Variegatusides: New Non-Sulphated Triterpene Glycosides from the Sea Cucumber Stichopus variegates Semper

Abstract
:1. Introduction
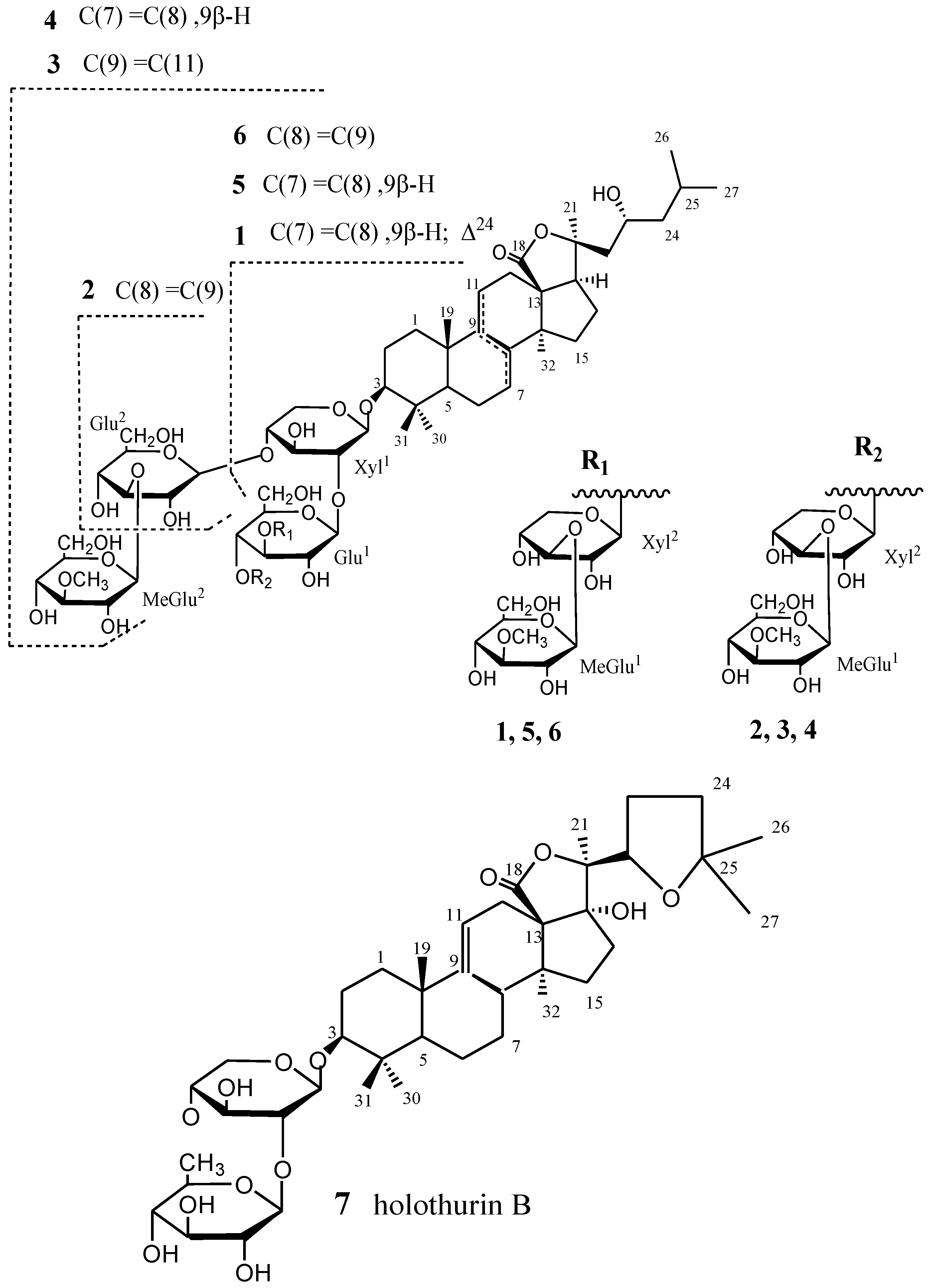
2. Results and Discussion


| No. | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δH mult. (J in Hz) | δC | HMBC (H→C) | δH mult. (J in Hz) | δC | HMBC (H→C) | |
| 1 | 1.20 m; 1.70 m | 36.2, CH2 | 1.17 m; 1.72 m | 36.2, CH2 | ||
| 2 | 1.94 m; 2.21 m | 27.2, CH2 | 1.90 m; 2.10 m | 27.1, CH2 | ||
| 3 | 3.31 dd (4.8, 9.6) | 88.9, CH | 4, 30, 31, 1 (Xyl1) | 3.23 dd (4.8, 10.4) | 88.7, CH | 4, 30, 31, 1 (Xyl1) |
| 4 | 39.8, C | 39.6, C | ||||
| 5 | 1.26 m | 51.1, CH | 3, 4, 10, 19, 30 | 1.13 m | 51.1, CH | 3, 4, 10, 19, 30, 31 |
| 6 | 1.65 m; 1.86 m | 22.1, CH2 | 1.67 m | 18.4, CH2 | ||
| 7 | 5.62 m | 120.3, CH | 5, 6, 8 | 2.14 m; 2.20 m | 27.1, CH2 | 5, 6, 8 |
| 8 | 143.6, C | 131.1, C | ||||
| 9 | 3.72 m | 48.7, CH | 8, 10, 11, 19 | 135.6, C | 8, 10, 11, 19 | |
| 10 | 37.2, C | 37.1, C | ||||
| 11 | 1.32 m; 1.46 m | 23.3, CH2 | 2.22 m; 2.34 m | 21.8, CH2 | ||
| 12 | 2.05 m | 33.2, CH2 | 9,11,13,18 | 2.12 m; 2.19 m | 28.2, CH2 | 9,11,13,18 |
| 13 | 59.0, C | 58.9, C | ||||
| 14 | 51.1, C | 49.0, C | ||||
| 15 | 1.80 m | 33.6, CH2 | 1.48 m; 1.60 m | 33.2, CH2 | ||
| 16 | 1.62 m; 1.74 m | 28.2, CH2 | 1.96 m; 2.10 m | 25.1, CH2 | ||
| 17 | 2.48 m | 52.7, CH | 16, 20, 21 | 2.41 m | 51.4, CH | 16, 20, 21 |
| 18 | 177.6, C | 177.4, C | ||||
| 19 | 1.11 s | 22.3, CH3 | 1, 5, 9, 10 | 1.26 s | 19.2, CH3 | 1, 5, 9, 10 |
| 20 | 83.7, CH | 84.0, C | ||||
| 21 | 1.67 s | 27.4, CH3 | 17, 20, 22 | 1.81s | 28.5, CH3 | 17, 20, 22 |
| 22 | 1.64 m; 2.34 m | 48.9, CH2 | 2.02 m, 2.15 m | 47.9, CH2 | ||
| 23 | 4.20 m | 66.0, CH | 24, 25 | 4.02 m | 65.7, CH | 24, 25 |
| 24 | 5.01 d (7.8 ) | 123.2,CH | 23, 25, 26,27 | 1 34 m; 1.68 m | 49.2, CH2 | 23, 25, 26, 27 |
| 25 | 131.2, C | 2.04 m | 24.7, CH | |||
| 26 | 0.96 d (6.6) | 26.4, CH3 | 24, 25, 27 | 1.00 d (6.8) | 23.8, CH3 | 24, 25, 27 |
| 27 | 1.30 d (6.0) | 19.4, CH3 | 24, 25, 26 | 1.01 d (6.8) | 22.1, CH3 | 24, 25, 26 |
| 30 | 1.10 s | 16.7, CH3 | 3, 4, 5, 31 | 1.07s | 16.5, CH3 | 3, 4, 5, 31 |
| 31 | 1.26 s | 28.2, CH3 | 3, 4, 5, 30 | 1.23 s | 28.0, CH3 | 3, 4, 5, 30 |
| 32 | 1.04 s | 28.3, CH3 | 8, 13, 14, 15 | 1.05 s | 25.4, CH3 | 8, 13, 14, 15 |
| No. | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δH mult. (J in Hz) | δC | HMBC (H→C) | δH mult. (J in Hz) | δC | HMBC (H→C) | |
| Xyl1 | ||||||
| 1 | 4. 83 d (7.2) | 105.8, CH | 3 (Aglycone) | 4.72 d (7.6) | 104.9, CH | 3 (Aglycone) |
| 2 | 4.17 m | 83.7, CH | 4.08 m | 82.9, CH | ||
| 3 | 4.10 m | 78.0, CH | 4.18 m | 75.5, CH | ||
| 4 | 3.99 m | 71.0, CH | 3.95 m | 77.4, CH | ||
| 5 | 3.70 m, 4.34 m | 66.6, CH2 | 3.66 m; 4.40 m | 63.6, CH2 | ||
| Glu1 | ||||||
| 1 | 5.28 d (7.8) | 105.9, CH | 2 (Xyl1) | 5.20 d (6.8) | 105.6, CH | 2 (Xyl1) |
| 2 | 3.94 m | 76.9, CH | 4.02 m | 75.6, CH | ||
| 3 | 4.41 m | 80.4, CH | 4.04 m | 69.1, CH | ||
| 4 | 3.74 m | 69.2, CH | 4.32 m | 80.5, CH | ||
| 5 | 4.21 m | 78.4, CH | 3.98 m | 75.7, CH | ||
| 6 | 4.26 m; 4.44 m | 62.2, CH2 | 4, 5 (Glu1) | 4.20 m; 4.44 m | 62.3, CH2 | C-4, 5 (Glu1) |
| Xyl2 | ||||||
| 1 | 5.11 d (7.8) | 105.0, CH | 3 (Glu1) | 5.05 d (6.8) | 105.2, CH | 4 (Glu1) |
| 2 | 3.83 m | 73.8, CH | 3.96 m | 73.1, CH | ||
| 3 | 4.07 m | 87.6, CH | 4.02 m | 87.7, CH | ||
| 4 | 4.17 m | 70.6, CH | 4.06 m | 69.8, CH | ||
| 5 | 3.58 m; 4.16 m | 66.8, CH2 | 3.55 m; 4.15m | 66.5, CH2 | ||
| Meglu1 | ||||||
| 1 | 5.25 d (7.2) | 105.6, CH | 3 (Xyl2) | 5.18 d (7.6) | 105.4, CH | C-3 (Xyl2) |
| 2 | 4.05 m | 76.8, CH | 3.98 m | 73.6, CH | ||
| 3 | 3.73 m | 88.1, CH | 3.68 m | 87.8, CH | ||
| 4 | 4.14 m | 75.2, CH | 4.09 m | 70.7, CH | 3, 5 (Meglu1) | |
| 5 | 3.82 m | 75.7, CH | 3 (Meglu1) | 3.82 m | 76.6, CH | |
| 6 | 4.42 m; 4.60 m | 61.2, CH2 | 4.38 m; 4.54 m | 61.3, CH2 | ||
| OMe | 3.87 s | 60.9, CH3 | 3 (Meglu1) | 3.85 s | 60.6, CH3 | 3 (Meglu1) |
| Glu2 | ||||||
| 1 | 4.96 d (7.6) | 102.9, CH | 4 (Xyl1) | |||
| 2 | 3.98 m | 75.1, CH | ||||
| 3 | 3.94 m | 78.2, CH | ||||
| 4 | 4.10 m | 70.7, CH | 5 (Glu2) | |||
| 5 | 4.20 m | 77.7, CH | ||||
| 6 | 4.22 m; 4.42 m | 62.3, CH2 | ||||
| MeGlu2 | ||||||
| 1 | ||||||
| 2 | ||||||
| 3 | ||||||
| 4 | ||||||
| 5 | ||||||
| 6 | ||||||
| OMe | ||||||

| No. | 3 | 4 | ||||
|---|---|---|---|---|---|---|
| δH mult. (J in Hz) | δC | HMBC (H→C) | δH mult. (J in Hz) | δC | HMBC (H→C) | |
| 1 | 1.21m; 1.67 m | 36.1, CH2 | 1.47 m | 36.3, CH2 | ||
| 2 | 1.95 m | 26.6, CH2 | 1.95 m | 27.2, CH2 | ||
| 3 | 3.24 dd (6.0,12.0) | 88.5, CH | 4, 30, 31, 1 (Xyl1) | 3.27 dd (4.2, 11.4) | 89.0, CH | 4, 30, 31, 1 (Xyl1) |
| 4 | 39.6, C | 39.6, C | ||||
| 5 | 0.90 m | 52.6, CH | 3, 4, 10, 19, 30, 31 | 1.01 m | 48.1, CH | 3, 4, 10, 19, 30 |
| 6 | 1.20 m; 1.40 m | 20.8, CH2 | 1.97 m | 23.1, CH2 | ||
| 7 | 1.26 m | 27.8, CH2 | 5.69 m | 120.0, CH | 5, 6, 8 | |
| 8 | 3.24 m | 39.7, CH | 147.1, C | |||
| 9 | 151.2, C | 8, 10, 11, 19 | 3.44 m | 47.6, CH | 8, 10, 11, 19 | |
| 10 | 39.2, C | 35.6, C | ||||
| 11 | 5.60 brd (4.8) | 111.0, CH | 1.80 m | 23.4, CH2 | ||
| 12 | 2.04 m | 29.2, CH2 | 9, 11, 13, 18 | 1.90 m; 2.10 m | 30.6, CH2 | 9, 11, 13, 18 |
| 13 | 58.0, C | 58.7, C | ||||
| 14 | 47.2, C | 51.4, C | ||||
| 15 | 1.33 m; 1.67 m | 35.3, CH2 | 1.70 m; 1.82 m | 34.4, CH2 | ||
| 16 | 1.90 m | 24.5, CH2 | 1.88 m; 2.10 m | 24.7, CH2 | ||
| 17 | 2.50 dd (4.8, 10.8) | 51.7, CH | 2.44 dd (4.8, 9.6) | 53.8, CH | 16, 20, 21 | |
| 18 | 178.9, C | 180.7, C | ||||
| 19 | 1.38 s | 21.7, CH3 | 1, 5, 9, 10 | 1.21 s | 28.9, CH3 | 1, 5, 9, 10 |
| 20 | 84.5, C | 85.0, C | ||||
| 21 | 1.80 s | 28.3, CH3 | 17, 20, 22 | 1.83 s | 28.1, CH3 | 17, 20, 22 |
| 22 | 2.02 m; 2.19 m | 47.0; CH2 | 2.02 m; 2.13 m | 47.5, CH2 | ||
| 23 | 4.08 m | 65.0, CH | 25 | 4.01 m | 65.5, CH | 24, 25 |
| 24 | 1.26 m; 1.63 m | 48.8, CH2 | 23, 25, 26, 27 | 1.28 m; 1.64 m | 49.3, CH2 | 23, 25, 26, 27 |
| 25 | 2.03 m | 24.3, CH | 1.91 m | 25.3, CH | ||
| 26 | 1.02 s | 23.5, CH3 | 24, 25, 27 | 1.00 d (6.0) | 22.2, CH3 | 24, 25, 27 |
| 27 | 0.98 s | 21.9, CH3 | 24, 25, 26 | 0.96 d (6.6) | 23.8, CH3 | 24, 25, 26 |
| 30 | 1.07 s | 16.4, CH3 | 3, 4, 5, 31 | 1.10 s | 17.5, CH3 | 3, 4, 5, 31 |
| 31 | 1.22 s | 27.8, CH3 | 3, 4, 5, 30 | 1.12 s | 30.0, CH3 | 3, 4, 5, 30 |
| 32 | 0.87 s | 19.6, CH3 | 8, 13, 14, 15 | 1.11 s | 31.0, CH3 | 8, 13, 14, 15 |
| No. | 3 | 4 | ||||
|---|---|---|---|---|---|---|
| δH mult. (J in Hz) | δC | HMBC (H→C) | δH mult. (J in Hz) | δC | HMBC (H→C) | |
| Xyl1 | ||||||
| 1 | 4.65 d (7.2) | 104.9, CH | 3 (Aglycone) | 4.77 d (7.2) | 105.3, CH | 3 (Aglycone) |
| 2 | 4.06 m | 82.0, CH | 4.11 m | 83.2, CH | ||
| 3 | 4.12 m | 75.3, CH | 4.18 m | 75.7, CH | ||
| 4 | 3.95 m | 77.2, CH | 4.00 m | 76.8, CH | ||
| 5 | 3.60 m; 4.12 m | 64.7, CH2 | 3.64 m; 4.40 m | 64.2, CH2 | ||
| Glu1 | ||||||
| 1 | 5.11 d (7.8) | 105.0, CH | 2 (Xyl1) | 5.23 d (7.8) | 105.7, CH | 2 (Xyl1) |
| 2 | 3.96 m | 73.2, CH | 3.98 m | 73.2, CH | ||
| 3 | 3.99 m | 68.8, CH | 4.06 m | 69.2, CH | ||
| 4 | 4.20 m | 80.2, CH | 4.38 m | 80.3, CH | ||
| 5 | 4.18 m | 75.4, CH | 4.22 m | 75.4, CH | ||
| 6 | 4.11 m; 4.43 m | 61.9, CH2 | 4, 5 (Glu1) | 4.26 m; 4.42 m | 62.1, CH2 | 4, 5 (Glu1) |
| Xyl2 | ||||||
| 1 | 4.95 d (7.2) | 104.5, CH | 4 (Glu1) | 5.11 d (7.8) | 105.0, CH | 4 (Glu1) |
| 2 | 3.95 m | 73.5, CH | 4.02 m | 73.1, CH | ||
| 3 | 4.12 m | 87.4, CH | 1 (Meglu1) | 4.10 m | 87.0, CH | 1 (Meglu1) |
| 4 | 4.00 m | 69.3, CH | 4.04 m | 68.6, CH | ||
| 5 | 3.56 m; 4.10 m | 66.1, CH2 | 3.56 m; 4.19 m | 66.6, CH2 | ||
| Meglu1 | ||||||
| 1 | 5.26 d (7.8) | 103.1, CH | 3 (Xyl2) | 5.03 d (7.2) | 105.8, CH | 3 (Xyl2) |
| 2 | 4.01 m | 76.2, CH | 3 (Meglu1) | 3.98 m | 75.2, CH | 3 (Meglu1) |
| 3 | 3.73 m | 87.6, CH | 3.76 m | 88.1, CH | ||
| 4 | 3.95 m | 70.4, CH | 4.13 m | 70.6, CH | ||
| 5 | 3.90 m | 77.9, CH | 4 (Meglu1) | 3.95 m | 78.4, CH | 4 (Meglu1) |
| 6 | 4.10 m; 4.32 m | 61.9, CH2 | 4.26 m; 4.46 m | 62.2, CH2 | ||
| OMe | 3.87 s | 60.5, CH3 | 3 (Meglu1) | 3.86 s | 60.8, CH3 | 3 (Meglu1) |
| Glu2 | ||||||
| 1 | 4.88 d (7.2) | 102.3, CH | 4 (Xyl1) | 4.98 d (7.2) | 102.9, CH | 4 (Xyl1) |
| 2 | 3.93 m | 75.9, CH | 4.00 m | 73.7, CH | ||
| 3 | 4.08 m | 87.0, CH | 1 (Meglu2) | 4.06 m | 87.6, CH | 1 (Meglu2) |
| 4 | 4.03 m | 70.4, CH | 4.08 m | 69.7, CH | ||
| 5 | 3.94 m | 77.6, CH | 3.94 m | 78.4, CH | ||
| 6 | 4.10 m; 4.40 m | 61.0, CH2 | 4.24 m; 4.48 m | 62.2, CH2 | ||
| MeGlu2 | ||||||
| 1 | 5.28 d (7.2) | 105.2, CH | 3 (Glu2) | 5.25 d (7.8) | 105.6, CH | 3 (Glu2) |
| 2 | 4.02 m | 76.1, CH | 3.99 m | 75.2, CH | ||
| 3 | 3.72 m | 87.5, CH | 2, -4 (Meglu2,OMe) | 3.74 m | 88.1, CH | 2, 4 (Meglu2,OMe) |
| 4 | 4.05 m | 71.0, CH | 4.15 m | 70.6, CH | ||
| 5 | 3.80 m | 78.0, CH | 3.96 m | 77.5, CH | ||
| 6 | 4.10 m; 4.44 m | 61.9, CH2 | 4.20 m; 4.41 m | 61.1, CH2 | ||
| OMe | 3.86 s | 60.5, CH3 | 3 (Meglu2) | 3.85 s | 60.8, CH3 | 3 (Meglu2) |
| C. albicans | C. pseudotropicalis | C. neoformans | C. parapsilosis | M. gypseum | C. tropicalis | |
|---|---|---|---|---|---|---|
| 1 | 12.5 | 25 | 50 | 25 | 12.5 | 12.5 |
| 2 | 3.4 | 3.4 | 6.8 | 3.4 | 3.4 | 13.6 |
| 3 | 25 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
| 4 | 25 | 12.5 | 25 | 12.5 | 12.5 | 12.5 |
| 5 | 25 | 25 | 12.5 | 12.5 | 12.5 | 12.5 |
| 6 | 100 | 125 | 50 | 25 | 25 | >125 |
| 7 | 18.8 | 18.8 | 9.4 | >125 | >125 | 37.6 |
| FCZ a | 1 | 4 | 4 | 1 | >64 | 1 |
| ICZ a | 0.25 | 0.125 | 0.5 | 0.125 | 0.125 | 0.5 |
| KCZ a | 1 | 0.125 | 0.5 | 0.125 | 0.125 | 0.125 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.3.1. Variegateside C (1)
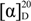
{kind=link}
{kind=link}
3.3.2. Variegateside D (2)
3.3.3. Variegateside E (3)
3.3.4. Variegateside F (4)
3.4. Acid Hydrolysis of Compounds 1–4
3.5. Bioassay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maier, M.S.; Roccatagliata, A.J.; Kuriss, A.; Cludil, H.; Seldes, A.M.; Pujol, C.A.; Damonte, E.B. Two new cytotoxic and virucidal trisulfated triterpene glycosides from the Antarctic sea cucumber Staurocucumis liouvillei. J. Nat. Prod. 2001, 64, 732–736. [Google Scholar] [CrossRef]
- Kalinin, V.A.; Anisimov, M.M.; Prokofieva, N.G.; Avilov, S.A.; Afiyatullov, S.S.; Stonik, V.A. Biological Activities and Biological Role of Triterpene Glycosides from Holothuroids (Echinodermata). In Echinoderm Studies; Jangoux, M., Lawrence, J.M., Eds.; A.A. Balkema: Rotterdam, The Netherlands, 1996; Volume 5, pp. 139–181. [Google Scholar]
- Stonik, V.A.; Elyakov, G.B. Secondary Metabolites from Echinoderms as Chemotaxonomic Markers. In Bioorganic Marine Chemistry; Scheuer, P.J., Ed.; Springer: Berlin, Germany, 1988; pp. 43–86. [Google Scholar]
- Zou, Z.-R.; Yi, Y.-H.; Wu, H.-M.; Yao, X.-S.; Du, L.-J.; Wu, J.-H.; Liaw, C.C.; Lee, K.H. Intercedensides A–C, three new cytotoxic triterpene glycosides from the sea cucumber Mensamaria intercedens Lampert. J. Nat. Prod. 2003, 66, 1055–1060. [Google Scholar] [CrossRef]
- Zhang, S.-L.; Li, L.; Yi, Y.-H.; Zou, Z.-R.; Sun, P. Philinopgenin A, B, and C, three new triterpenoid aglycones from the sea cucumber Pentacta quadrangulasis. Mar. Drugs 2004, 2, 185–191. [Google Scholar] [CrossRef]
- Zou, Z.-R.; Yi, Y.-H.; Wu, H.-M.; Wu, J.-H.; Liaw, C.C.; Lee, K.H. Intercedensides D–I, cytotoxic triterpene glycosides from the sea cucumber Mensamaria intercedens Lampert. J. Nat. Prod. 2005, 68, 540–547. [Google Scholar] [CrossRef]
- Yi, Y.-H.; Xu, Q.-Z.; Li, L.; Zhang, S.-L.; Wu, H.-M.; Ding, J.; Tong, Y.-G.; Tan, W.-F.; Li, M.-H.; Tian, F.; et al. Philinopsides A and B, two new sulfated triterpene glycosides from the sea cucumber Pentacta quadrangularis. Helv. Chim. Acta 2006, 89, 54–64. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Yi, Y.-H.; Tang, H.-F. Bioactive triterpene glycosides from the sea cucumber Holothuria fuscocinerea. J. Nat. Prod. 2006, 69, 1492–1495. [Google Scholar] [CrossRef]
- Han, H.; Yi, Y.-H.; Xu, Q.-Z.; La, M.-P.; Zhang, H.-W. Two new cytotoxic triterpene glycosides from the sea cucumber Holothuria scabra. Planta Med. 2009, 75, 1680–1612. [Google Scholar]
- Han, H.; Xu, Q.-Z.; Tang, H.-F.; Yi, Y.-H.; Gong, W. Cytotoxic holostane-type triterpene glycosides from the sea cucumber Pentacta quadrangularis. Planta Med. 2010, 76, 1900–1904. [Google Scholar] [CrossRef]
- Liao, Y.-L. Chinese Fauna Echinodermata Holothuroidea; Science Press: Beijing, China, 1997; pp. 154–156. [Google Scholar]
- Kitagawa, I.; Kobayashi, M.; Inamoto, T.; Yasuzawa, T.; Kyogoku, Y.M. The structure of six antifungal oligoglycosides, stichloroside-A1, stichloroside-A2, stichloroside-B1, stichloroside-B2, stichloroside-C1, and stichloroside-C2, from the sea cucumber Stichopus chloronotus (Brandt). Chem. Pharm. Bull. 1981, 29, 2387–2391. [Google Scholar] [CrossRef]
- Wang, X.-H.; Li, L.; Yi, Y.-H.; Sun, P.; Yan, B.; Pan, M.-X.; Han, H.; Wang, X.-D. Two new triterpene glycosides from sea cucumber Stichopus variegatus Semper. Chin. J. Nat. Med. 2006, 4, 177–180. [Google Scholar]
- Nurettin, Y.; John, A.F. A triterpenoid saponin from Cucumaria frondosa. Phytochemistry 1999, 50, 135–138. [Google Scholar] [CrossRef]
- Kitagawa, I.; Kobayashi, M.; Hori, M.; Kyogoku, Y. Marine natural products. XVIII. Four lanostane-type triterpene oligoglycosides, bivittosides A, B, C, and D, from the Okinawan sea cucumber Bohadschia bivittata Mitsukuri. Chem. Pharm. Bull. 1989, 37, 61–67. [Google Scholar] [CrossRef]
- Shashkov, A.S.; Chizhov, O.S. C13 NMR spectroscopy in chemistry of carbohydrates and related compounds. Bioorg. Khim. 1976, 2, 437–497. [Google Scholar]
- Greta, M.; Peter, C.N.; Vladimir, I.K.; Sergey, A.A.; Alexandra, S.S.; Pavel, S.D.; Valentin, A.S.; Valery, S.L. Structure of the major triterpene glycosiden from the sea cucumber Stichopus mollis and evidence to reclassify this species into the new genus Australostichopus. Biochem. Syst. Ecol. 2004, 32, 637–650. [Google Scholar] [CrossRef]
- Zhang, J.-D.; Xu, Z.; Cao, Y.-B.; Chen, H.; Yan, L.; An, M.-M.; Gao, P.-H.; Wang, Y.; Jia, X.-M.; Jiang, Y.-Y. Antifungal activities and action mechanisms of compounds from Tribubuls terrestris L. J. Ethnopharmacol. 2006, 103, 76–84. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, X.-H.; Zou, Z.-R.; Yi, Y.-H.; Han, H.; Li, L.; Pan, M.-X. Variegatusides: New Non-Sulphated Triterpene Glycosides from the Sea Cucumber Stichopus variegates Semper. Mar. Drugs 2014, 12, 2004-2018. https://doi.org/10.3390/md12042004
Wang X-H, Zou Z-R, Yi Y-H, Han H, Li L, Pan M-X. Variegatusides: New Non-Sulphated Triterpene Glycosides from the Sea Cucumber Stichopus variegates Semper. Marine Drugs. 2014; 12(4):2004-2018. https://doi.org/10.3390/md12042004
Chicago/Turabian StyleWang, Xiao-Hua, Zheng-Rong Zou, Yang-Hua Yi, Hua Han, Ling Li, and Min-Xiang Pan. 2014. "Variegatusides: New Non-Sulphated Triterpene Glycosides from the Sea Cucumber Stichopus variegates Semper" Marine Drugs 12, no. 4: 2004-2018. https://doi.org/10.3390/md12042004
APA StyleWang, X.-H., Zou, Z.-R., Yi, Y.-H., Han, H., Li, L., & Pan, M.-X. (2014). Variegatusides: New Non-Sulphated Triterpene Glycosides from the Sea Cucumber Stichopus variegates Semper. Marine Drugs, 12(4), 2004-2018. https://doi.org/10.3390/md12042004