escherichia coli: Requirement of a nitrile hydratase activator and post-translational modification of a ligand cystein. J. Biochem. 1999, 125, 696–704. [Google Scholar] [CrossRef]
- Liebeton, K.; Eck, J. Identification and expression in E. coli of novel nitrile hydratases from the metagenome. Eng. Life Sci. 2004, 4, 57–62. [Google Scholar]
- Komeda, H.; Kobayashi, M.; Shimizu, S. Characterization of the gene cluster of high-molecular-mass nitrile hydratase (H–NHase) induced by its reaction product in Rhodococcus rhodochrous J1. Proc. Natl. Acad. Sci. USA 1996, 93, 4267–4272. [Google Scholar] [CrossRef]
- Fallon, R.D.; Stieglitz, B.; Turner, I., Jr. A Pseudomonas putida capable of stereoselective hydrolysis of nitriles. Appl. Microbiol. Biotechnol. 1997, 47, 156–161. [Google Scholar] [CrossRef]
- Zhou, Z.; Hashimoto, Y.; Shiraki, K.; Kobayashi, M. Discovery of posttranslational maturation by self-subunit swapping. Proc. Natl. Acad. Sci. USA 2008, 105, 14849–14854. [Google Scholar] [CrossRef]
- Rodolfo-Metalpa, R.; Lombardi, C.; Cocito, S.; Hall-Spencer, J.M.; Gambi, M.C. Effects of ocean acidification and high temperatures on the bryozoan Myriapora truncata at natural CO2 vents. Mar. Ecol. 2010, 31, 447–456. [Google Scholar]
- Nagasawa, T.; Nanba, H.; Ryuno, K.; Takeuchi, K.; Yamada, H. Nitrile hydratase of Pseudomonas chlororaphis B23. Purification and characterization. Eur. J. Biochem. 1987, 162, 691–698. [Google Scholar] [CrossRef]
- Cramp, R.A.; Cowan, D.A. Molecular characterisation of a novel thermophilic nitrile hydratase. Biochim. Biophys. Acta 1999, 1431, 249–260. [Google Scholar] [CrossRef]
- Thomas, S.M.; DiCosimo, R.; Nagarajan, V. Biocatalysis: Applications and potentials for the chemical industry. Trends Biotechnol. 2002, 20, 238–242. [Google Scholar] [CrossRef]
- Wang, M.-X. Enantioselective biotransformations of nitriles in organic synthesis. Top. Catal. 2005, 35, 117–130. [Google Scholar] [CrossRef]
- Thompson, J.E.; Barrow, K.D.; Faulkner, D.J. Localization of two brominated metabolites, aerothionin and homoaerothionin, in spherulous cells of the marine sponge Aplysina fistularis (=Verongia thiona). Acta Zool. 1983, 64, 199–210. [Google Scholar] [CrossRef]
- Turon, X.; Becerro, M.A.; Uriz, M.J. Distribution of brominated compounds within the sponge Aplysina aerophoba: Coupling of X-ray microanalysis with cryofixation techniques. Cell Tissue Res. 2000, 301, 311–322. [Google Scholar] [CrossRef]
- Fendert, T. Characterization of the enzymatic defence reaction in sponges from the genus Aplysina and isolation of bromotyrosine alkaloids from Aplysina insularis. Ph.D. Thesis, University Würzburg, Würzburg, Germany, 2000. [Google Scholar]
- Weiss, B.; Ebel, R.; Elbraechter, M.; Kirchner, M.; Proksch, P. Defense metabolites from the marine sponge Verongia aerophoba. Biochem. Syst. Ecol. 1996, 24, 1–12. [Google Scholar] [CrossRef]
- Teeyapant, R.; Woerdenbag, H.J.; Kreis, P.; Hacker, J.; Wray, V.; Witte, L.; Proksch, P. Antibiotic and cytotoxic activity of brominated compounds from the marine sponge Verongia aerophoba. Z. Naturforsch. C 1993, 48, 939–945. [Google Scholar]
- Debitus, C.; Guella, G.; Mancini, I.; Waikedre, J.; Guemas, J.-P.; Nicolas, J.L.; Pietra, F. Quinolones from a bacterium and tyrosine metabolites from its host sponge, Suberea creba from the Coral Sea. J. Mar. Biotechnol. 1998, 6, 136–141. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Schaegger, H.; Von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar] [CrossRef]
- Farkas, L.; Gottsegen, A.; Nogradi, M.; Antus, S. Synthesis of the natural isoflavanones ferreirin, dalbergioidin, and ougenin. J. Chem. Soc. C 1971, 1971, 1994–2000. [Google Scholar]
- Andersen, R.J.; Faulkner, D.J. Synthesis of aeroplysinin-1 and related compounds. J. Am. Chem. Soc. 1975, 97, 936–937. [Google Scholar] [CrossRef]
- Lee, S.-S.; Su, M.-J. Aporphine and oxoaporphine compounds and pharmaceutical use thereof. WO2007134485A1, 29 November 2007. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lipowicz, B.; Hanekop, N.; Schmitt, L.; Proksch, P. An Aeroplysinin-1 Specific Nitrile Hydratase Isolated from the Marine Sponge Aplysina cavernicola. Mar. Drugs 2013, 11, 3046-3067. https://doi.org/10.3390/md11083046
Lipowicz B, Hanekop N, Schmitt L, Proksch P. An Aeroplysinin-1 Specific Nitrile Hydratase Isolated from the Marine Sponge Aplysina cavernicola. Marine Drugs. 2013; 11(8):3046-3067. https://doi.org/10.3390/md11083046
Chicago/Turabian StyleLipowicz, Bartosz, Nils Hanekop, Lutz Schmitt, and Peter Proksch. 2013. "An Aeroplysinin-1 Specific Nitrile Hydratase Isolated from the Marine Sponge Aplysina cavernicola" Marine Drugs 11, no. 8: 3046-3067. https://doi.org/10.3390/md11083046