
Avarol Induces Apoptosis in Pancreatic Ductal Adenocarcinoma Cells by Activating PERK–eIF2α–CHOP Signaling

Abstract
:
1. Introduction
2. Results
2.1. Avarol Selectively Induces Apoptosis in Pancreatic Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 values (μM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MEF | IMR90 | HFL1 | HEK293 | A549 | MCF7 | U2OS | HCT116 | AGS | KLM1 | Panc-1 | PK1 | |
| Avarol | >100 | >100 | 78 ± 12 | 92 ± 9 | 82 ± 8 | 70 ± 12 | 42 ± 7 | 29 ± 5 | 19 ± 4 | 37 ± 9 | 20 ± 3 | 23 ± 2 |
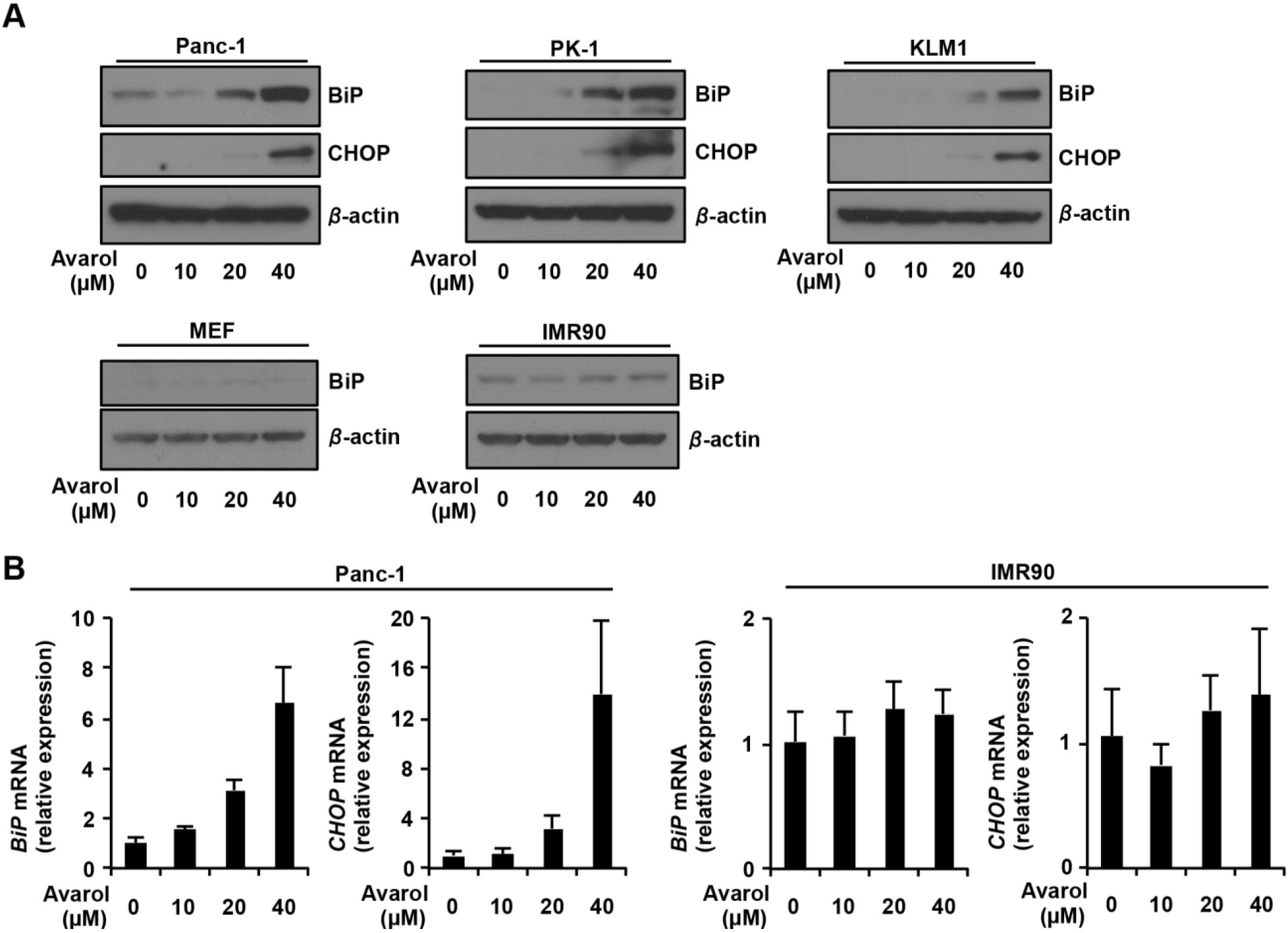
2.2. Avarol Induces ER Stress Responses and Upregulates BiP and CHOP Expression
2.3. Avarol Activates PERK–eIF2α Signaling Pathway


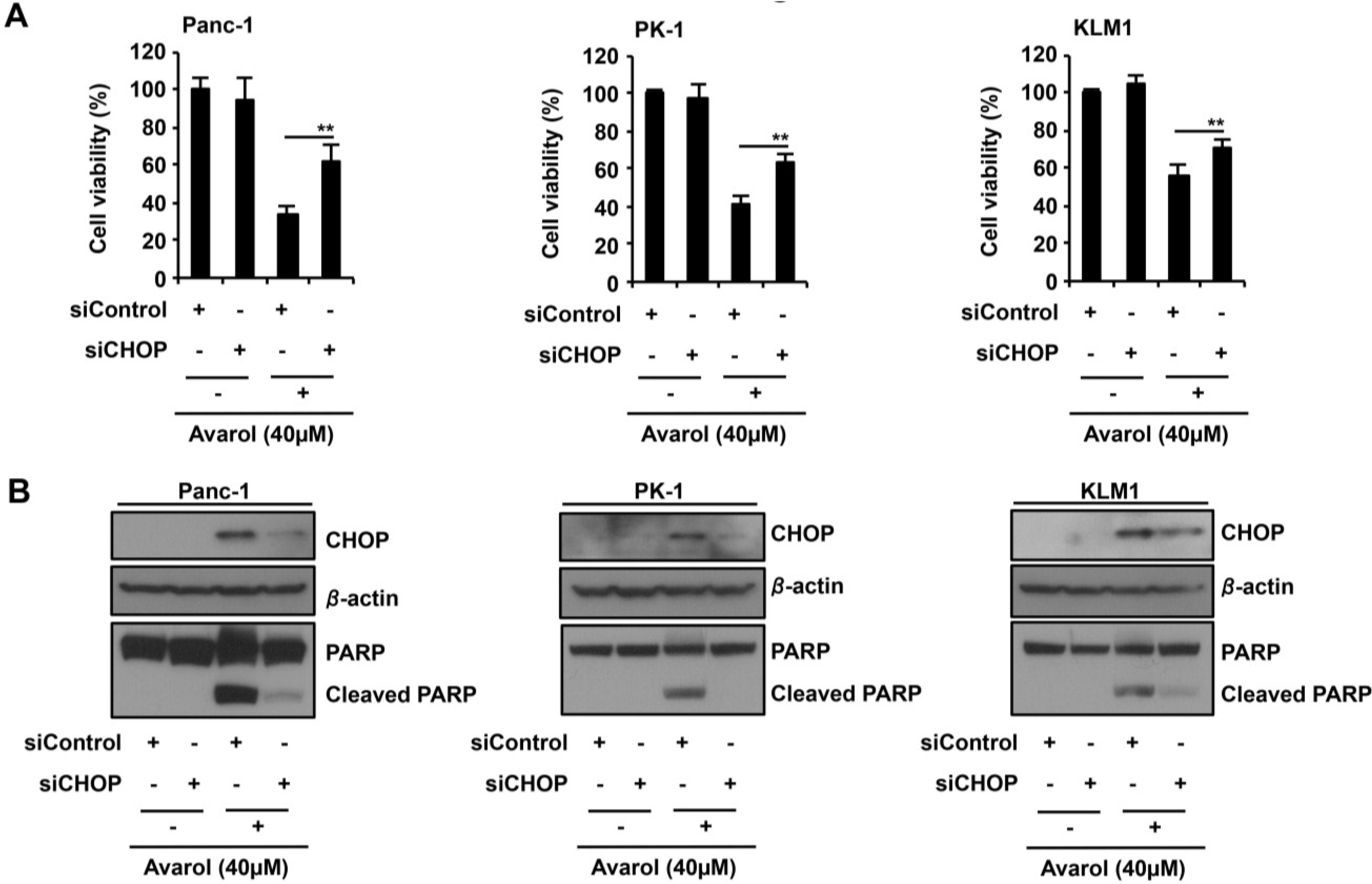
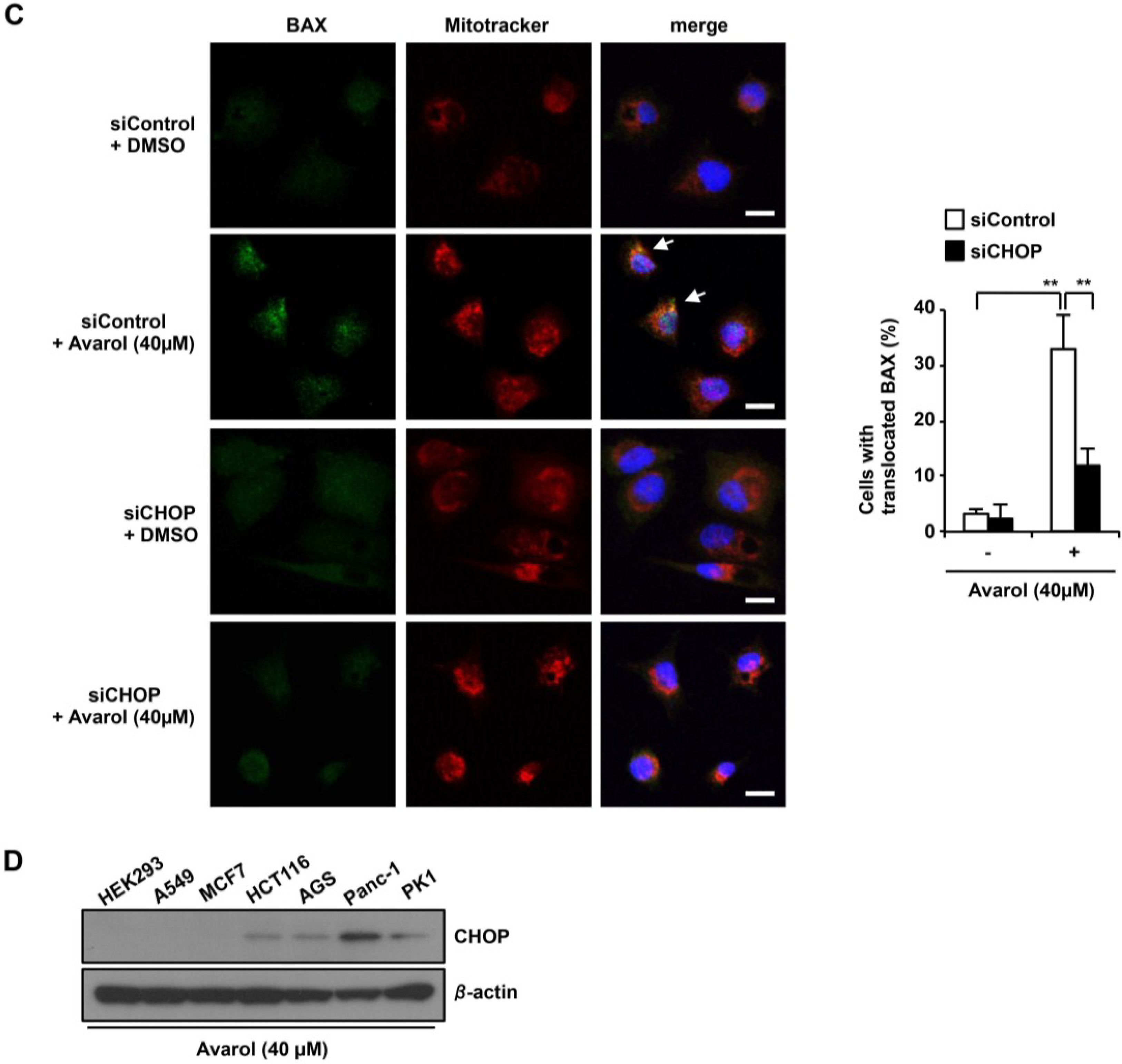
2.4. CHOP Is the Key Factor in Avarol-Induced Apoptosis


3. Discussion

4. Materials and Methods
4.1. Cell Lines
4.2. Immunoblotting Analysis
4.3. Cell Viability Assays
4.4. Real-Time Quantitative PCR and RT-PCR
4.5. Luciferase Assay
4.6. siRNA Experiments
4.7. Immunostaining
4.8. Statistical Analysis
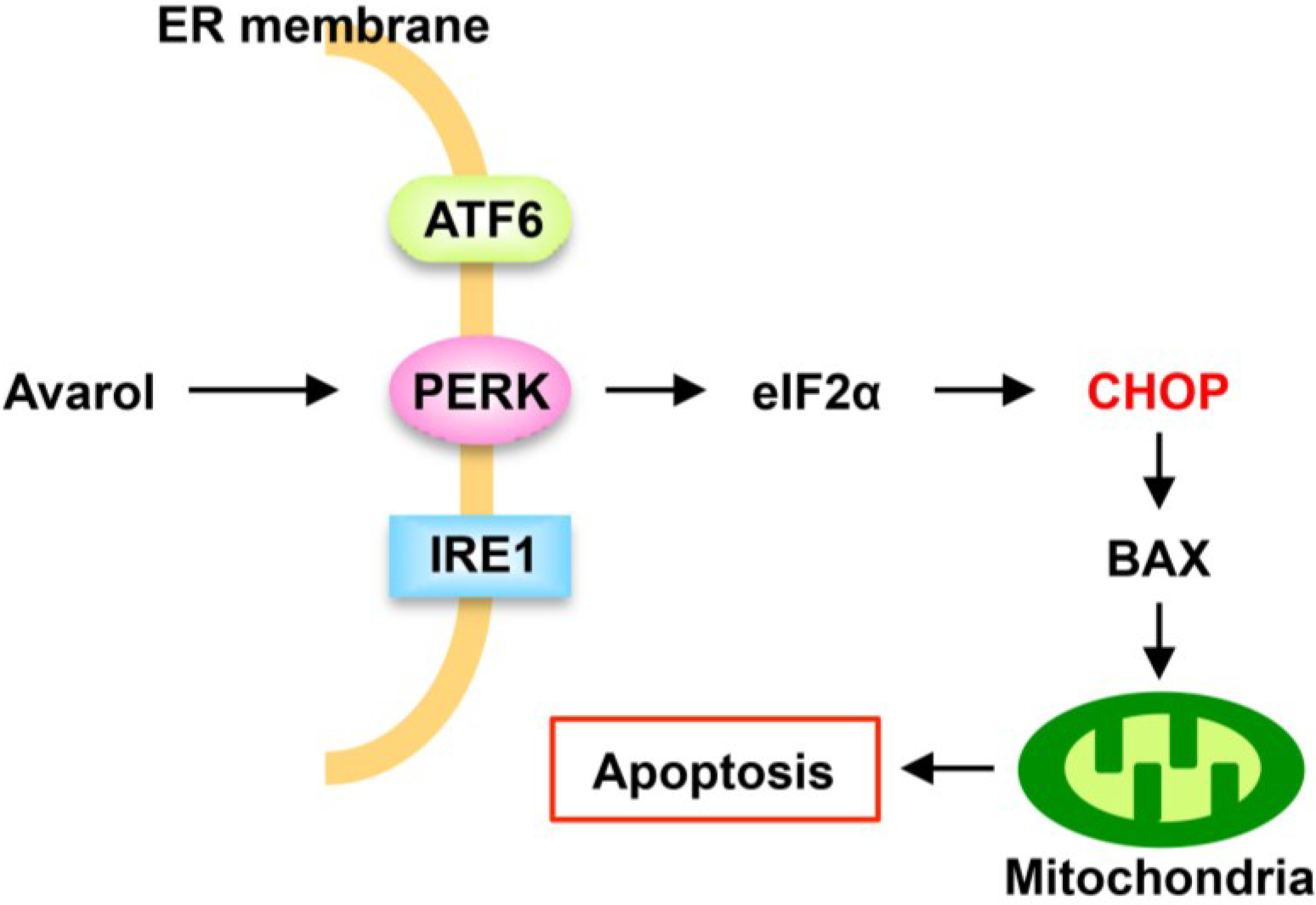
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Abbreviations
| ATF6 | activating transcription factor 6 |
| BiP | binding immunoglobulin protein |
| ER | endoplasmic reticulum |
| ERSE | ER stress response element |
| CHOP | C/EBP homologous transcription factor |
| IRE1 | protein-kinase and site-specific endoribonuclease |
| PERK | protein kinase R-like ER kinase/pancreatic eIF2 kinase |
| PDAC | pancreatic ductal adenocarcinoma |
| UPR | unfolded protein response |
Conflicts of Interest
References
- Cariello, L.; Zanetti, L.; Cuomo, V.; Vanzanella, F. Antimicrobial activity of avarol, a sesquiterpenoid hydroquinone from the marine sponge, Dysidea avara. Comp. Biochem. Physiol. B Comp. Biochem. 1982, 71, 281–283. [Google Scholar] [CrossRef]
- Sarin, P.S.; Sun, D.; Thornton, A.; Muller, W.E. Inhibition of replication of the etiologic agent of acquired immune deficiency syndrome (human T-lymphotropic retrovirus/lymphadenopathy-associated virus) by avarol and avarone. J. Natl. Cancer Inst. 1987, 78, 663–666. [Google Scholar] [PubMed]
- Amigo, M.; Paya, M.; Braza-Boils, A.; de Rosa, S.; Terencio, M.C. Avarol inhibits TNF-alpha generation and NF-kappaB activation in human cells and in animal models. Life Sci. 2008, 82, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.E.; Sladic, D.; Zahn, R.K.; Bassler, K.H.; Dogovic, N.; Gerner, H.; Gasic, M.J.; Schroder, H.C. Avarol-induced DNA strand breakage in vitro and in Friend erythroleukemia cells. Cancer Res. 1987, 47, 6565–6571. [Google Scholar] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Lennon, A.M.; Wolfgang, C.L.; Canto, M.I.; Klein, A.P.; Herman, J.M.; Goggins, M.; Fishman, E.K.; Kamel, I.; Weiss, M.J.; Diaz, L.A.; et al. The early detection of pancreatic cancer: What will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res. 2014, 74, 3381–3389. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Trepel, J.; Neckers, L. Ras, ROS and proteotoxic stress: A delicate balance. Cancer Cell 2011, 20, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Oyadomari, S.; Mori, K.; Mori, M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J. Biol. Chem. 2002, 277, 12343–12350. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Pae, H.O.; Zheng, M.; Park, R.; Kim, Y.M.; Chung, H.T. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef]
- Lee do, Y.; Lee, K.S.; Lee, H.J.; Kim do, H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K., Jr.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Hoshino, T.; Tanaka, K.; Tsutsumi, S.; Ishihara, T.; Mima, S.; Suzuki, K.; Ogawa, S.; Mizushima, T. Up-regulation of 150-kDa oxygen-regulated protein by celecoxib in human gastric carcinoma cells. Mol. Pharmacol. 2007, 71, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Tian, F.; Chu, K.; Hwang, S.Y.; Yoon, K.W.; Byun, S.; Hiraki, M.; Mandinova, A.; Lee, S.W. CDIP1-BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep. 2013, 5, 331–339. [Google Scholar] [CrossRef]
- Namba, T.; Hoshino, T.; Suemasu, S.; Takarada-Iemata, M.; Hori, O.; Nakagata, N.; Yanaka, A.; Mizushima, T. Suppression of expression of endoplasmic reticulum chaperones by Helicobacter pylori and its role in exacerbation of non-steroidal anti-inflammatory drug-induced gastric lesions. J. Biol. Chem. 2010, 285, 37302–37313. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namba, T.; Kodama, R. Avarol Induces Apoptosis in Pancreatic Ductal Adenocarcinoma Cells by Activating PERK–eIF2α–CHOP Signaling. Mar. Drugs 2015, 13, 2376-2389. https://doi.org/10.3390/md13042376
Namba T, Kodama R. Avarol Induces Apoptosis in Pancreatic Ductal Adenocarcinoma Cells by Activating PERK–eIF2α–CHOP Signaling. Marine Drugs. 2015; 13(4):2376-2389. https://doi.org/10.3390/md13042376
Chicago/Turabian StyleNamba, Takushi, and Rika Kodama. 2015. "Avarol Induces Apoptosis in Pancreatic Ductal Adenocarcinoma Cells by Activating PERK–eIF2α–CHOP Signaling" Marine Drugs 13, no. 4: 2376-2389. https://doi.org/10.3390/md13042376