
Chamigrane Sesquiterpenes from a Basidiomycetous Endophytic Fungus XG8D Associated with Thai Mangrove Xylocarpus granatum

Abstract
:
1. Introduction
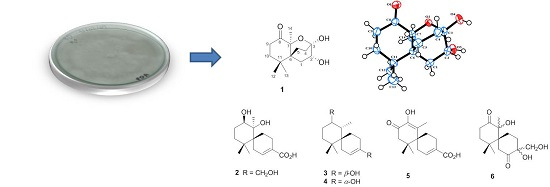
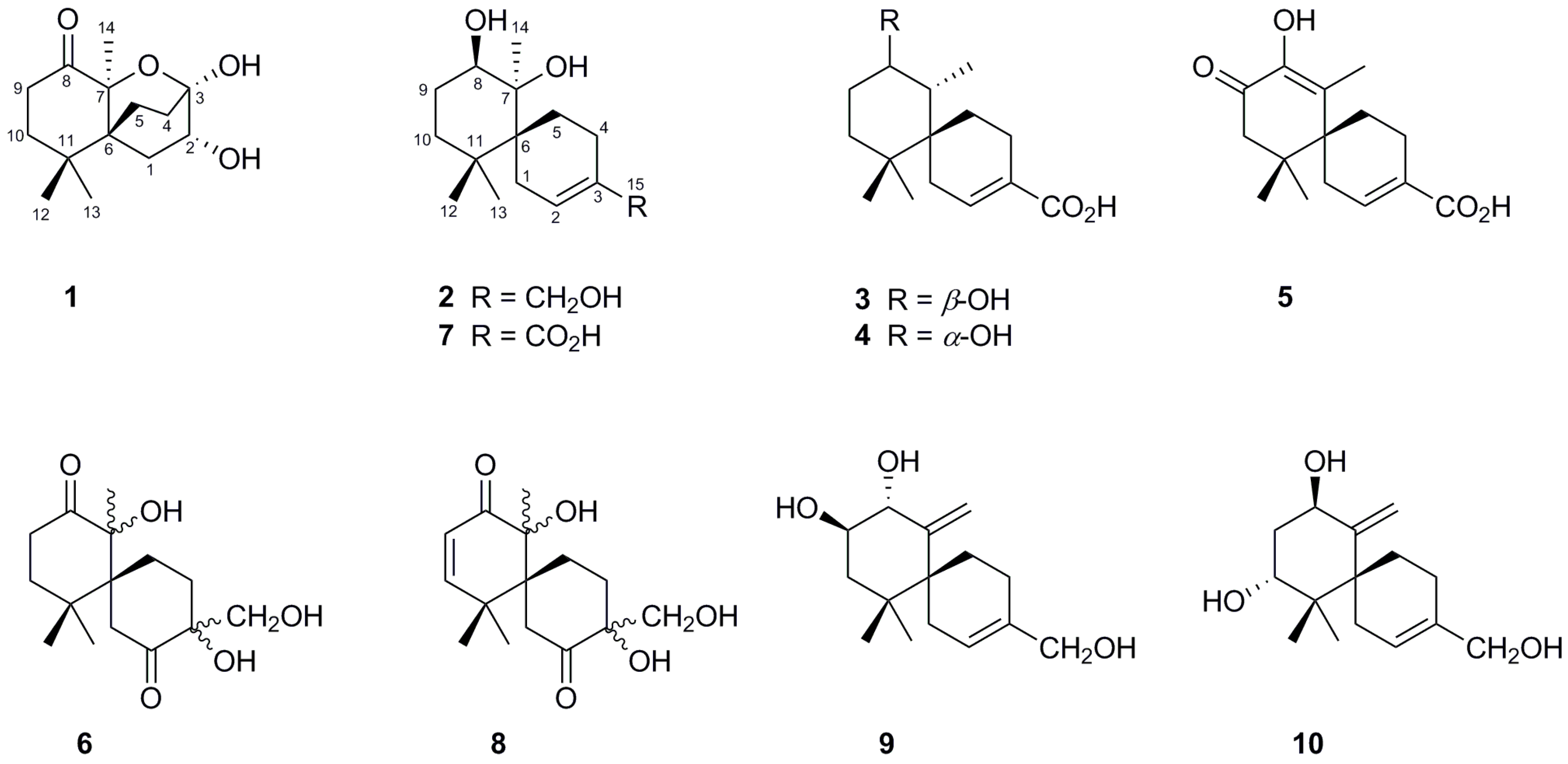
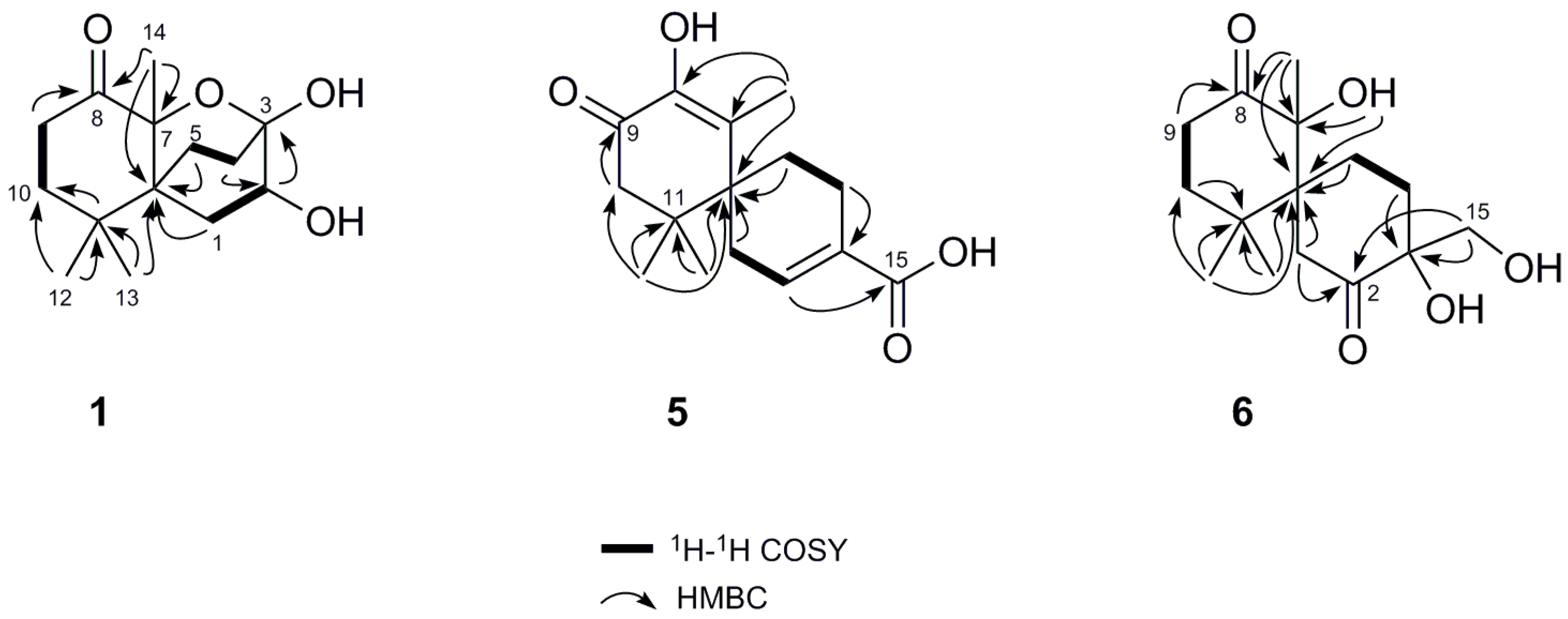
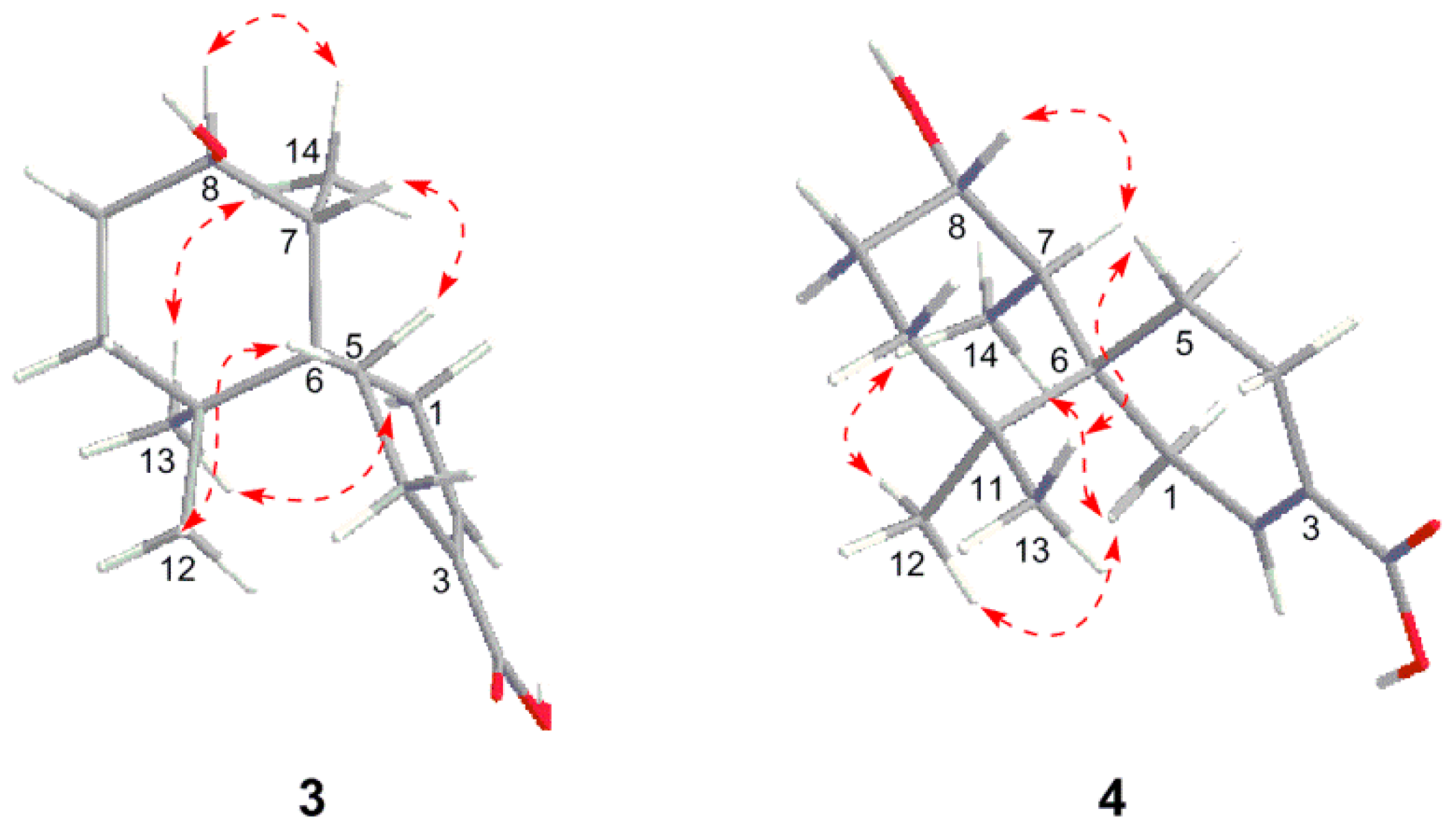
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
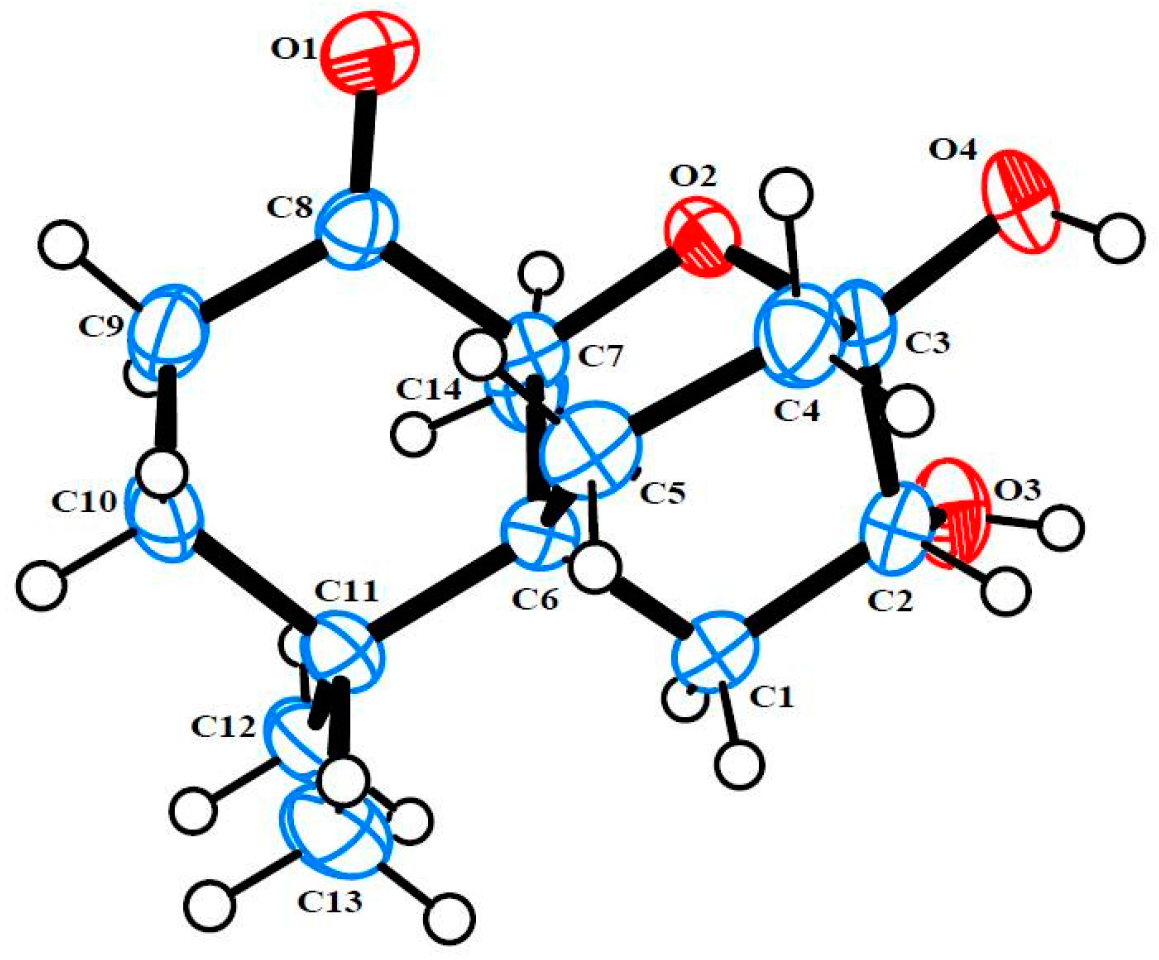
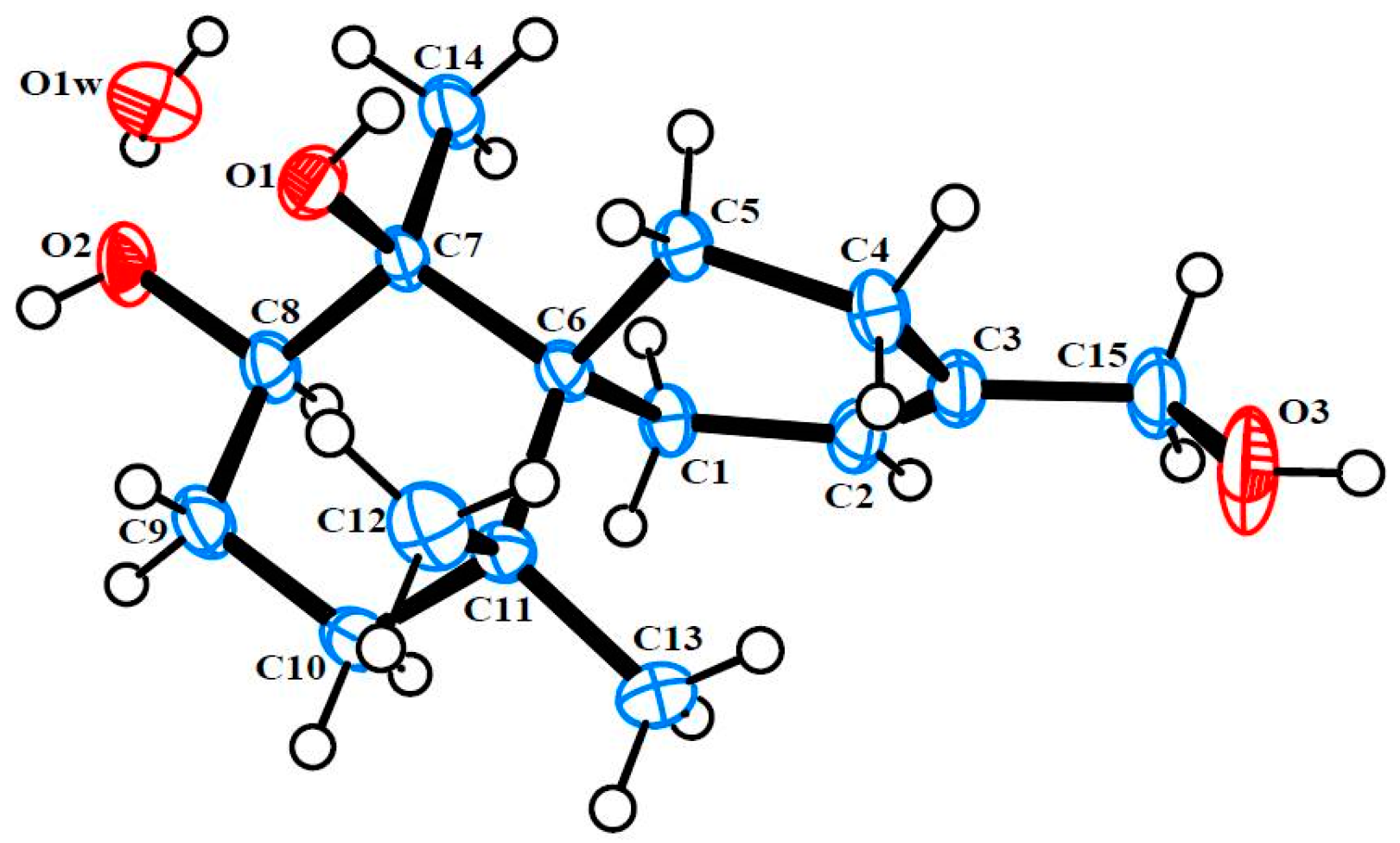
3.4. Single X-ray Crystallograpic Analysis of Merulinols A (1) and B (2)
3.5. Cell Culture
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, H.F.; Pei, Y.H. Secondary metabolites from marine-derived microorganisms. J. Asian Nat. Prod. Res. 2014, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Anada, K.; Sridhar, K.R. Diversity of endophytic fungi in the roots of mangrove species on the west coast of India. Can. J. Microbiol. 2002, 48, 871–878. [Google Scholar] [CrossRef]
- Debbab, A.; Aly, A.H.; Proksch, P. Mangrove derived fungal endophytes—A chemical and biological perception. Fungal Divers. 2013, 61, 1–27. [Google Scholar] [CrossRef]
- Zhou, Z.F.; Kurtán, T.; Yang, X.H.; Mándi, A.; Geng, M.Y.; Ye, B.P.; Taglialatela-Scafati, O.; Guo, Y.W. Penibruguieramine A, a novel pyrrolizidine alkaloid from the endophytic fungus Penicillium sp. GD6 associated with Chinese Mangrove Bruguiera gymnorrhiza. Org. Lett. 2014, 16, 1390–1393. [Google Scholar] [PubMed]
- Meng, L.H.; Li, X.M.; Lv, C.T.; Huang, C.G.; Wang, B.G. Brocazines A–F, cytotoxic bisthiodiketopiperazine derivatives from Penicillium brocae MA-231, an endophytic fungus derived from the marine mangrove plant Avicennia marina. J. Nat. Prod. 2014, 77, 1921–1927. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Lin, T.; Wang, W.; Xin, Z.; Zhu, T.; Gu, Q.; Li, D. Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013, 76, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.Y.; Wei, W.; Guo, Y.; Wang, T.; Jiao, R.H.; Ng, S.W.; Tan, R.X.; Ge, H.M. Sesquiterpenoids from the mangrove-derived endophytic fungus Diaporthe sp. J. Nat. Prod. 2012, 75, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Pudhom, K.; Teerawatananond, T.; Chookpaiboon, S. Spirobisnaphthalenes from the mangrove-derived fungus Rhytidhysteron sp. AS21B. Mar. Drugs 2014, 12, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Pudhom, K.; Teerawatananond, T. Rhytidenones A–F, spirobisnaphthalenes from Rhytidhysteron sp. AS21B, an endophytic fungus. J. Nat. Prod. 2014, 77, 1962–1966. [Google Scholar] [CrossRef] [PubMed]
- Chokpaiboon, S.; Sommit, D.; Teerawatnanond, T.; Muangsin, N.; Bunyapaiboonsri, T.; Pudhom, K. Cytotoxic nor-chamigrane and chamigrane endoperoxides from a basidiomycetous fungus. J. Nat. Prod. 2010, 73, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Chokpaiboon, S.; Sommit, D.; Bunyapaiboonsri, T.; Matsubara, K.; Pudhom, K. Antiangiogenic effect of chamigrane endoperoxides from a Thai mangrove-derived fungus. J. Nat. Prod. 2011, 74, 2290–2294. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Daitoh, M.; Vairappan, C.S.; Abe, T.; Masuda, M. Novel halogenated metabolites from the Malaysian Laurencia pannosa. J. Nat. Prod. 2001, 64, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Brito, I.; Cueto, M.; Díaz-Marrero, A.R.; Darias, J.; Martín, A.S. Oxachamigrenes, new halogenated sesquiterpenes from Laurencia obtusa. J. Nat. Prod. 2002, 65, 946–948. [Google Scholar] [CrossRef] [PubMed]
- Davyt, D.; Fernandez, R.; Suescun, L.; Mombrú, A.W.; Saldaña, J.; Domínguez, L.; Coll, J.; Fujii, M.T.; Manta, E. New sesquiterpene derivatives from the red alga Laurencia scoparia. Isolation, structure determination, and anthelmintic activity. J. Nat. Prod. 2001, 64, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Kimura, J.; Kamada, N.; Tsujimoto, Y. Fourteen chamigrane derivatives from a red alga, Laurencia nidifica. Bull. Chem. Soc. Jpn. 1999, 72, 289–292. [Google Scholar] [CrossRef]
- Al-Massarani, S.M. Phytochemical and biological properties of sesquiterpene constituents from the marine red seaweed Laurencia: A review. Nat. Prod. Chem. Res. 2014, 2, 1000147. [Google Scholar]
- Ebel, R. Terpenes from marine-derived fungi. Mar. Drugs 2010, 8, 2340–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.Z.; Dong, Z.J.; Wang, F.; Liu, J.K. Two novel norsesquiterpene peroxides from basidiomycete Steccherinum ochraceum. Tetrahedron Lett. 2010, 51, 3152–3153. [Google Scholar] [CrossRef]
- Li, H.; Huang, H.; Shao, C.; Huang, H.; Jiang, J.; Zhu, X.; Liu, Y.; Liu, L.; Lu, Y.; Li, M.; et al. Cytotoxic norsesquiterpene peroxides from the endophytic fungus Talaromyces flavus isolated from the mangrove plant Sonneratia apetala. J. Nat. Prod. 2011, 74, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, M.; Prachyawarakorn, V.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Cytotoxic sesquiterpenes from the endophytic fungus Pseudolagrobasidium acaciicola. Phytochemistry 2016, 122, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.N. Rediscovering natural product biodiversity. Drug Discov. Today 1999, 4, 449–451. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.M.; Shang, Z.; Li, C.S.; Ji, N.Y.; Wang, B.G. Meroterpenoid and diphenyl ether derivatives from Penicillium sp. MA-37, a fungus isolated from marine mangrove rhizospheric soil. J. Nat. Prod. 2012, 75, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Zheng, C.J.; Chen, G.Y.; Song, X.P.; Han, C.R.; Li, G.N.; Fu, Y.H.; Chen, W.H.; Niu, Z.G. Bioactive anthraquinone derivatives from the mangrove-derived fungus Stemphylium sp. 33231. J. Nat. Prod. 2014, 74, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Hewage, R.T.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. One strain-many compounds (OSMAC) method for production of polyketides, azaphilones and an isochromanone using the endophytic fungus Dothideomycete sp. Phytochemistry 2014, 108, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELX-97, Program for Crystal Structure Solution; University of Göttingen: Göttingen Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELX-97, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 8 July 2016).
- Xiao, D.; Zhu, S.P.; Gu, Z.L. Quercetin induced apoptosis in human leukemia HL-60 cells. Acta Pharmacol. Sin. 1997, 18, 280–283. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 b | 3 a | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 32.4, CH2 | 1.99, m | 29.2, CH2 | 2.19, d (20.0) | 26.9, CH2 | 2.15, m |
| 2.11, dt (14.4, 2.8) | 2.34, d (20.0) | |||||
| 2 | 69.4, CH | 3.85, dd (9.6, 2.0) | 124.0, CH | 5.16, br s | 144.1, CH | 7.18, br s |
| 3 | 94.5, qC | 137.7, qC | 130.0, qC | |||
| 4 | 29.5, CH2 | 1.66, m | 25.1, CH2 | 1.89, m | 23.3, CH2 | 2.24, m |
| 1.94, m | 2.44, m | |||||
| 5 | 25.7, CH2 | 1.64, m | 25.8, CH2 | 2.05, m | 29.1, CH2 | 1.63, m |
| 1.70, m | ||||||
| 6 | 43.5, qC | 44.3, qC | 41.3, qC | |||
| 7 | 86.0, qC | 78.3, qC | 44.9, CH | 1.65, m | ||
| 8 | 212.0, qC | 76.9, CH | 3.56, br s | 72.5, CH | 3.33, td (12.0, 4.0) | |
| 9 | 35.1, CH2 | 2.31, m | 26.9, CH2 | 1.38, dq (12.0, 4.0) | 31.6, CH2 | 1.48,m |
| 2.75, m | 2.10, m | 1.79, dq (12.0, 4.0) | ||||
| 10 | 37.3, CH2 | 1.61, m | 34.7, CH2 | 1.00, m | 36.6, CH2 | 1.23, dt (12.0, 4.0) |
| 1.92, m | 1.86, m | 1.60, m | ||||
| 11 | 37.1, qC | 38.3, qC | 37.3, qC | |||
| 12 | 27.3, CH3 | 0.94, s | 30.5, CH3 | 0.85, s | 27.9, CH3 | 0.78, s |
| 13 | 25.1, CH | 1.24, s | 26.3, CH3 | 1.17, s | 22.5, CH3 | 0.99, s |
| 14 | 25.8, CH3 | 1.68, s | 25.2, CH3 | 1.33, s | 12.8, CH3 | 1.01, d (6.8) |
| 15 | 66.9, CH2 | 3.89, br s | 171.3, qC | |||
| 7-OH | 3.08, s | |||||
| Position | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 29.9, CH2 | 2.17, m | 30.1, CH2 | 2.27, d (20.0) | 38.9, CH2 | 2.70, d (14.4) |
| 2.40, m | 2.43, d (20.0) | 2.79, d (14.4) | ||||
| 2 | 142.7, CH | 7.12, br s | 142.4, CH | 7.26, br s | 215.8, qC | |
| 3 | 129.1, qC | 130.6, qC | 75.9, qC | |||
| 4 | 22.6, CH2 | 2.27, m | 22.3, CH2 | 2.10, m | 33.3, CH2 | 1.76, m |
| 2.35, m | 2.58, d (18.0) | 2.24, m | ||||
| 5 | 27.5, CH2 | 1.50, m | 30.7, CH2 | 1.88, m | 23.5, CH2 | 1.54, m |
| 1.82, m | 1.80, m | |||||
| 6 | 39.5, qC | 43.0, qC | 55.2, qC | |||
| 7 | 36.8, CH | 1.95, m | 143.8, qC | 81.8, qC | ||
| 8 | 70.8, CH | 3.91, dd (12.0, 6.8) | 135.9, qC | 213.8, qC | ||
| 9 | 27.7, CH2 | 1.63,m | 192.3, qC | 33.7, CH2 | 2.41,m | |
| 2.74, m | ||||||
| 10 | 35.6, CH2 | 1.48, m | 47.1, CH2 | 2.22, d (18.0) | 36.5, CH2 | 1.61, m |
| 2.78, d (18.0) | 1.91, m | |||||
| 11 | 36.6, qC | 40.8, qC | 39.0, qC | |||
| 12 | 25.7, CH3 | 0.84, s | 23.8, CH3 | 1.01, s | 25.0, CH3 | 1.28, s |
| 13 | 26.3, CH3 | 0.95, s | 24.8, CH3 | 1.05, s | 28.6, CH3 | 1.00, s |
| 14 | 11.4, CH3 | 0.98, d (6.8) | 15.1, CH3 | 1.85, s | 25.4, CH3 | 1.42, s |
| 15 | 171.4, qC | 170.9, qC | 67.1, CH2 | 3.48, d (11.6) | ||
| 3.85, d (11.6) | ||||||
| 7-OH | 4.15, br s | |||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choodej, S.; Teerawatananond, T.; Mitsunaga, T.; Pudhom, K. Chamigrane Sesquiterpenes from a Basidiomycetous Endophytic Fungus XG8D Associated with Thai Mangrove Xylocarpus granatum. Mar. Drugs 2016, 14, 132. https://doi.org/10.3390/md14070132
Choodej S, Teerawatananond T, Mitsunaga T, Pudhom K. Chamigrane Sesquiterpenes from a Basidiomycetous Endophytic Fungus XG8D Associated with Thai Mangrove Xylocarpus granatum. Marine Drugs. 2016; 14(7):132. https://doi.org/10.3390/md14070132
Chicago/Turabian StyleChoodej, Siwattra, Thapong Teerawatananond, Tohru Mitsunaga, and Khanitha Pudhom. 2016. "Chamigrane Sesquiterpenes from a Basidiomycetous Endophytic Fungus XG8D Associated with Thai Mangrove Xylocarpus granatum" Marine Drugs 14, no. 7: 132. https://doi.org/10.3390/md14070132