
Protective Effect of Diphlorethohydroxycarmalol against Ultraviolet B Radiation-Induced DNA Damage by Inducing the Nucleotide Excision Repair System in HaCaT Human Keratinocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
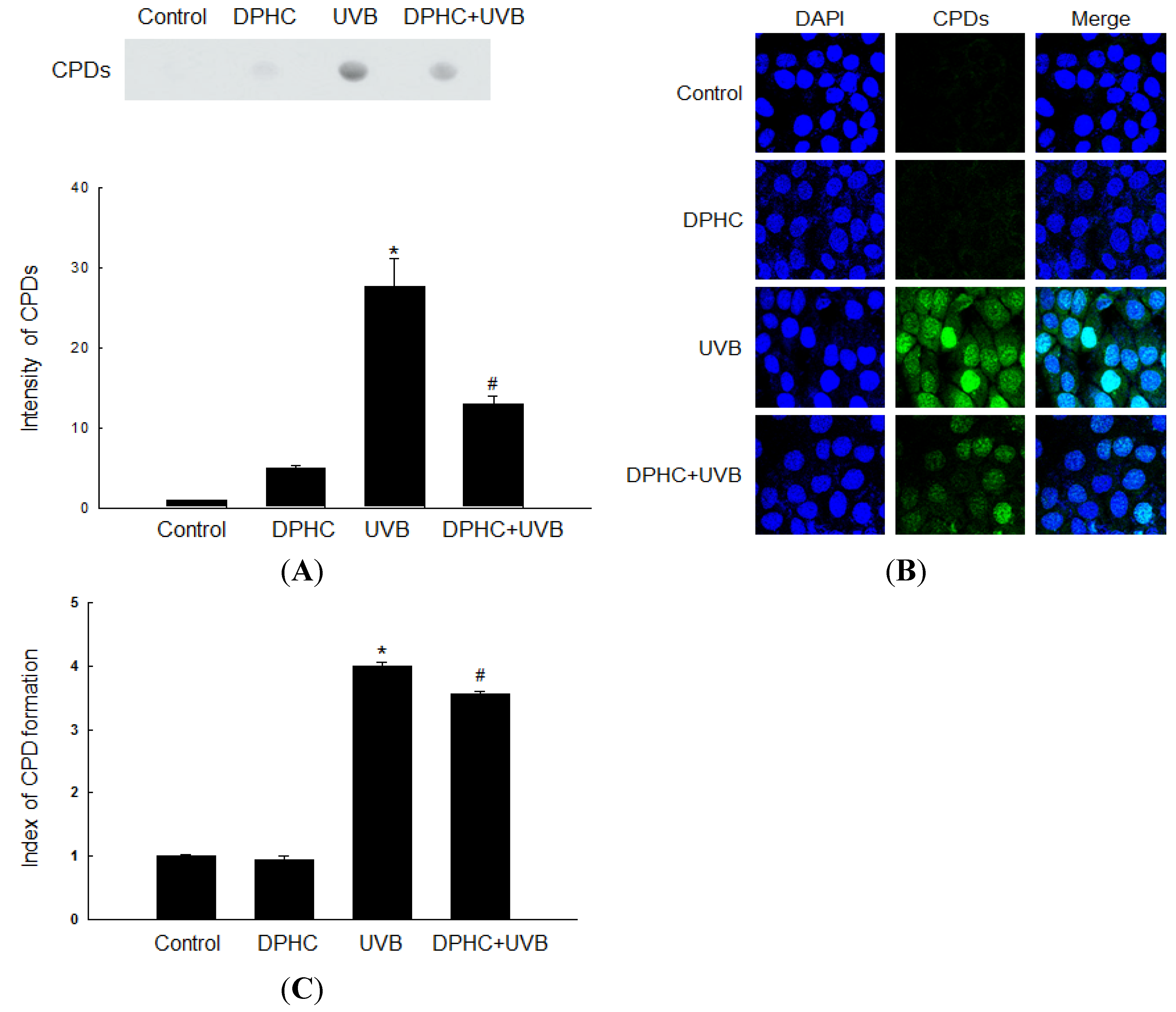
2.1. Effect of DPHC against UVB-Induced CPDs Formation

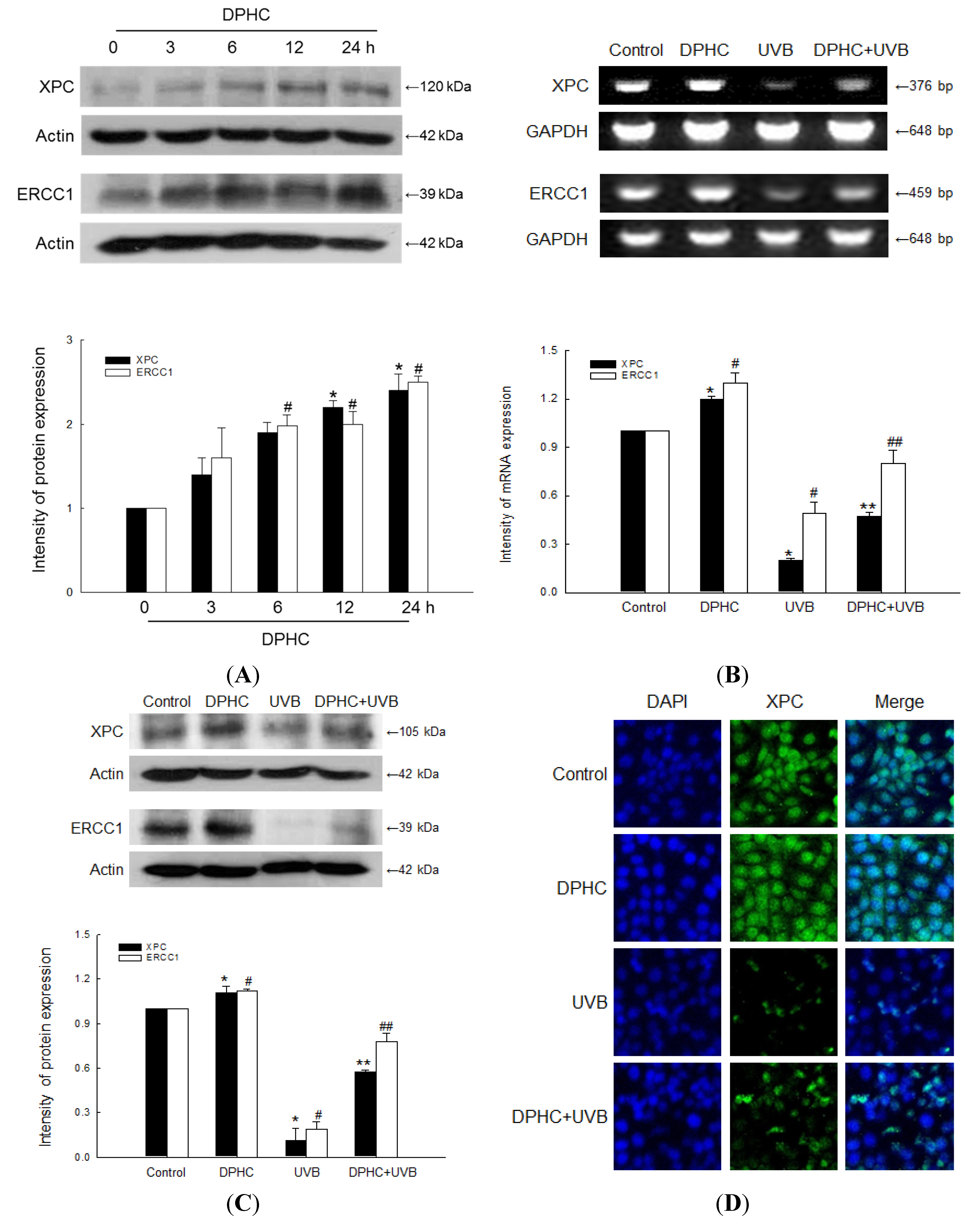
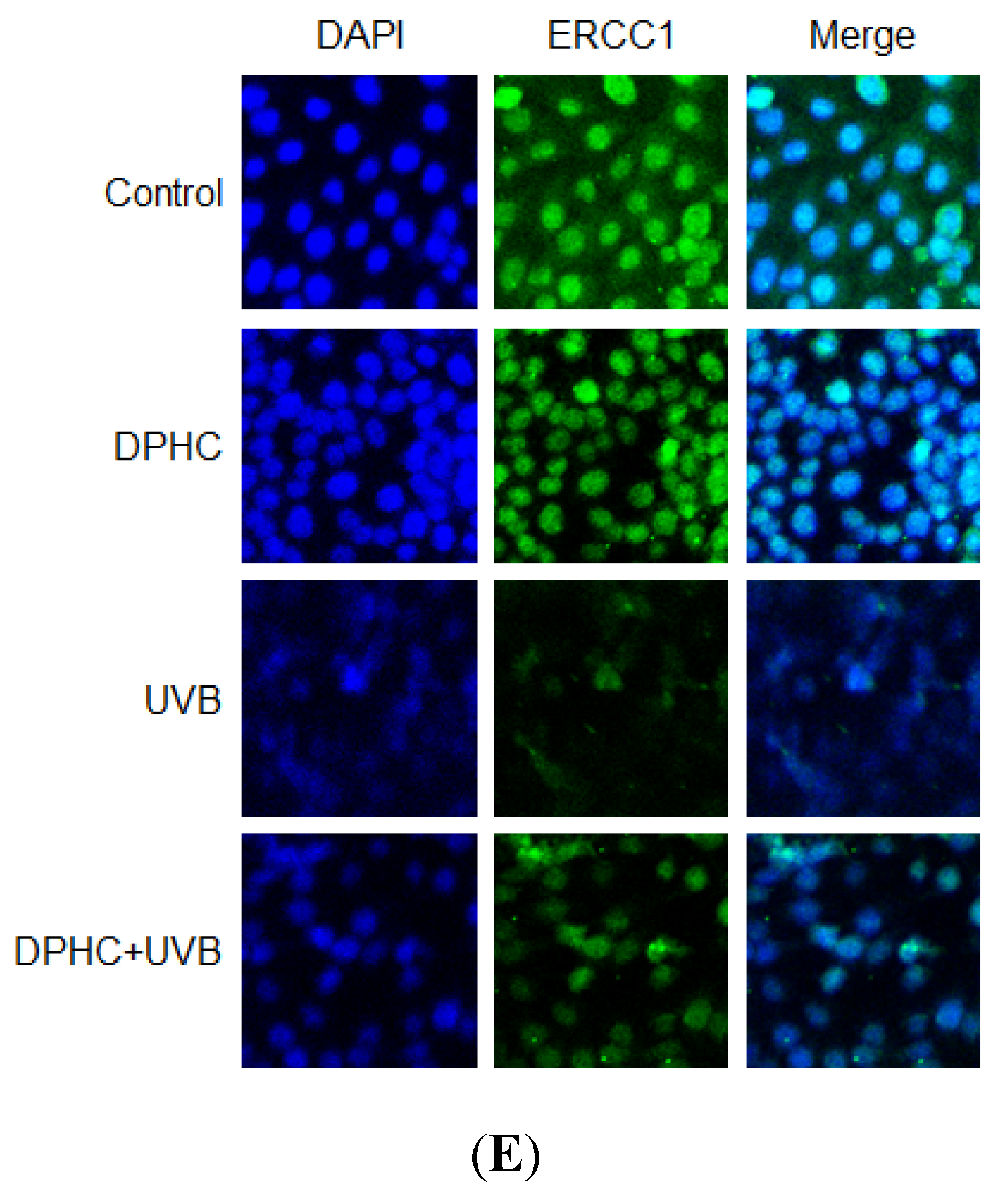
2.2. Effect of DPHC against UVB-Reduced NER System


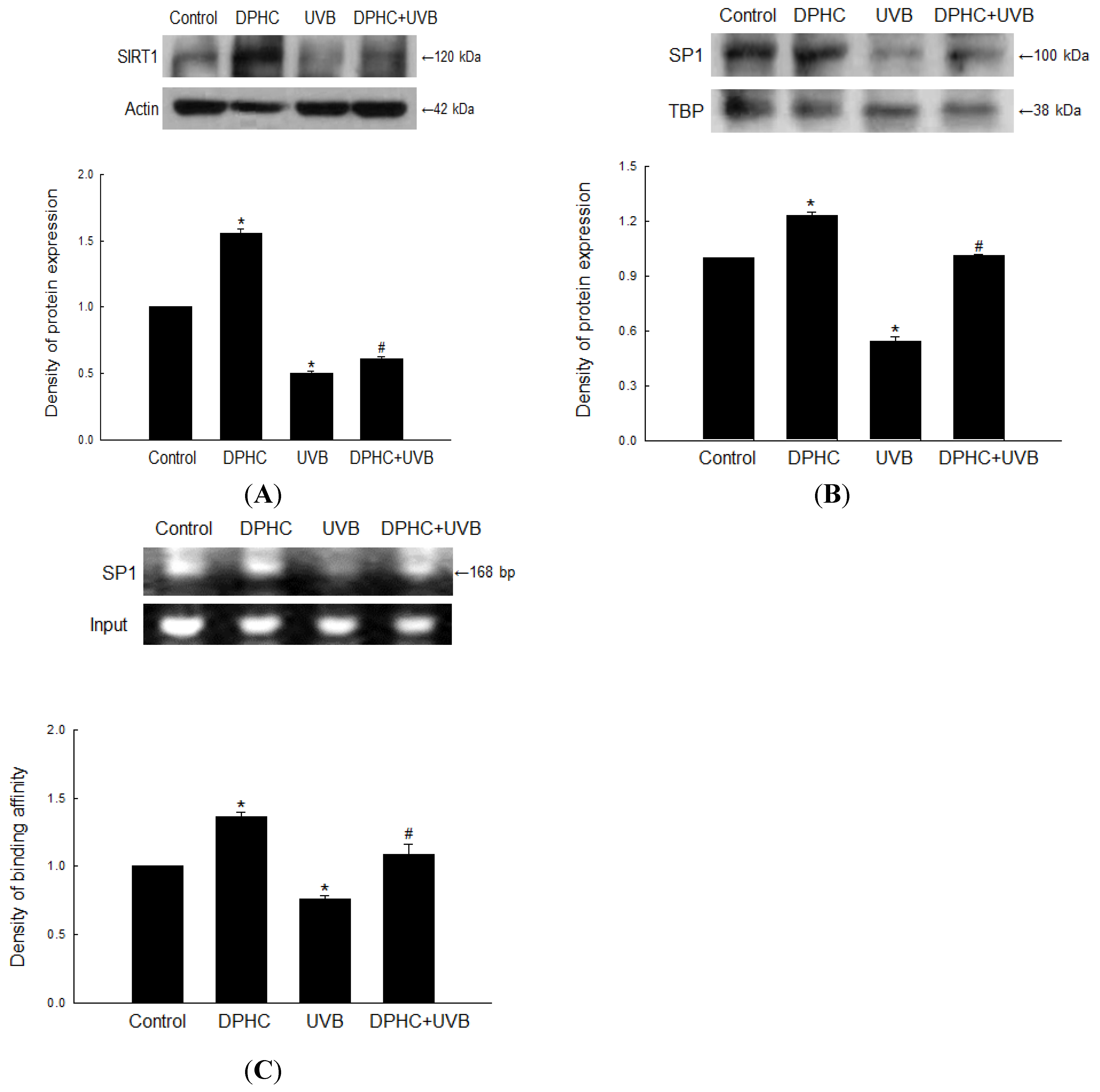
2.3. Effect of DPHC on Sirtuin 1 (SIRT1) and Specificity Protein 1 (SP1) Levels in UVB-Exposed Cells

3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Cell Culture
4.3. UVB Radiation
4.4. Dot Blot and ELISA Analysis
4.5. Immune-Cytochemical Analysis
4.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.7. Western Blot Analysis
4.8. Chromatin Immune-Precipitation (ChIP)
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bowden, G.T. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signaling. Nat. Rev. Cancer 2004, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.P.; Richa Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Budden, T.; Bowden, N.A. The role of altered nucleotide excision repair and UVB-induced DNA damage in melanomagenesis. Int. J. Mol. Sci. 2013, 14, 1132–1151. [Google Scholar] [CrossRef] [PubMed]
- Legerski, R.; Peterson, C. Expression cloning of a human DNA repair gene involved in xeroderma pigmentosum group C. Nature 1992, 359, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Shimizu, Y.; Iwai, S.; Hanaoka, F. A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair 2002, 1, 95–107. [Google Scholar] [CrossRef]
- Emmert, S.; Kobayashi, N.; Khan, S.G.; Kraemer, K.H. The xeroderma pigmentosum group C gene leads to selective repair of cyclobutane pyrimidine dimers rather than 6-4 photoproducts. Proc. Natl. Acad. Sci. USA 2000, 97, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C. Subpathways of nucleotide excision repair and their regulation. Oncogene 2002, 21, 8949–8956. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T. DNA repair in mammalian cells: So DNA repair really is that important? Cell. Mol. Life Sci. 2009, 66, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Kim, J.P.; Jung, W.K.; Lee, N.H.; Kang, H.S.; Jun, E.M.; Park, S.H.; Kang, S.M.; Lee, Y.J.; Park, P.J.; et al. Identification of chemical structure and free radical scavenging activity of diphlorethohydroxycarmalol isolated from a brown alga, Ishige okamurae. J. Microbiol. Biotechnol. 2008, 18, 676–681. [Google Scholar] [PubMed]
- Zou, Y.; Qian, Z.J.; Li, Y.; Kim, M.M.; Lee, S.H.; Kim, S.K. Antioxidant effects of phlorotannins isolated from Ishige okamurae in free radical mediated oxidative systems. J. Agric. Food Chem. 2008, 56, 7001–7009. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.J.; Kang, K.A.; Kim, K.C.; Chae, S.; Kim, G.O.; Shin, T.; Kim, H.S.; Hyun, J.W. Diphlorethohydroxycarmalol attenuated cell damage against UVB radiation via enhancing antioxidant effects and absorbing UVB ray in human HaCaT keratinocytes. Environ. Toxicol. Pharmacol. 2013, 36, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Yoon, K.D.; Kim, C.Y.; Kim, J.H.; Shin, C.G.; Kim, J. Inhibitory activity on HIV-1 reverse transcriptase and integrase of a carmalol derivative from a brown alga, Ishige okamurae. Phytother. Res. 2006, 20, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Hwang, J.Y.; Choi, J.I.; Han, J.S.; Kim, H.J.; Jeon, Y.J. Diphlorethohydroxycarmalol isolated from Ishige okamurae, a brown algae, a potent α-glucosidase and α-amylase inhibitor, alleviates postprandial hyperglycemia in diabetic mice. Eur. J. Pharmacol. 2009, 615, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Ko, S.C.; Kang, S.M.; Cha, S.H.; Lee, S.H.; Kang, D.H.; Jung, W.K.; Affan, A.; Oh, C.; Jeon, Y.J. Inhibitory effect of diphlorethohydroxycarmalol on melanogenesis and its protective effect against UV-B radiation-induced cell damage. Food Chem. Toxicol. 2010, 48, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.J.; Han, S.C.; Koh, Y.S.; Jeon, Y.J.; Yoo, E.S. Diphlorethohydroxycarmalol, isolated from Ishige okamurae, increases prostaglandin E2 through the expression of cyclooxygenase-1 and -2 in HaCaT human keratinocytes. Biomol. Ther. 2012, 20, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Moon, C.; Yang, W.; Ko, E.J.; Hyun, J.W.; Joo, H.G.; Jee, Y.; Lee, N.H.; Park, J.W.; Ko, R.K.; et al. Diphlorethohydroxycarmalol, isolated from the brown algae Ishige okamurae, protects against radiation-induced cell damage in mice. Food Chem. Toxicol. 2011, 49, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Shea, C.R.; Guo, X.; Li, X.; Soltani, K.; Han, W.; He, Y.Y. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc. Natl. Acad. Sci. USA 2010, 107, 22623–22628. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, H.R.; Mahfouf, W.; Ali, N.; Chemin, C.; Ged, C.; Kim, A.L.; de Verneuil, H.; Taïeb, A.; Bickers, D.R.; Mazurier, F. Hypoxia-inducible factor-1α regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res. 2010, 38, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 2001, 1, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Fleck, O.; Nielsen, O. DNA repair. J. Cell Sci. 2004, 117, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.E.; Praetorius-Ibba, M.; Zhu, Q.; El-Mahdy, M.A.; Wani, G.; Zhao, Q.; Qin, S.; Patnaik, S.; Wani, A.A. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res. 2007, 35, 5338–5350. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Ständer, S.; Berneburg, M.; Böhm, M.; Kulms, D.; van Steeg, H.; Grosse-Heitmeyer, K.; Krutmann, J.; Schwarz, T. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat. Cell Biol. 2002, 4, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Doig, J.; Anderson, C.; Lawrence, N.J.; Selfridge, J.; Brownstein, D.G.; Melton, D.W. Mice with skin-specific DNA repair gene (Ercc1) inactivation are hypersensitive to ultraviolet irradiation-induced skin cancer and show more rapid actinic progression. Oncogene 2006, 25, 6229–6238. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Soltani, K.; Shea, C.R.; Li, X.; He, Y.Y. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene 2015, 34, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Wang, W.J.; Song, X.; Chu, W.M.; et al. SIRT1 confers protection against UVB- and H2O2-induced cell death via modulation of p53 and JNK in cultured skin keratinocytes. J. Cell. Mol. Med. 2009, 13, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Mantena, S.K.; Meeran, S.M. Silymarin protects epidermal keratinocytes from ultraviolet radiation-induced apoptosis and DNA damage by nucleotide excision repair mechanism. PLoS ONE 2011, 6, e21410. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.X.; Jin, S.L.; Luo, D.; Lin, X.F.; Gao, J. Ginsenoside Rb1 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Biol. Pharm. Bull. 2009, 32, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.A.; Knaapen, A.M.; Houben, J.M.; van Kempen, F.C.; de Hoon, J.P.; Gottschalk, R.W.; Godschalk, R.W.; van Schooten, F.J. The role of glutathione in the regulation of nucleotide excision repair during oxidative stress. Toxicol. Lett. 2007, 168, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Güngör, N.; Godschalk, R.W.; Pachen, D.M.; van Schooten, F.J.; Knaapen, A.M. Activated neutrophils inhibit nucleotide excision repair in human pulmonary epithelial cells: Role of myeloperoxidase. FASEB J. 2007, 21, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Nakane, M.; Hattori, T.; Matsunaga, T.; Ihara, M.; Nikaido, O. Simultaneous establishment of monoclonal antibodies specific for either cyclobutane pyrimidine dimer or (6-4) photoproduct from the same mouse immunized with ultraviolet-irradiated DNA. Photochem. Photobiol. 1991, 54, 225–232. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piao, M.J.; Hewage, S.R.K.M.; Han, X.; Kang, K.A.; Kang, H.K.; Lee, N.H.; Hyun, J.W. Protective Effect of Diphlorethohydroxycarmalol against Ultraviolet B Radiation-Induced DNA Damage by Inducing the Nucleotide Excision Repair System in HaCaT Human Keratinocytes. Mar. Drugs 2015, 13, 5629-5641. https://doi.org/10.3390/md13095629
Piao MJ, Hewage SRKM, Han X, Kang KA, Kang HK, Lee NH, Hyun JW. Protective Effect of Diphlorethohydroxycarmalol against Ultraviolet B Radiation-Induced DNA Damage by Inducing the Nucleotide Excision Repair System in HaCaT Human Keratinocytes. Marine Drugs. 2015; 13(9):5629-5641. https://doi.org/10.3390/md13095629
Chicago/Turabian StylePiao, Mei Jing, Susara Ruwan Kumara Madduma Hewage, Xia Han, Kyoung Ah Kang, Hee Kyoung Kang, Nam Ho Lee, and Jin Won Hyun. 2015. "Protective Effect of Diphlorethohydroxycarmalol against Ultraviolet B Radiation-Induced DNA Damage by Inducing the Nucleotide Excision Repair System in HaCaT Human Keratinocytes" Marine Drugs 13, no. 9: 5629-5641. https://doi.org/10.3390/md13095629