
Identification and Bioactivity of Compounds from the Mangrove Endophytic Fungus Alternaria sp.

Abstract
:1. Introduction

2. Results and Discussion
2.1. Chemical Structure Elucidation

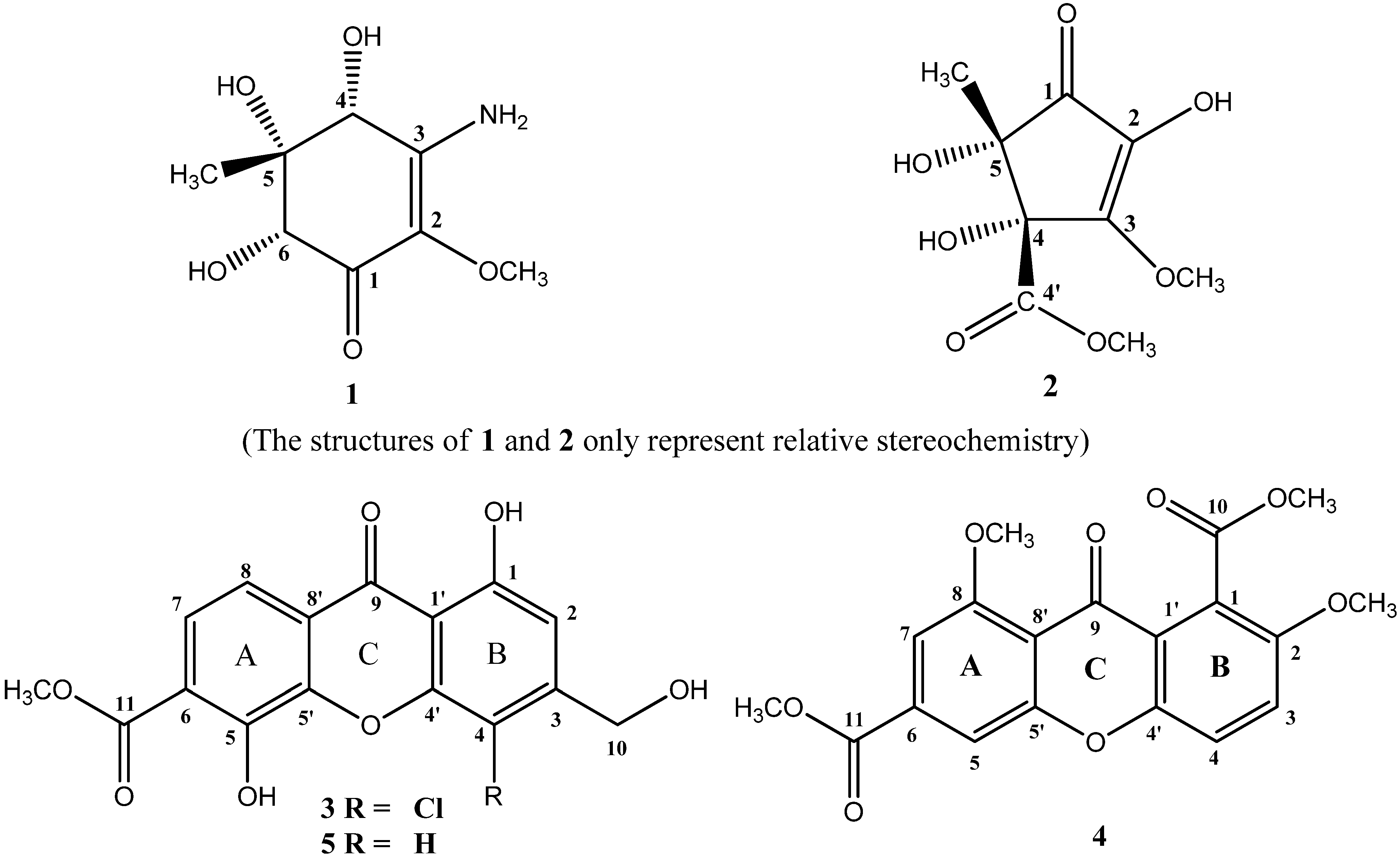{kind=link}
{kind=link}
| Position | 1 a | 2 a | ||
|---|---|---|---|---|
| δC, mult. | δH | δC, mult. | δH | |
| 1 | 185.62, C | - | 197.95, C | - |
| 2 | 126.93, C | - | 132.96, C | - |
| 3 | 156.36, C | - | 157.68, C | - |
| 4 | 71.55, CH | 4.22 (s) | 81.97, C | - |
| 5 | 77.97, C | - | 78.15, C | - |
| 6 | 76.99, CH | 3.72 (s) | - | - |
| 2-OCH3 | 58.13, CH3 | 3.46 (s) | - | - |
| 2-OH | - | - | - | 9.00 (s) |
| 3-NH2 | - | 6.30 (s), 6.83 (s) | - | - |
| 3-OCH3 | - | - | 58.55, CH3 | 3.99 (s) |
| 4-OH | - | 5.94 (s) | - | 6.10 (s) |
| 5-CH3 | 13.39, CH3 | 0.83 (s) | 22.16, CH3 | 1.16 (s) |
| 5-OH | - | 4.94 (s) | - | 5.63 (s) |
| 6-OH | - | 4.50 (s) | - | - |
| 4′ | - | - | 171.13, C | - |
| 4′-OCH3 | - | - | 52.06, CH3 | 3.59 (s) |
| Position | 3 a | 4 b | ||
|---|---|---|---|---|
| δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | |
| 1 | 159.55, C | - | 120.71, C | - |
| 2 | 108.59, CH | 6.98 (s) | 152.78, C | - |
| 3 | 150.57, C | - | 119.49, CH | 7.52 (d, 9.6) |
| 4 | 107.95, C | - | 119.07, CH | 7.38 (d, 8.4) |
| 5 | 149.15, C | - | 111.29, CH | 7.72 (d, 1.2) |
| 6 | 117.63, C | - | 135.66, C | - |
| 7 | 126.08, CH | 7.49 (d, 9.0) | 105.52, CH | 7.41 (d, 1.2) |
| 8 | 120.74, CH | 7.68 (d, 9.0) | 160.89, C | - |
| 9 | 180.73, C | - | 175.06, C | - |
| 10 | 61.03, CH2 | 4.65 (d, 5.4) | 167.74, C | - |
| 11 | 167.09, C | - | 165.52, C | - |
| 1′ | 107.11, C | - | 121.40, C | - |
| 4′ | 150.78, C | - | 149.18, C | - |
| 5′ | 151.61, C | - | 157.43, C | - |
| 8′ | 117.57, C | - | 114.18, C | - |
| 1-OH | - | 12.20 (s) | - | - |
| 2-OCH3 | - | - | 56.96, CH3 | 3.92 (s) |
| 5-OH | - | 10.59 (s) | - | - |
| 8-OCH3 | - | - | 56.79, CH3 | 4.06 (s) |
| 10-OH | - | 5.71 (s) | - | - |
| 10-OCH3 | - | - | 53.03, CH3 | 4.09 (s) |
| 11-OCH3 | 52.78, CH3 | - | 52.83, CH3 | 4.00 (s) |

2.2. Biological Activity
| Compounds | 1 | 2 | 3 | 4 | Ascorbic Acid b |
|---|---|---|---|---|---|
| EC50 | 8.19 ± 0.15 | 16.09 ± 0.01 | >500.00 | >500.00 | 17.14 ± 0.11 |
| Fusarium graminearum | Calletotrichum musae | Escherichia coil | Staphyloccocus aureus | |
|---|---|---|---|---|
| 1 | 738.92 | >1970.44 | 1970.44 | 1970.44 |
| 2 | 215.52 | 862.07 | 1724.14 | 1724.14 |
| 3 | 107.14 | 214.29 | NT | NT |
| 5 | 474.68 | 474.68 | >1265.82 | >1265.82 |
| Cefradine a | NT | NT | 143.10 | 17.89 |
| Kanamycin a | NT | NT | 1.54 | 1.54 |
| Triadimefon a | 510.64 | 340.43 | NT | NT |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material and Fermentation
3.3. Extraction and Isolation
3.4. The ABTS Radical Scavenging Activity Assay
3.5. Antimicrobial Activity Assay
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kjer, J.; Debbab, A.; Aly, A.H.; Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 2010, 5, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.K.; Guo, S.X. Fungal endophyte communities in four Rhizophoraceae mangrove species on the south coast of China. Ecol. Res. 2011, 26, 403–409. [Google Scholar] [CrossRef]
- Nowicki, M.; Nowakowska, M.; Niezgoda, A.; Kozik, E. Alternaria black spot of crucifers: Symptoms, importance of disease, and perspectives of resistance breeding. Veg. Crop. Res. Bull. 2012, 76, 5–19. [Google Scholar] [CrossRef]
- Xu, X.L.; Zhao, S.J.; Wei, J.L.; Fang, N.Q.; Yin, L.Y.; Sun, J.S. Porric acid D from marine-derived fungus Alternaria sp. isolated from Bohai Sea. Chem. Nat. Compd. 2012, 47, 893–895. [Google Scholar] [CrossRef]
- Shaaban, M.; Shaaban, K.A.; Abdel-Aziz, M.S. Seven naphtho-gamma-pyrones from the marine-derived fungus Alternaria alternata: Structure elucidation and biological properties. Org. Med. Chem. Lett. 2012, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Kjer, J.; Wray, V.; Edrada-Ebel, R.; Ebel, R.; Pretsch, A.; Lin, W.H.; Proksch, P. Xanalteric acids I and II and related phenolic compounds from an endophytic Alternaria sp. isolated from the mangrove plant Sonneratia alba. J. Nat. Prod. 2009, 72, 2053–2057. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Jin, H.; Song, B.; Zhu, X.; Zhao, H.X.; Cai, J.Y.; Lu, Y.J.; Chen, B.; Lin, Y.C. The cytotoxicity and anticancer mechanisms of alterporriol L, a marine bianthraquinone, against MCF-7 human breast cancer cells. Appl. Microbiol. Biotechnol. 2012, 93, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Pan, J.H.; Chen, B.; Yu, M.; Huang, H.B.; Zhu, X.; Lu, Y.J.; She, Z.G.; Lin, Y.C. Three bianthraquinone derivatives from the mangrove endophytic fungus Alternaria sp. ZJ9-6B from the South China Sea. Mar. Drugs 2011, 9, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Cox, D.G.; Ding, W.J.; Huang, G.H.; Lin, Y.C.; Li, C.Y. Three new resveratrol derivatives from the mangrove endophytic fungus Alternaria sp. Mar. Drugs 2014, 12, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.P.; Li, J.; Li, H.X.; Long, Y.H.; Lin, S.E.; Lu, Y.J.; He, L.; Lin, Y.C.; Liu, L.; She, Z.G. Alterporriol-type dimers from the mangrove endophytic fungus, Alternaria sp. (SK11), and their MptpB inhibitions. Mar. Drugs 2014, 12, 2953–2969. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.W.; Ouyang, M.A.; Shen, S.; Li, W. Bioactive metabolites from a marine-derived strain of the fungus. Nat. Prod. Res. 2012, 26, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, G.; Banumathi, B.; Suresh, G. Evaluation of the antifungal activity of natural xanthones from Garcinia mangostana and their synthetic derivatives. J. Nat. Prod. 1997, 60, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.; Ali, M.; Akhtar, N. New antifungal xanthones from the seeds of Rhus coriaria L. Z. Naturforsch. C 2011, 66, 17–23. [Google Scholar] [CrossRef] [PubMed]
- El-Shanawany, M.A.; Mohamed, G.A.; Nafady, A.M.; Ibrahim, S.R.M.; Radwan, M.M.; Ross, S.A. A new xanthone from the roots of Centaurium spicatum. Phytochem. Lett. 2011, 4, 126–128. [Google Scholar] [CrossRef]
- Shiono, Y.; Murayama, T.; Takahashi, K.; Okada, K.; Katohda, S.; Ikeda, M. Three oxygenated cyclohexenone derivatives produced by an endophytic fungus. Biosci. Biotechnol. Biochem. 2005, 69, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Lubken, T.; Schmidt, J.; Porzel, A.; Arnold, N.; Wessjohann, L. Hygrophorones A–G: Fungicidal cyclopentenones from Hygrophorus species (Basidiomycetes). Phytochemistry 2004, 65, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Zhao, M.Y.; Liu, F.; Zeng, S.T.; Hu, J. Antioxidants in volatile Maillard reaction products: Identification and interaction. LWT Food Sci. Technol. 2013, 53, 22–28. [Google Scholar] [CrossRef]
- Braca, A.; De Tommasi, N.; Di Bari, L.; Pizza, C.; Politi, M.; Morelli, I. Antioxidant principles from Bauhinia tarapotensis. J. Nat. Prod. 2001, 64, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shao, C.L.; Wang, K.L.; Xu, Y.; She, Z.G.; Wang, C.Y. Dihydroisocoumarin derivatives with antifouling activities from a gorgonian-derived Eurotium sp. fungus. Tetrahedron 2014, 70, 9132–9138. [Google Scholar] [CrossRef]
- Finefield, J.M.; Sherman, D.H.; Kreitman, M.; Williams, R.M. Enantiomeric natural products: Occurrence and biogenesis. Angew. Chem. Int. Ed. Engl. 2012, 51, 4802–4836. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.L.; Zhang, X.Y.; Zheng, Z.H.; Xu, X.Y.; Qi, S.H. Three new polyketides from marine-derived fungus Penicillium citrinum SCSGAF 0167. Nat. Prod. Res. 2014, 28, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Wang, K.; Wan, L.L.; Li, A.F.; Hu, Q.; Zhang, C.W. Production, characterization, and antioxidant activity of fucoxanthin from the marine diatom Odontella aurita. Mar. Drugs 2013, 11, 2667–2681. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.L.; Ma, Y.M. Isolation and anti-phytopathogenic activity of secondary metabolites from Alternaria sp. FL25, an endophytic fungus in Ficus carica. Chin. J. Appl. Environ. Biol. 2010, 16, 76–78. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Ding, W.; Wang, R.; Du, Y.; Liu, H.; Kong, X.; Li, C. Identification and Bioactivity of Compounds from the Mangrove Endophytic Fungus Alternaria sp. Mar. Drugs 2015, 13, 4492-4504. https://doi.org/10.3390/md13074492
Wang J, Ding W, Wang R, Du Y, Liu H, Kong X, Li C. Identification and Bioactivity of Compounds from the Mangrove Endophytic Fungus Alternaria sp. Marine Drugs. 2015; 13(7):4492-4504. https://doi.org/10.3390/md13074492
Chicago/Turabian StyleWang, Jinhua, Weijia Ding, Ruimin Wang, Yipeng Du, Huanliang Liu, Xuehua Kong, and Chunyuan Li. 2015. "Identification and Bioactivity of Compounds from the Mangrove Endophytic Fungus Alternaria sp." Marine Drugs 13, no. 7: 4492-4504. https://doi.org/10.3390/md13074492
APA StyleWang, J., Ding, W., Wang, R., Du, Y., Liu, H., Kong, X., & Li, C. (2015). Identification and Bioactivity of Compounds from the Mangrove Endophytic Fungus Alternaria sp. Marine Drugs, 13(7), 4492-4504. https://doi.org/10.3390/md13074492