
Chitin and Chitosan as Direct Compression Excipients in Pharmaceutical Applications


Abstract
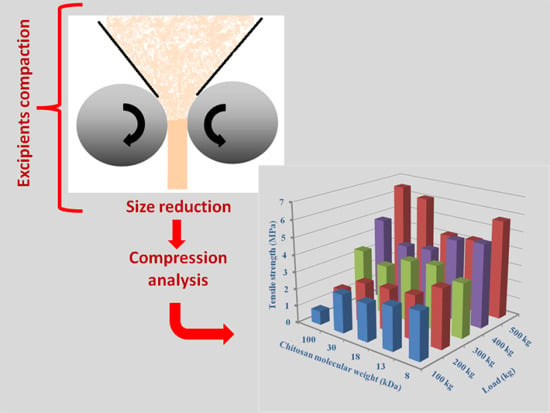
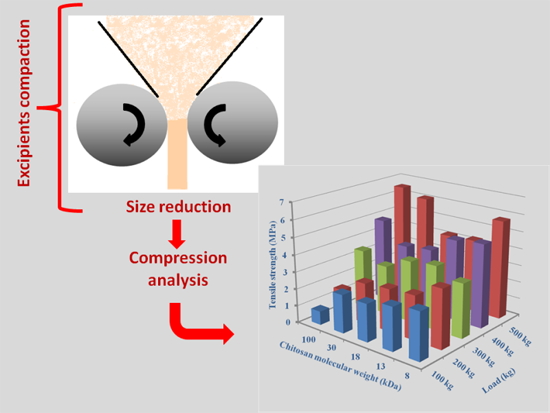
:
1. Introduction
1.1. Chitin and Chitosan Production
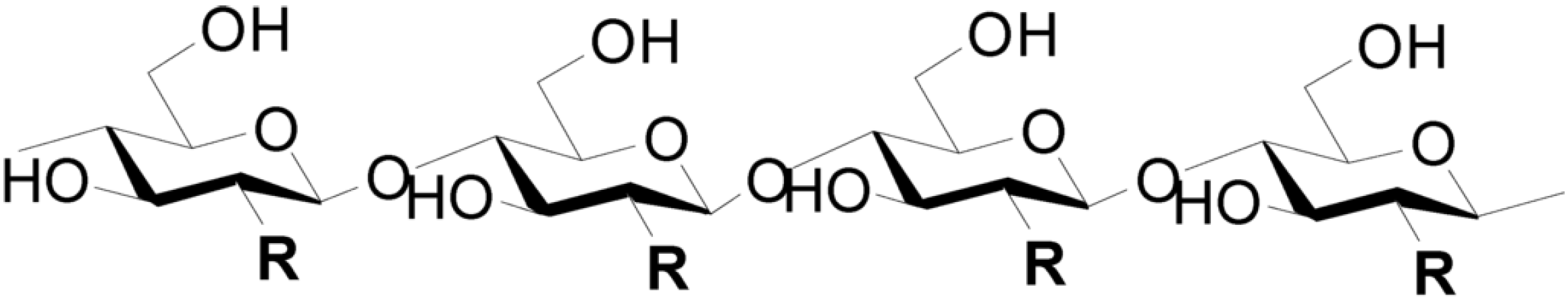
1.2. Chitin and Chitosan: Physical and Chemical Properties

1.3. Applications of Chitin and Chitosan
2. Chitin and Chitosan for Direct Compression Processing


| Excipient | Specific Surface Area (m2/g) | Pore Volume (cm3/g) | References |
|---|---|---|---|
| Lactose H2O | 0.26 | 0.090 | [54] |
| Microcrystalline cellulose | 0.42 | 1.67 | [55,56] |
| Maize starch | 0.58 | 0.0012 | [57] |
| Synthesized CaHPO4 | 3.31 | 0.0065 | [58] |
| Chitosan | 330 | 15 | [59] |
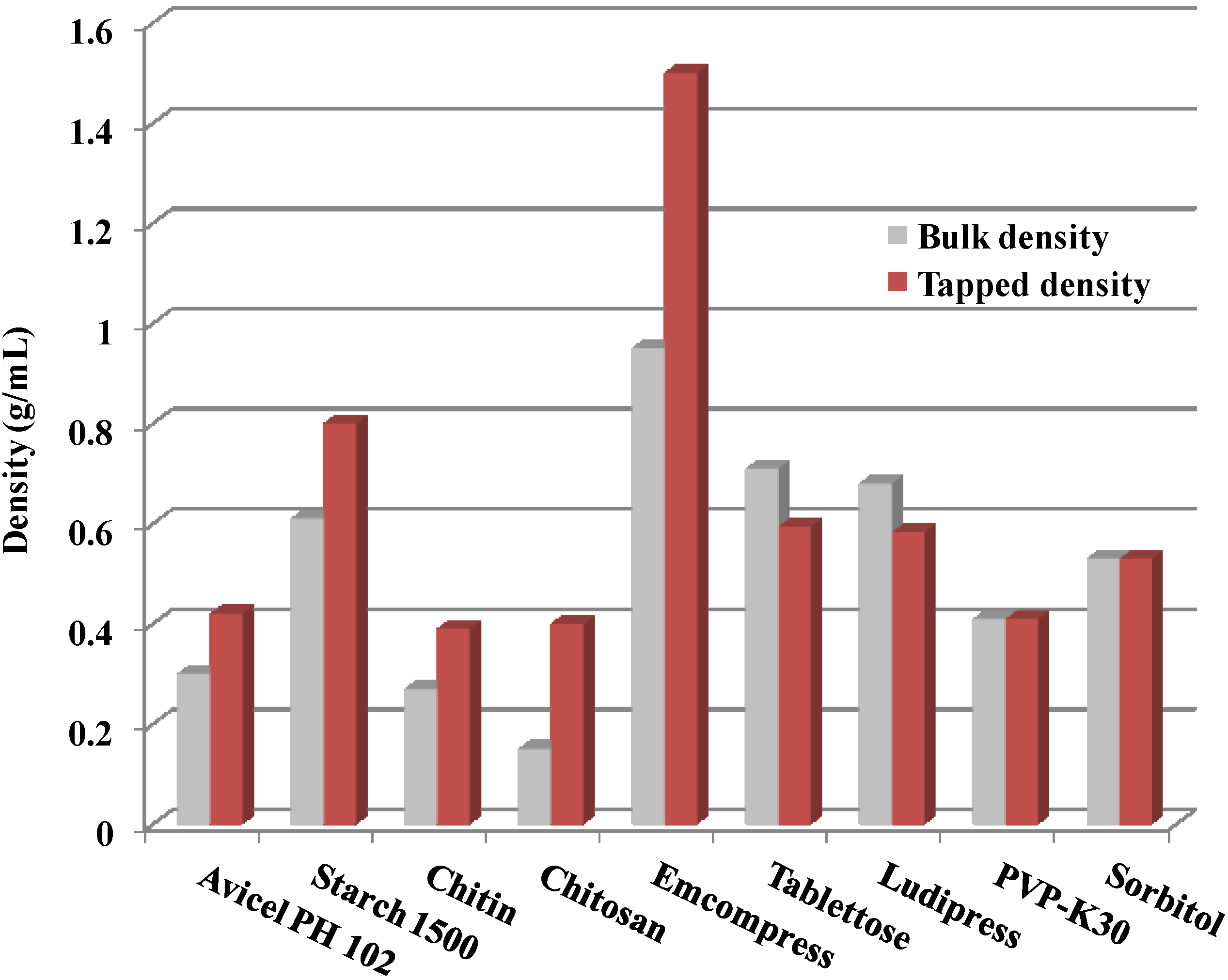
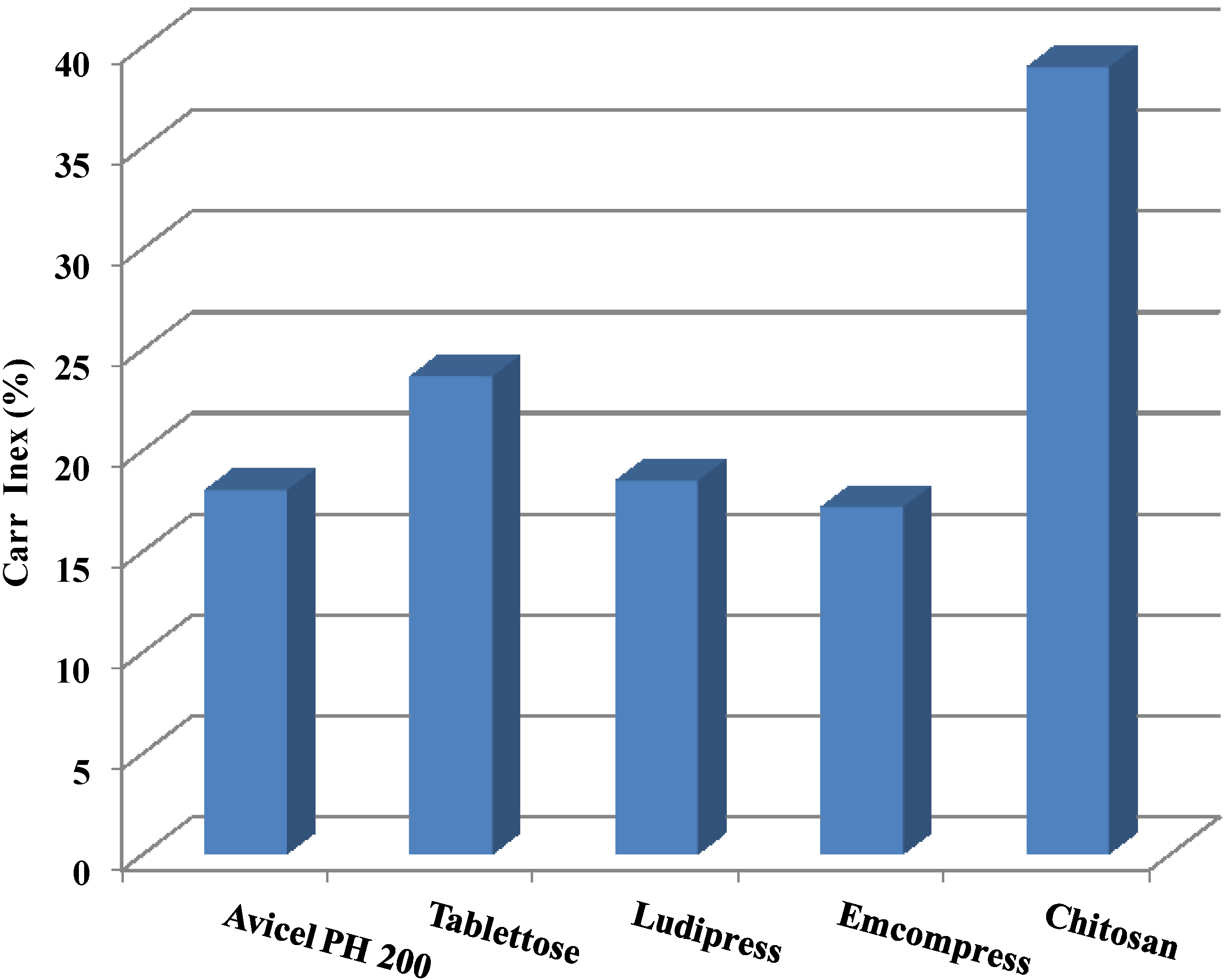
2.2. Powder Flow

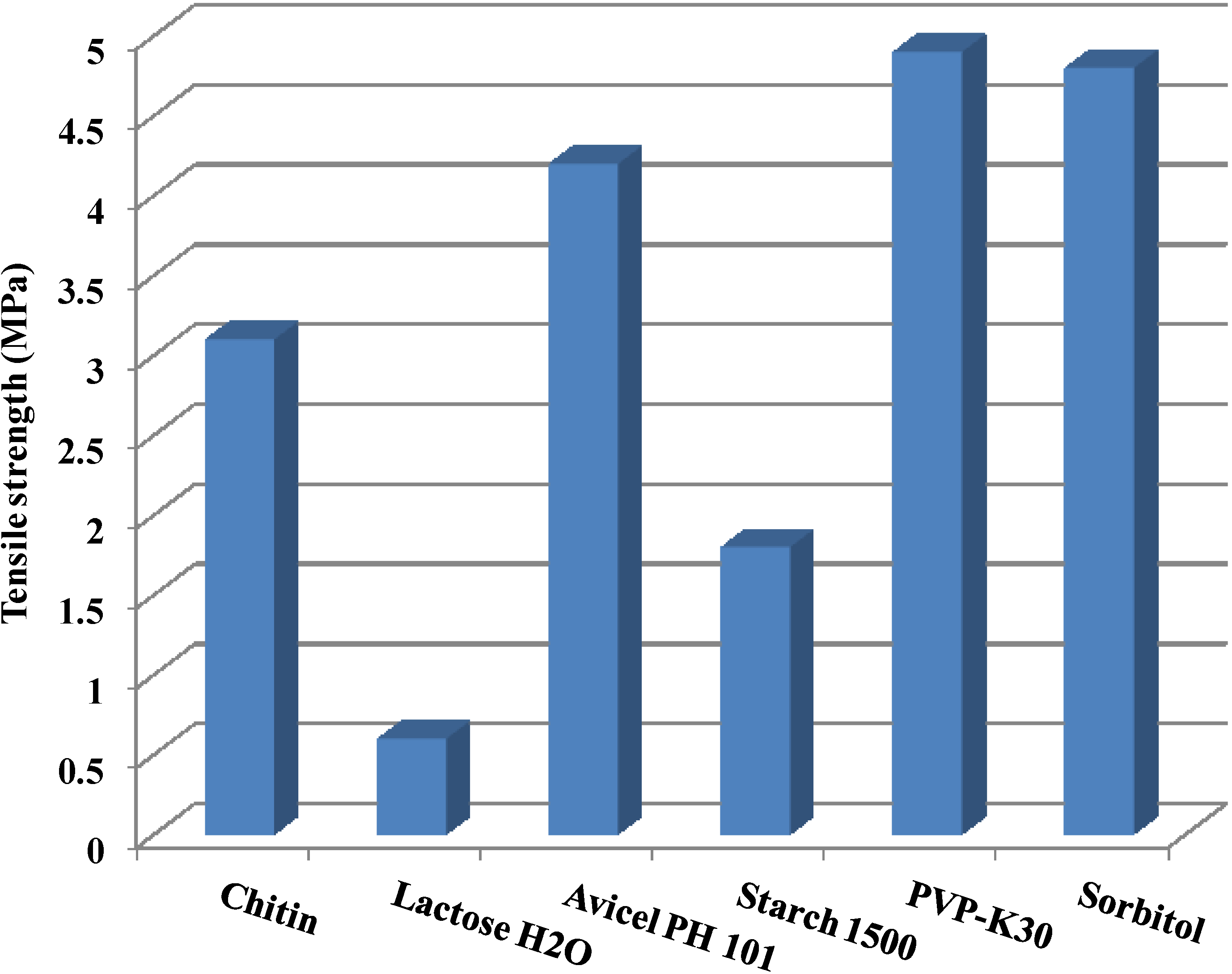
2.3. Tensile Strength

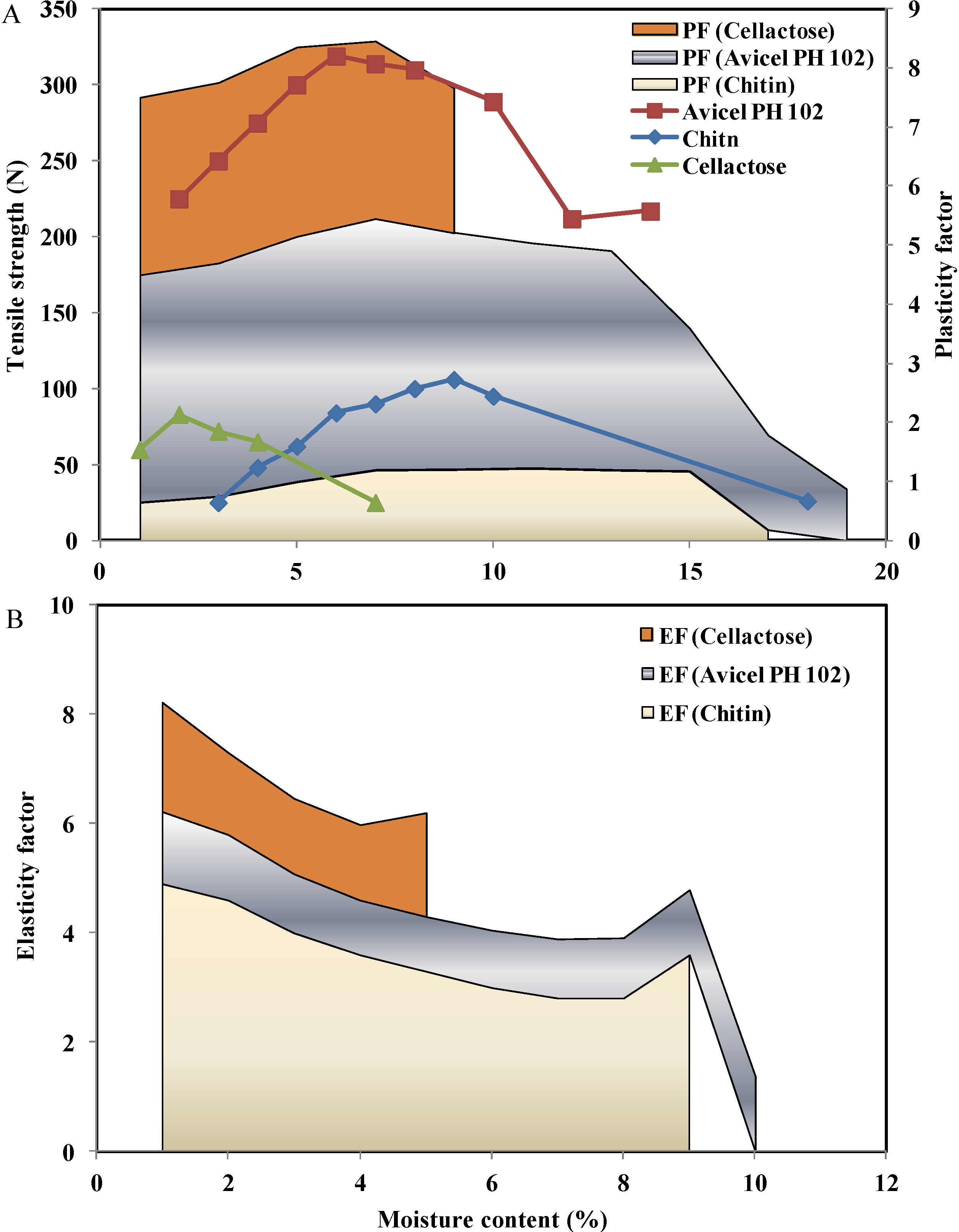
2.3.1. Effect of Moisture Content
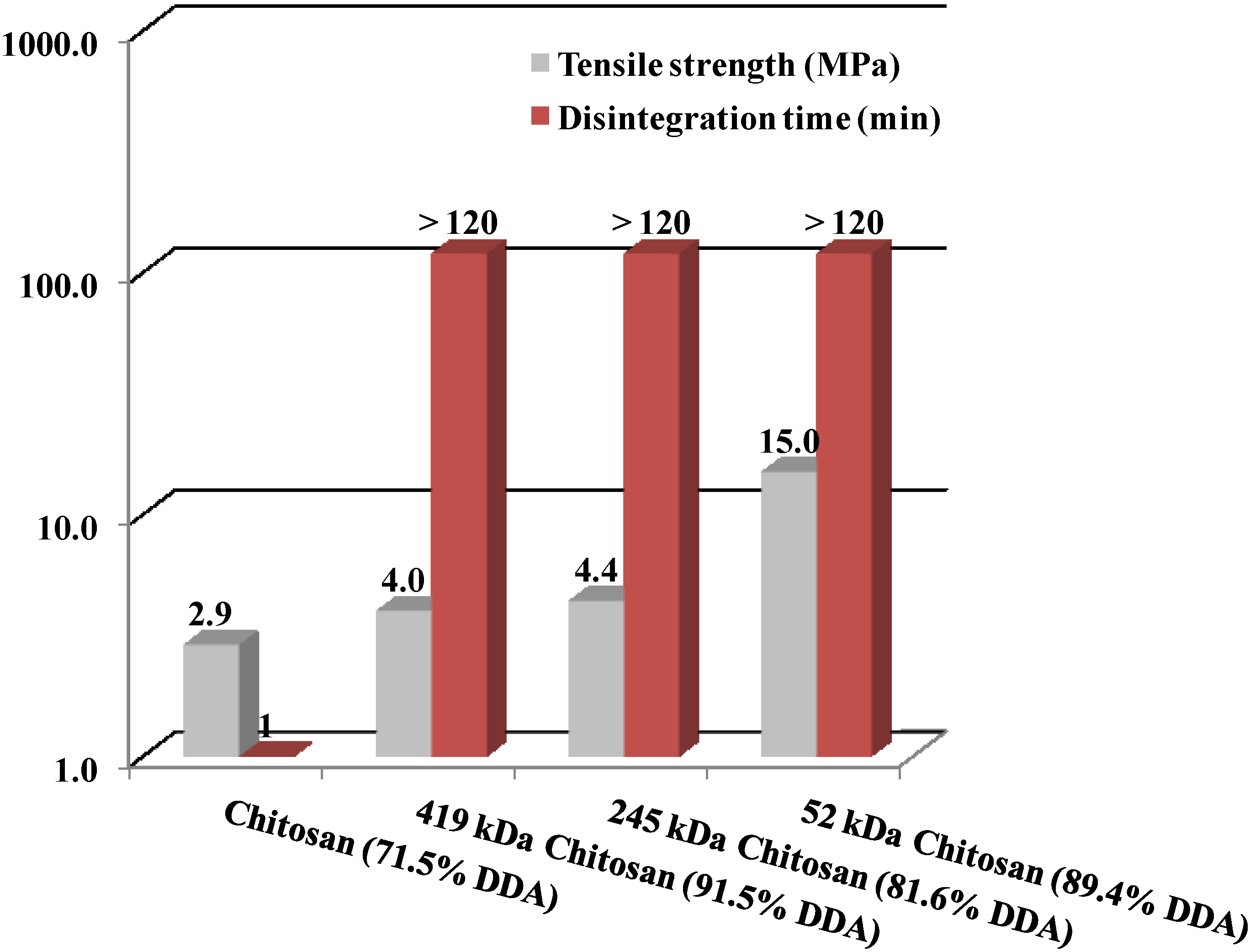
2.3.2. Effect of Degree of Deacetylation
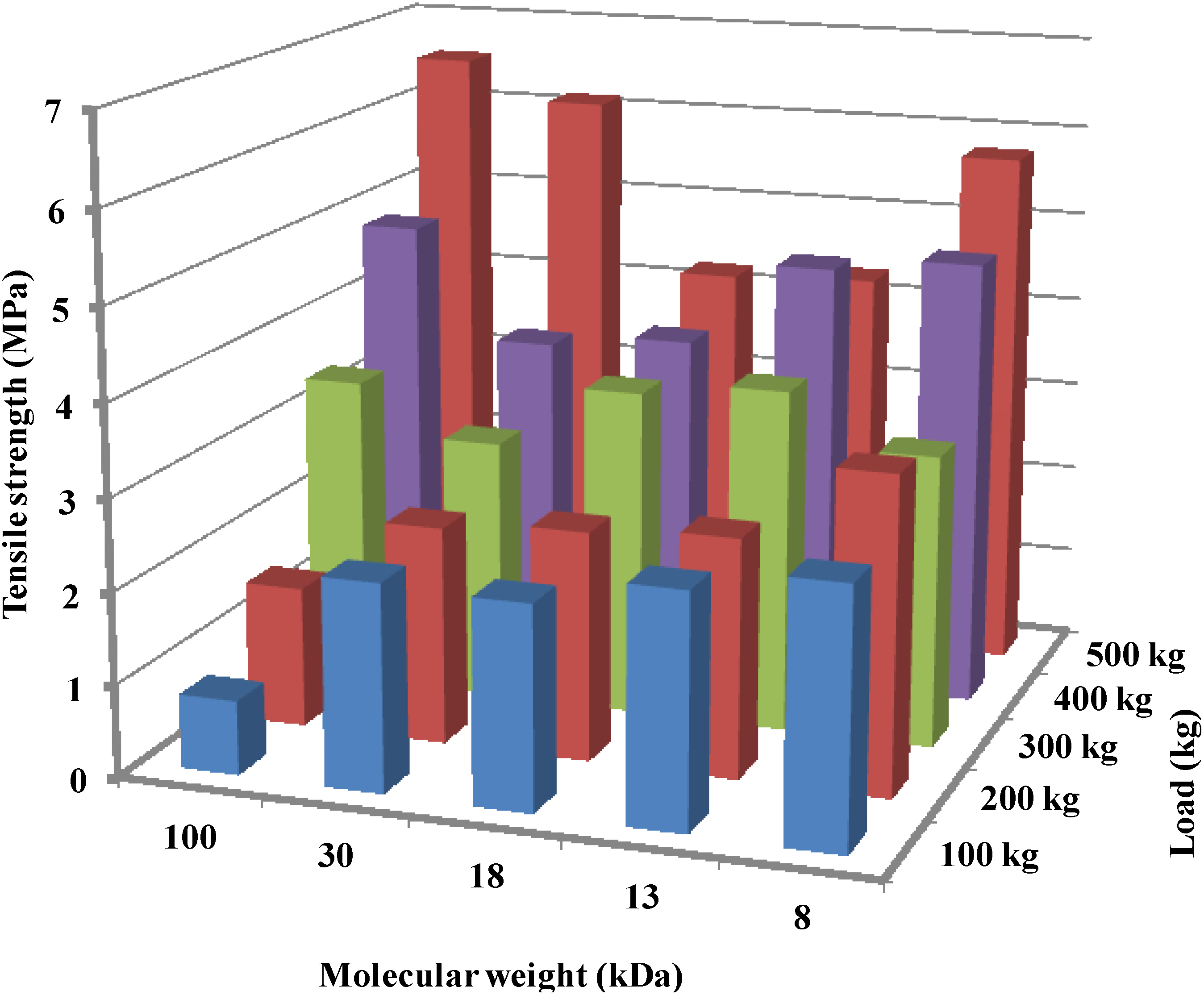
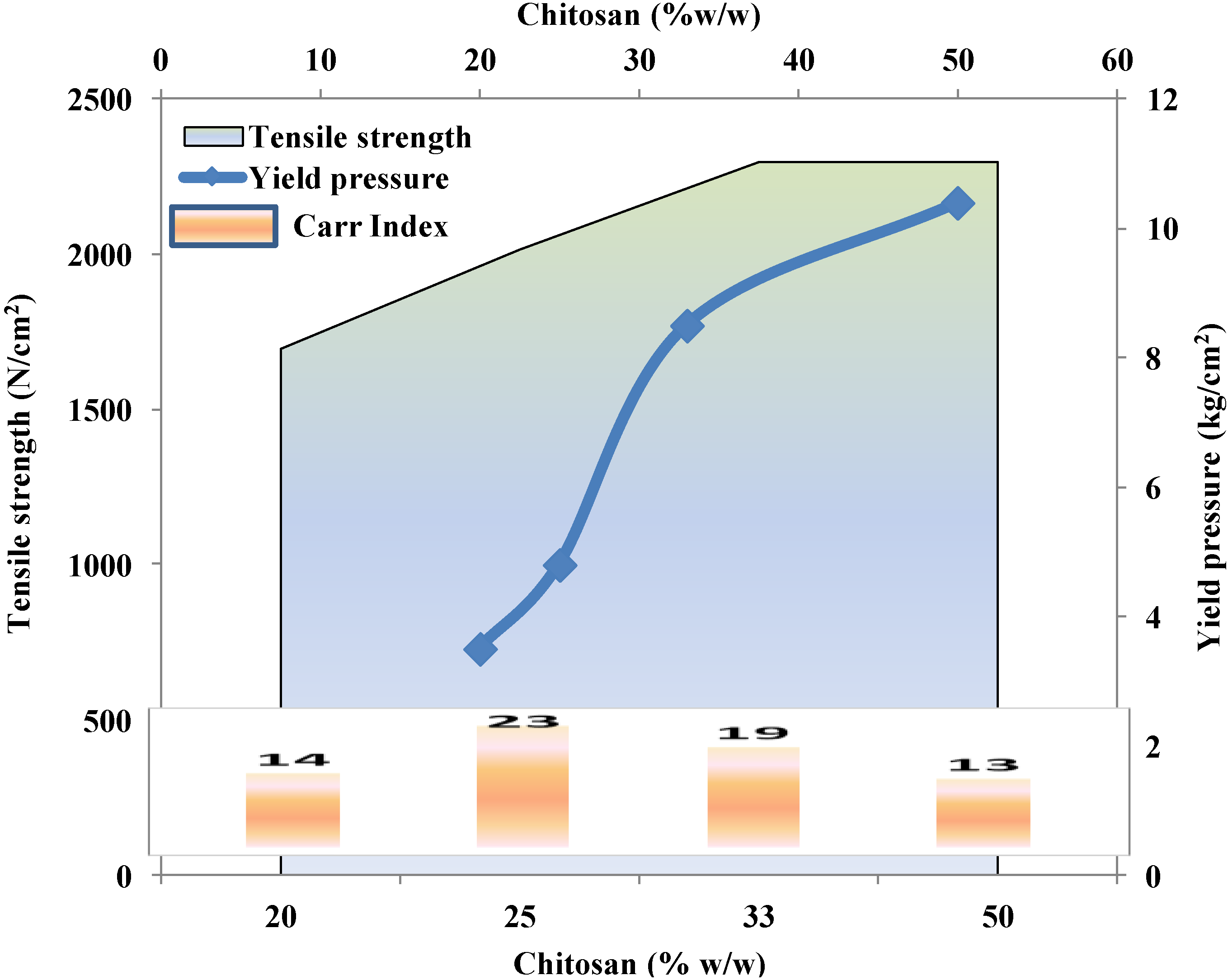
2.3.3. Effect of Molecular Weight


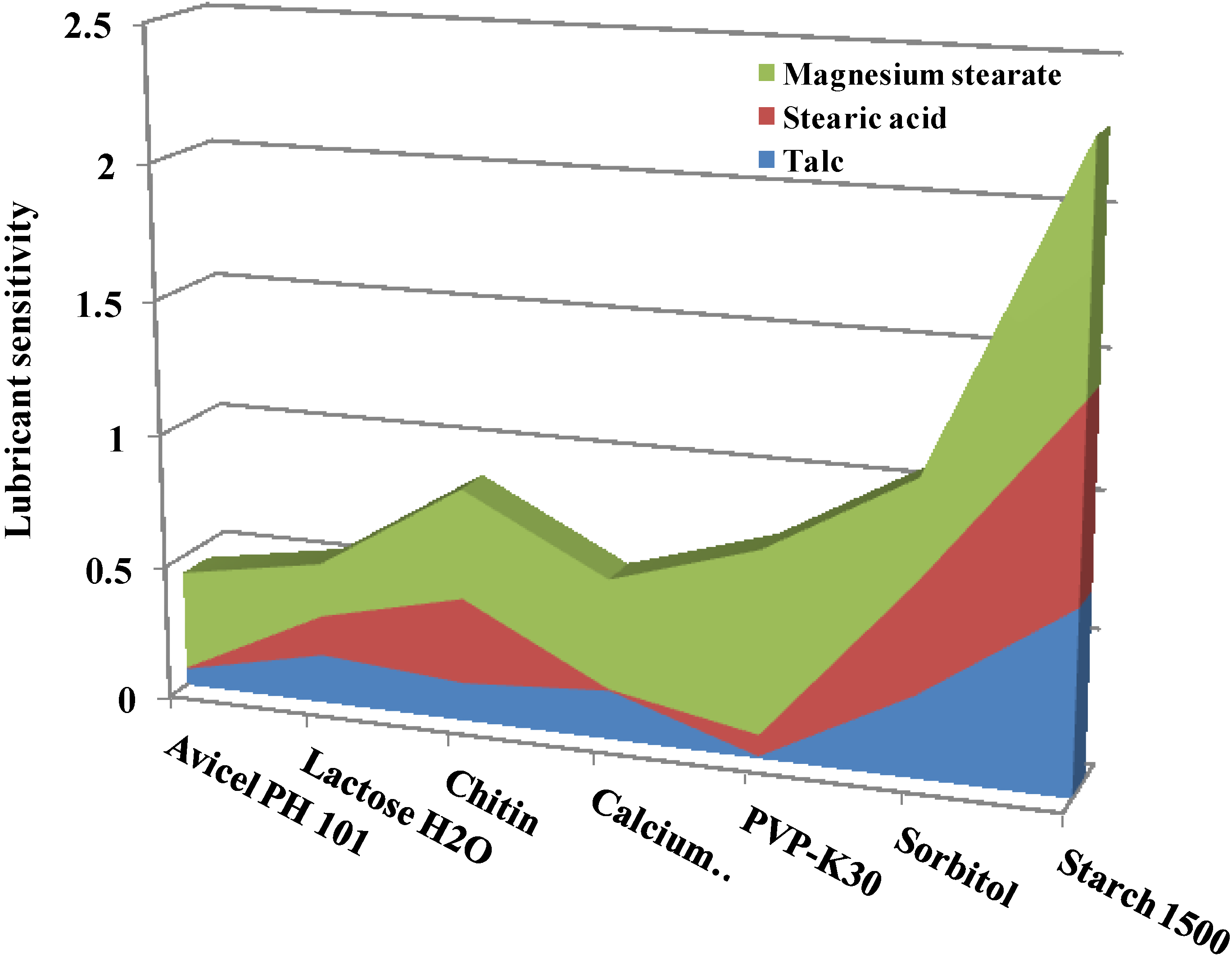
2.3.4. Effect of Lubricant

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.4. Compressibility of Chitin and Chitosan
2.4.1. Heckel Analysis
| Parameter | Calcium Diphosphate | Chitin | Lactose H2O | Avicel PH 101 | Starch 1500 | PVP K30 | Sorbitol |
|---|---|---|---|---|---|---|---|
| PY | 250.1 | 122 | 150 | 62.5 | 75.1 | 35.7 | 48.4 |
| D0 | 0.36 | 0.12 | 0.38 | 0.23 | 0.33 | 0.27 | 0.39 |
| DA | 0.49 | 0.52 | 0.69 | 0.44 | 0.48 | 0.72 | 0.79 |
| DB | 0.13 | 0.31 | 0.31 | 0.21 | 0.15 | 0.46 | 0.4 |

2.4.2. Kawakita Analysis
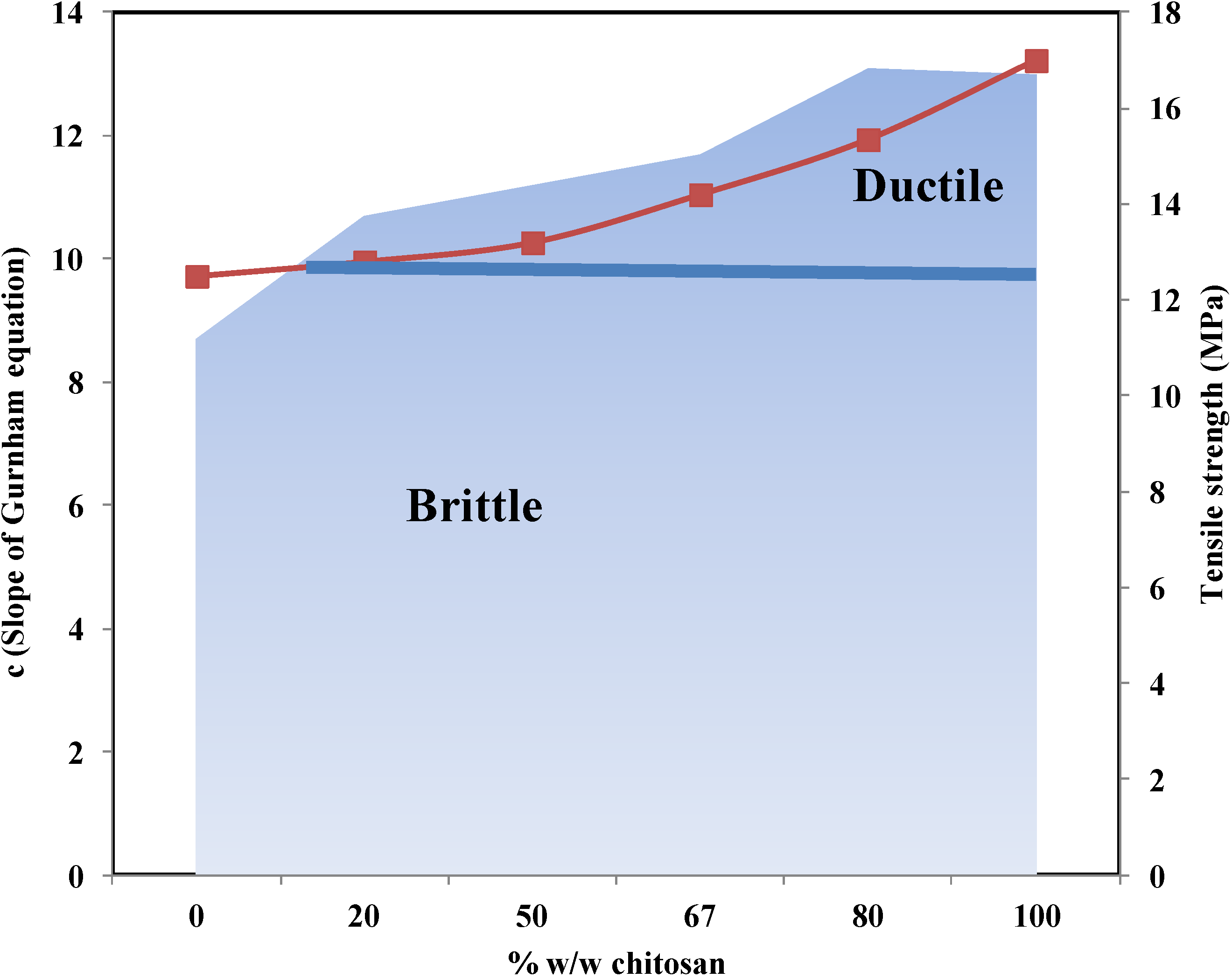
2.4.3. The Gurnham Equation
2.4.4. Plasticity/Elasticity Factors
2.5. Factors Contributing to the Powder Compressibility Properties of Chitin/Chitosan
2.5.1. Moisture Content
2.5.2. Degree of Deacetylation
2.5.3. Molecular Weight
| Material/MW (kDa) | Kawakita Parameter | |||||
|---|---|---|---|---|---|---|
| a | ab | b | 1/b | PY | A | |
| Chitin | 0.818 | 0.077 | 0.094 | 10.57 | - | - |
| Chitosan/100 | 0.75 | 0.092 | 0.12 | 8.15 | 72.5 | 0.42 |
| Chitosan/30 | 0.54 | 0.066 | 0.12 | 8.14 | 98.0 | 0.46 |
| Chitosan/18 | 0.63 | 0.084 | 0.13 | 25.55 | 106.4 | 0.47 |
| Chitosan/8 | 0.52 | 0.024 | 0.046 | 21.55 | 153.9 | 0.60 |
2.6. Compressibility Changes upon Formulation and/or Modification of Chitosan
2.6.1. Physical Mixing

2.6.2. Spray-Drying

2.6.3. Co-Precipitation of Silicon Derivatives on Chitin/Chitosan
| Mixture | BD | TD | % Comp. | Heckel Parameters | Kawakita Parameters | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| PY | DA | DB | a | b | 1/b | ab | ||||
| Chitosan–silica (50% chitosan) | 0.38 | 0.41 | 7.32 | – | – | – | – | – | – | – |
| Chitin–silica (50% chitin) | 0.45 | 0.5 | 10 | 98 | 0.165 | 0.588 | – | – | – | – |
| Chitin–Mg silicate (68% chitin) | – | – | – | – | – | – | 0.75 | – | 17.37 | 0.043 |
| Chitin | 0.27 | 0.39 | 30.77 | – | – | – | 0.82 | 1.67 | 0.6 | 0.077 |
| Avicel® 200 | – | – | – | 81.3 | 0.09 | 0.611 | – | – | – | – |
2.6.4. Co-Processing of Chitin/Chitosan by Compaction
3. Conclusions
Author Contributions
Conflicts of Interest
References
- Gohel, M.C.; Jogani, P.D. A review of co-processed directly compressible excipients. J. Pharm. Pharm. Sci. 2005, 8, 76–93. [Google Scholar] [PubMed]
- Michoel, A.; Rombaut, P.; Verhoye, A. Comparative evaluation of co-processed lactose and microcrystalline cellulose with their physical mixtures in the formulation of folic acid tablets. Pharm. Dev. Technol. 2002, 7, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Bindhumadhavan, G.; Seville, J.P.K.; Adams, M.J.; Greenwood, R.W.; Fitzpatrick, S. Roll compaction of a pharmaceutical excipient: Experimental validation of rolling theory for granular solids. Chem. Eng. Sci. 2005, 60, 3891–3897. [Google Scholar] [CrossRef]
- Pingali, K.; Mendez, R.; Lewis, D.; Michniak-Kohn, B.; Cuitino, A.; Muzzio, F. Mixing order of glidant and lubricant—Influence on powder and tablet properties. Int. J. Pharm. 2011, 409, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Haware, R.V.; Tho, I.; Bauer-Brandl, A. Evaluation of a rapid approximation method for the elastic recovery of tablets. Powder Technol. 2010, 202, 71–77. [Google Scholar] [CrossRef]
- Rashid, I.; al Omari, M.M. H.; Badwan, A.A. From native to multifunctional starch-based excipients designed for direct compression formulation. Starch Stärke 2013, 65, 552–571. [Google Scholar] [CrossRef]
- Daraghmeh, N.H.; Chowdhry, B.Z.; Leharne, S.A.; al Omari, M.M.; Badwan, A.A. Chapter 2—Chitin. In Profiles of Drug Substances Excipients, and Related Methodology; Harry, G.B., Ed.; Academic Press: Waltham, MA, USA, 2011; Volume 36, pp. 35–102. [Google Scholar]
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Aranaz, I.; Mengibar, M.; Harris, R.; Panos, I.; Miralles, B.; Acosta, N.; Galed, G.; Heras, A. Functional characterization of chitin and chitosan. Curr. Chem. Biol. 2009, 3, 203–230. [Google Scholar]
- Hamid, R.A.; Al-Akayleh, F.; Shubair, M.; Rashid, I.; Remawi, M.A.; Badwan, A. Evaluation of three chitin metal silicate co-precipitates as a potential multifunctional single excipient in tablet formulations. Mar. Drugs 2010, 8, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, C.; Xue, Y.; Gao, R.; Zhang, X. Determination of the degree of deacetylation of chitin and chitosan by X-ray powder diffraction. Carbohydr. Res. 2005, 340, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Sweidan, K.; Jaber, A.M.; Al-jbour, N.; Obaidat, R.; Al-Remawi, M.; Badwan, A. Further investigation on the degree of deacetylation of chitosan determined by potentiometric titration. J. Excip. Food Chem. 2011, 2, 16–25. [Google Scholar]
- Pillai, C.K.S.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Dutta, P.K.; Dutta, J.; Tripathi, V.S. Chitin and chitosan: Chemistry, properties and applications. J. Sci. Ind. Res. 2004, 63, 20–31. [Google Scholar]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Qandil, A.; Obaidat, A.; Ali, M.M.; Al-Taani, B.; Tashtoush, B.; Al-Jbour, N.; al Remawi, M.; Al-Sou’od, K.; Badwan, A. Investigation of the interactions in complexes of low molecular weight chitosan with ibuprofen. J. Sol. Chem. 2009, 38, 695–712. [Google Scholar] [CrossRef]
- Kumar, M.N.; Muzzarelli, R.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.A.; Tubertini, O. Chitin and chitosan as chromatographic supports and adsorbents for collection of metal ions from organic and aqueous solutions and sea-water. Talanta 1969, 16, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Basu, P.S.; Datta, T.K. Isolation and characterization of Vicia faba lectin affinity purified on chitin column. Prep. Biochem. 1984, 14, 373–387. [Google Scholar] [PubMed]
- Songkroah, C.; Nakbanpote, W.; Thiravetyan, P. Recovery of silver-thiosulphate complexes with chitin. Proc. Biochem. 2004, 39, 1553–1559. [Google Scholar] [CrossRef]
- Krajewska, B. Application of chitin- and chitosan-based materials for enzyme immobilizations: A review. Enzyme Microb. Technol. 2004, 35, 126–139. [Google Scholar] [CrossRef]
- Lim, S.H.; Hudson, S.M. Synthesis and antimicrobial activity of a water-soluble chitosan derivative with a fiber-reactive group. Carbohydr. Res. 2004, 339, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Albarghouthi, M.; Fara, D.A.; Saleem, M.; el-Thaher, T.; Matalka, K.; Badwan, A. Immobilization of antibodies on alginate-chitosan beads. Int. J. Pharm. 2000, 206, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Yusof, N.L.; Wee, A.; Lim, L.Y.; Khor, E. Flexible chitin films as potential wound-dressing materials: Wound model studies. J. Biomed. Mater. Res. A 2003, 66, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Rathke, T.D.; Hudson, S.M. Review of chitin and chitosan as fiber and film formers. J. Macromol. Sci. C 1994, 34, 375–437. [Google Scholar] [CrossRef]
- Pierson, Y.; Chen, X.; Bobbink, F.D.; Zhang, J.; Yan, N. Acid-catalyzed chitin liquefaction in ethylene glycol. ACS Sustain. Chem. Eng. 2014, 2, 2081–2089. [Google Scholar] [CrossRef]
- Chen, X.; Chew, S.L.; Kerton, F.M.; Yan, N. Direct conversion of chitin into a N-containing furan derivative. Green Chem. 2014, 16, 2204–2212. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.; Barontini, G.; Rocchetti, R. Immobilized enzymes on chitosan columns: α-chymotrypsin and acid phosphatase. Biotechnol. Bioeng. 1976, 18, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.; Baldassarre, V.; Conti, F.; Ferrara, P.; Biagini, G.; Gazzanelli, G.; Vasi, V. Biological activity of chitosan: Ultrastructural study. Biomaterials 1988, 9, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.; Tarsi, R.; Filippini, O.; Giovanetti, E.; Biagini, G.; Varaldo, P.E. Antimicrobial properties of N-carboxybutyl chitosan. Antimicrob. Agents Chemother. 1990, 34, 2019–2023. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.A.A.; Mattioli-Belmonte, M.; Tietz, C.; Biagini, R.; Ferioli, G.; Brunelli, M.A.; Fini, M.; Giardino, R.; Ilari, P.; Biagini, G. Stimulatory effect on bone formation exerted by a modified chitosan. Biomaterials 1994, 15, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, C.; Stanic, V.; Gobbi, L.; Tosi, G.; Muzzarelli, R.A.A. Spray-drying of solutions containing chitosan together with polyuronans and characterisation of the microspheres. Carbohydr. Polym. 2004, 57, 73–82. [Google Scholar] [CrossRef]
- Sabnis, S.; Rege, P.; Block, L.H. Use of chitosan in compressed tablets of diclofenac sodium: Inhibition of drug release in an acidic environment. Pharm. Dev. Technol. 1997, 2, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Remuñán-López, C.; Portero, A.; Vila-Jato, J.L.; Alonso, M.A.J. Design and evaluation of chitosan/ethylcellulose mucoadhesive bilayered devices for buccal drug delivery. J. Control. Release 1998, 55, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Giunchedi, P.; Juliano, C.; Gavini, E.; Cossu, M.; Sorrenti, M. Formulation and in vivo evaluation of chlorhexidine buccal tablets prepared using drug-loaded chitosan microspheres. Eur. J. Pharm. Biopharm. 2002, 53, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Athamneh, N.A.; Tashtoush, B.M.; Qandil, A.M.; Al-Tanni, B.M.; Obaidat, A.A.; Al-Jbour, N.D.; Qinna, N.A.; Al-Sou’od, K.; Al-Remawi, M.M.; Badwan, A.A. A new controlled-release liquid delivery system based on diclofenac potassium and low molecular weight chitosan complex solubilized in polysorbates. Drug Dev. Ind. Pharm. 2013, 39, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, A.; Al-Remawi, M.; Qinna, N.; Farouk, A.; Al-Sou’od, K.A.; Badwan, A.A. Chitosan-sodium lauryl sulfate nanoparticles as a carrier system for the in vivo delivery of oral insulin. AAPS PharmSciTech 2011, 12, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Assaf, S.M.; al-Jbour, N.D.; Eftaiha, A.A.F.; Elsayed, A.M.; al-Remawi, M.M.; Qinna, N.A.; Chowdhry, B.; Leharne, S.; Badwan, A.A. Factors involved in formulation of oily delivery system for proteins based on PEG-8 caprylic/capric glycerides and polyglyceryl-6 dioleate in a mixture of oleic acid with chitosan. J. Dispers. Sci. Technol. 2011, 32, 623–633. [Google Scholar] [CrossRef]
- Qinna, N.A.; Akayleh, F.T.; al Remawi, M.M.; Kamona, B.S.; Taha, H.; Badwan, A.A. Evaluation of a functional food preparation based on chitosan as a meal replacement diet. J. Funct. Foods 2013, 5, 1125–1134. [Google Scholar] [CrossRef]
- Sinha, V.R.; Singla, A.K.; Wadhawan, S.; Kaushik, R.; Kumria, R.; Bansal, K.; Dhawan, S. Chitosan microspheres as a potential carrier for drugs. Int. J. Pharm. 2004, 274, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; de la Luz Reus-Medina, M.; Yang, D. Preparation, characterization, and tabletting properties of a new cellulose-based pharmaceutical aid. Int. J. Pharm. 2002, 235, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Rege, P.R.; Shukla, D.J.; Block, L.H. Chitinosans as tableting excipients for modified release delivery systems. Int. J. Pharm. 1999, 181, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Aucamp, M.E.; Campus, N.W.U.P. Assessment of the Tableting Properties of Chitosan Through Wet Granulation and Direct Compression Formulations. M.Sc. thesis, North-West University, Potchefstroom Campus, South Africa, 2004. [Google Scholar]
- De Kock, J.M. Chitosan as a Multipurose Excipien in Directly Compressed Minitablets. Ph.D. thesis, North-West University, Potchefsroom Campus, South Africa, 2005. [Google Scholar]
- Sawayanagi, Y.; Nambu, N.; Nagai, T. Directly compressed tablets containing chitin or chitosan in addition to lactose or potato starch. Chem. Pharm. Bull. (Tokyo) 1982, 30, 2935–2940. [Google Scholar] [CrossRef]
- Mir, V.G.; Heinamaki, J.; Antikainen, O.; Sandler, N.; Revoredo, O.B.; Colarte, A.I.; Nieto, O.M.; Yliruusi, J. Application of crustacean chitin as a co-diluent in direct compression of tablets. AAPS PharmSciTech 2010, 11, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Charlton, B.; Newton, J.M. Theoretical estimation of punch velocities and displacements of single-punch and rotary tablet machines. J. Pharm. Pharmacol. 1984, 36, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Buys, G.M.; Campus, N.W.U.P. Formulation of a Chitosan Multi-Unit Dosage Form for Drug Delivery to the Colon. Ph.D. thesis, North-West University, Potchefstroom Campus, South Africa, 2006. [Google Scholar]
- Krasilnikova, O.K.; Solovtsova, O.V.; Khozina, E.V.; Grankina, T.Y. Porous Structure and Adsorption Behaviours of Chitosan; Nova Science Publisher, Inc.: Hauppauge, NY, USA, 2011. [Google Scholar]
- Rashid, I.; Al-Remawi, M.; Eftaiha, A.A.; Badwan, A. Chitin–silicon dioxide coprecipitate as a novel superdisintegrant. J. Pharm. Sci. 2008, 97, 4955–4969. [Google Scholar] [CrossRef] [PubMed]
- Rashid, I.; Daraghmeh, N.; Al-Remawi, M.; Leharne, S.A.; Chowdhry, B.Z.; Badwan, A. Characterization of chitin-metal silicates as binding superdisintegrants. J. Pharm. Sci. 2009, 98, 4887–4901. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Daraghmeh, N.; Trujillo, D. Effect of the alkaline treatment conditions on the tableting performance of chitin obtained from shrimp. Afr. J. Pharm. Pharmacol. 2014, 8, 211–219. [Google Scholar]
- Sonnekus, J. Characterization of the flow and compression properties of chitosan. M.Sc.thesis, North-West University, Potchefstroom Campus, South Africa, 2008. [Google Scholar]
- De Boer, A.H.; Vromans, H.; Leur, C.F.; Bolhuis, G.K.; Kussendrager, K.D.; Bosch, H. Studies on tableting properties of lactose. Pharm. Weekbl. Sci. Ed. 1986, 8, 145–150. [Google Scholar]
- Gustafsson, C. Solid State Characterisation and Compaction Behaviour of Pharmaceutical Materials; Uppsala University/Acta Universitatis Uppsaliensis: Uppsala, Sweden, 2000. [Google Scholar]
- Koo, O.M.; Heng, P.W. The influence of microcrystalline cellulose grade on shape and shape distributions of pellets produced by extrusion-spheronization. Chem. Pharm. Bull. (Tokyo) 2001, 49, 1383–1387. [Google Scholar] [CrossRef]
- Sujka, M.; Jamroz, J. α-Amylolysis of native potato and corn starches—SEM, AFM, nitrogen and iodine sorption investigations. LWT Food Sci. Technol. 2009, 42, 1219–1224. [Google Scholar] [CrossRef]
- Luna-Zaragoza, D.; Romero-Guzmán, E.; Reyes-Gutiérrez, L. Surface and physicochemical characterization of phosphates vivianite, Fe2(PO4)3 and hydroxyapatite, Ca5(PO4)3OH. J. Min. Mater. Charact. Eng. 2009, 8, 591–609. [Google Scholar]
- Quingnard, F.; di Renzo, F.; Gubal, E. From natural polysacharide to materials from catalysis, adsorption, and remediation. In Carbohydrates in Sustainable Development I; Springer-Verlag Berlin Heidelberg: Heidelberg, Germany, 2010; pp. 165–197. [Google Scholar]
- Lindberg, N.O.; Palsson, M.; Pihl, A.C.; Freeman, R.; Freeman, T.; Zetzener, H.; Enstad, G. Flowability measurements of pharmaceutical powder mixtures with poor flow using five different techniques. Drug. Dev. Ind. Pharm. 2004, 30, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.L. Evaluating flow properties of solids. Chem. Eng. 1965, 72, 163–168. [Google Scholar]
- Iida, K.; Aoki, K.; Danjo, K.; Otsuka, A.; Chen, C.Y.; Horisawa, E. A comparative evaluation of the mechanical properties of various celluloses. Chem. Parm. Bull. 1997, 45, 217–220. [Google Scholar] [CrossRef]
- Zainuddin, I.M.; Yasuda, M.; Horio, T.; Matsusaka, S. Experimental study on powder flowability using vibration shear tube method. Part. Part. Syst. Charact. 2012, 29, 8–15. [Google Scholar] [CrossRef]
- Alonso, M.J.; Sanchez, A. The potential of chitosan in ocular drug delivery. J. Pharm. Pharmacol. 2003, 55, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Velasco, M.V.; Munoz-Ruiz, A.; Monedero, M.C.; Jiménez-Castellanos, M.R. Study of flowability of powders. effect of the addition of lubricants. Drug Dev. Ind. Pharm. 1995, 21, 2385–2391. [Google Scholar] [CrossRef]
- Buys, G.M.; du Plessis, L.H.; Marais, A.F.; Kotze, A.F.; Hamman, J.H. Direct compression of chitosan: Process and formulation factors to improve powder flow and tablet performance. Curr. Drug. Deliv. 2013, 10, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Karehill, P.G.; Glazer, M.; Nyström, C. Studies on direct compression of tablets. XXIII. The importance of surface roughness for the compactability of some directly compressible materials with different bonding and volume reduction properties. Int. J. Pharm. 1990, 64, 35–43. [Google Scholar] [CrossRef]
- Mohammed, H.; Briscoe, B.J.; Pitt, K.G. The interrelationship between the compaction behaviour and the mechanical strength of pure pharmaceutical tablets. Chem. Eng. Sci. 2005, 60, 3941–3947. [Google Scholar] [CrossRef]
- Rojas, J.; Ciro, Y.; Correa, L. Functionality of chitin as a direct compression excipient: An acetaminophen comparative study. Carbohydr. Polym. 2014, 103, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Garr, J.S.M.; Rubinstein, M.H. The influence of moisture content on the consolidation and compaction properties of paracetamol. Int. J. Pharm. 1992, 81, 187–192. [Google Scholar] [CrossRef]
- Khan, F.; Pilpel, N. The effect of particle size and moisture on the tensile strength of microcrystalline cellulose powder. Powder Technol. 1986, 48, 145–150. [Google Scholar] [CrossRef]
- Malamataris, S.; Goidas, P.; Dimitriou, A. Moisture sorption and tensile strength of some tableted direct compression excipients. Int. J. Pharm. 1991, 68, 51–60. [Google Scholar] [CrossRef]
- Nokhodchi, A.; Rubinstein, M.H.; Larhrib, H.; Guyot, J.C. The effect of moisture on the properties of ibuprofen tablets. Int. J. Pharm. 1995, 118, 191–197. [Google Scholar] [CrossRef]
- Shukla, A.J.; Price, J.C. Effect of moisture content on compression properties of two dextrose-based directly compressible diluents. Pharm. Res. 1991, 8, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Ahlneck, C.; Alderborn, G. Moisture adsorption and tabletting. I. Effect on volume reduction properties and tablet strength for some crystalline materials. Int. J. Pharm. 1989, 54, 131–141. [Google Scholar] [CrossRef]
- Agrawal, A.M.; Manek, R.V.; Kolling, W.M.; Neau, S.H. Water distribution studies within microcrystalline cellulose and chitosan using differential scanning calorimetry and dynamic vapor sorption analysis. J. Pharm. Sci. 2004, 93, 1766–1779. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Hatakeyama, T. Interaction between water and hydrophilic polymers. Thermochimica Acta 1998, 308, 3–22. [Google Scholar] [CrossRef]
- García Mir, V.; Heinämäki, J.; Antikainen, O.; Iraizoz Colarte, A.; Airaksinen, S.; Karjalainen, M.; Bilbao Revoredo, O.; Nieto, O.M.; Yliruusi, J. Effects of moisture on tablet compression of chitin. Carbohydr. Polym. 2011, 86, 477–483. [Google Scholar] [CrossRef]
- Ferrari, F.; Bertoni, M.; Bonferoni, M.C.; Rossi, S.; Caramella, C.; Nyström, C. Investigation on bonding and disintegration properties of pharmaceutical materials. Int. J. Pharm. 1996, 136, 71–79. [Google Scholar] [CrossRef]
- Kumar, G.; Ravi, M. Trends in controlled drug release formulations using chitin and chitosan. J. Sci. Ind. Res. 2000, 59, 201–213. [Google Scholar]
- Hwang, K.T.; Jung, S.T.; Lee, G.D.; Chinnan, M.S.; Park, Y.S.; Park, H.J. Controlling molecular weight and degree of deacetylation of chitosan by response surface methodology. J. Agric. Food Chem. 2002, 50, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Nunthanid, J.; Puttipipatkhachorn, S.; Yamamoto, K.; Peck, G.E. Physical properties and molecular behavior of chitosan films. Drug Dev. Ind. Pharm. 2001, 27, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Cervera, M.F.; Heinämäki, J.; Krogars, K.; Jörgensen, A.C.; Karjalainen, M.; Colarte, A.I.; Yliruusi, J. Solid-state and mechanical properties of aqueous chitosan-amylose starch films plasticized with polyols. AAPS PharmSciTech 2004, 5, 109–114. [Google Scholar] [CrossRef]
- Rashid, I.; Daraghmeh, N.; Al-Remawi, M.; Leharne, S.A.; Chowdhry, B.Z.; Badwan, A. Characterization of the impact of magnesium stearate lubrication on the tableting properties of chitin-Mg silicate as a superdisintegrating binder when compared to Avicel® 200. Powder Technol. 2010, 203, 609–619. [Google Scholar] [CrossRef]
- Nyström, C.; Alderborn, G.; Duberg, M.; Karehill, P.G. Bonding surface area and bonding mechanism—Two important factors for the understanding of powder compactibility. Drug Dev. Ind. Pharm. 1993, 19, 2143–2196. [Google Scholar] [CrossRef]
- Picker-Freyer, K.M.; Brink, D. Evaluation of powder and tableting properties of chitosan. AAPS PharmSciTech 2006, 7, E152–E161. [Google Scholar] [CrossRef]
- Perioli, L.; Ambrogi, V.; Pagano, C.; Scuota, S.; Rossi, C. FG90 chitosan as a new polymer for metronidazole mucoadhesive tablets for vaginal administration. Int. J. Pharm. 2009, 377, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Heckel, R.W. Density-pressure relationships in powder compaction. Trans. Metall. Soc. 1961, 221, 671–675. [Google Scholar]
- Krycer, I.; Pope, D.G.; Hersey, J.A. An evaluation of the techniques employed to investigate powder compaction behaviour. Int. J. Pharm. 1982, 12, 113–134. [Google Scholar] [CrossRef]
- Shivanand, P.; Sprockel, O.L. Compaction behavior of cellulose polymers. Powder Technol. 1992, 69, 177–184. [Google Scholar] [CrossRef]
- Lin, C.W.; Cham, T.M. Compression behavior and tensile strength of heat-treated polyethylene glycols. Int. J. Pharm. 1995, 118, 169–179. [Google Scholar] [CrossRef]
- Nordstrom, J.; Klevan, I.; Alderborn, G. A particle rearrangement index based on the Kawakita powder compression equation. J. Pharm. Sci. 2009, 98, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Burt, H.M.; Miller, R.A. The Gurnham equation in characterizing the compressibility of pharmaceutical materials. Int. J. Pharm. 2006, 317, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Antikainen, O.; Yliruusi, J. Determining the compression behaviour of pharmaceutical powders from the force–distance compression profile. Int. J. Pharm. 2003, 252, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.; Mallick, S. Correlation between compaction and dissolution of metoprolol tartarate tablets prepared by direct compression using different polymers. Int. J. Pharm. Pharma. Sci. 2012, 4, 77–88. [Google Scholar]
- Tye, C.K.; Sun, C.C.; Amidon, G.E. Evaluation of the effects of tableting speed on the relationships between compaction pressure, tablet tensile strength, and tablet solid fraction. J. Pharm. Sci. 2005, 94, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Rowe, R.C. Brittle/ductile behaviour in pharmaceutical materials used in tabletting. Int. J. Pharm. 1987, 36, 205–209. [Google Scholar] [CrossRef]
- Akin-Ajani, O.D.; Itiola, O.A.; Odeku, O.A. Effects of plantain and corn starches on the mechanical and disintegration properties of paracetamol tablets. AAPS PharmSciTech 2005, 6, E458–E463. [Google Scholar] [CrossRef] [PubMed]
- Belda, P.M.; Mielck, J.B. The tabletting machine as an analytical instrument: consequences of uncertainties in punch force and punch separation data on some parameters describing the course of the tabletting process. Eur. J. Pharm. Biopharm. 1999, 48, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Davies, W.L.; Gloor, W.T. Batch production of pharmaceutical granulations in a fluidized bed I: Effects of process variables on physical properties of final granulation. J. Pharm. Sci. 1971, 60, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Rong Huei, C.; Hwa, H.D. Effect of molecular weight of chitosan with the same degree of deacetylation on the thermal, mechanical, and permeability properties of the prepared membrane. Carbohydr. Polym. 1996, 29, 353–358. [Google Scholar] [CrossRef]
- Knapczyk, J. Excipient ability of chitosan for direct tableting. Int. J. Pharm. 1993, 89, 1–7. [Google Scholar] [CrossRef]
- Seetharaman, S.; Balya, H.; Abdul Ahad, H. Formulation and evaluation of sustained release matrix tablets of ciprofloxacin HCL using gum kondagogu and chitosan as matrix forming polymers. Int. J. Pharm. Sci. Rev. Res. 2014, 24, 115–119. [Google Scholar]
- Al-Akayleh, F.; Al Remawi, M.; Rashid, I.; Badwan, A. Formulation and in vitro assessment of sustained release terbutaline sulfate tablet made from binary hydrophilic polymer mixtures. Pharm. Dev. Technol. 2013, 18, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Eftaiha, A.A.F.; Qinna, N.; Rashid, I.S.; Al Remawi, M.M.; Al Shami, M.R.; Arafat, T.A.; Badwan, A.A. Bioadhesive controlled metronidazole release matrix based on chitosan and xanthan gum. Mar. Drugs 2010, 8, 1716–1730. [Google Scholar] [CrossRef] [PubMed]
- Eissens, A.C.; Bolhuis, G.K.; Hinrichs, W.L.J.; Frijlink, H.W. Inulin as filler-binder for tablets prepared by direct compaction. Eur. J. Pharm. Sci. 2002, 15, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Eftaiha, A.A.F.; El-Barghouthi, M.I.; Rashid, I.S.; Al-Remawi, M.M.; Saleh, A.I.; Badwan, A.A. Compressibility and compactibility studies of chitosan, xanthan gum, and their mixtures. J. Mater. Sci. 2009, 44, 1054–1062. [Google Scholar] [CrossRef]
- Broadhead, J.; Edmond Rouan, S.K.; Rhodes, C.T. The spray drying of pharmaceuticals. Drug Dev. Ind. Pharm. 1992, 18, 1169–1206. [Google Scholar] [CrossRef]
- Rege, P.R.; Garmise, R.J.; Block, L.H. Spray-dried chitinosans: Part II: In vitro drug release from tablets made from spray-dried chitinosans. Int. J. Pharm. 2003, 252, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Asada, M.; Takahashi, H.; Okamoto, H.; Tanino, H.; Danjo, K. Theophylline particle design using chitosan by the spray drying. Int. J. Pharm. 2004, 270, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hiorth, M.; Tho, I.; Sande, S.A. The formation and permeability of drugs across free pectin and chitosan films prepared by a spraying method. Eur. J. Pharm. Biopharm. 2003, 56, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Steckel, H.; Mindermann-Nogly, F. Production of chitosan pellets by extrusion/spheronization. Eur. J. Pharm. Biopharm. 2004, 57, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hugerth, A.; Caram-Lelham, N.; Sundelöf, L.O. The effect of charge density and conformation on the polyelectrolyte complex formation between carrageenan and chitosan. Carbohydr. Polym. 1997, 34, 149–156. [Google Scholar] [CrossRef]
- Tingstad, J.E. The theory and practice of industrial pharmacy. Second Ed. L. Lachman, H.A. Lieberman, and J.L. Kanig. Lea & Febiger, 600 Washington Square, Philadelphia. 1976; 65. [Google Scholar] [CrossRef]
- Rege, P.R.; Garmise, R.J.; Block, L.H. Spray-dried chitinosans. Part I: Preparation and characterization. Int. J. Pharm. 2003, 252, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bhise, K.S.; Dhumal, R.S.; Paradkar, A.R.; Kadam, S.S. Effect of drying methods on swelling, erosion and drug release from chitosan-naproxen sodium complexes. AAPS PharmSciTech 2008, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J. Effect of Polymorphism on the Particle and Compaction Properties of Microcrystalline Cellulose. In Cellulose-Medical, Pharmaceutical and Electronic Applications; van de Ven, T., Godbout, L., Eds.; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Kokil, S.; Patil, P.; Mahadik, K.; Paradkar, A. Studies on spray-dried mixtures of chitosan and hydrolyzed gelatin as tablet binder: A technical note. AAPS PharmSciTech 2005, 6, E437–E443. [Google Scholar] [CrossRef] [PubMed]
- El-Barghouthi, M.; Eftaiha, A.; Rashid, I.; al-Remawi, M.; Badwan, A. A novel superdisintegrating agent made from physically modified chitosan with silicon dioxide. Drug Dev. Ind. Pharm. 2008, 34, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Gana, F.Z.; Rashid, I.; Badwan, A.; Alkhamis, K.A. Determination of solid-state acidity of chitin-metal silicates and their effect on the degradation of cephalosporin antibiotics. J. Pharm. Sci. 2012, 101, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Rashid I., A.T.; Alkhamis, K.A.; Hassan, H.A.; Altalafha, T.H.; Badwan, A.A. Characterization of the Compression Properties of Compacted Chitosan as a Function of Molecular Weight; 5th Granulation Workshop: Sheffield, UK, 2013. [Google Scholar]
- Paronen, P.; Ilkka, J. Porosity-pressure functions. In Pharmaceutical Powder Compaction Technology. Drugs and the Pharmaceutical Sciences Series; Alderborn, G., Nystrom, C., Eds.; Marcel Dekker: New York, NY, USA, 1996; pp. 55–75. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badwan, A.A.; Rashid, I.; Omari, M.M.H.A.; Darras, F.H. Chitin and Chitosan as Direct Compression Excipients in Pharmaceutical Applications. Mar. Drugs 2015, 13, 1519-1547. https://doi.org/10.3390/md13031519
Badwan AA, Rashid I, Omari MMHA, Darras FH. Chitin and Chitosan as Direct Compression Excipients in Pharmaceutical Applications. Marine Drugs. 2015; 13(3):1519-1547. https://doi.org/10.3390/md13031519
Chicago/Turabian StyleBadwan, Adnan A., Iyad Rashid, Mahmoud M.H. Al Omari, and Fouad H. Darras. 2015. "Chitin and Chitosan as Direct Compression Excipients in Pharmaceutical Applications" Marine Drugs 13, no. 3: 1519-1547. https://doi.org/10.3390/md13031519