
Another Look at Pyrroloiminoquinone Alkaloids—Perspectives on Their Therapeutic Potential from Known Structures and Semisynthetic Analogues


Abstract
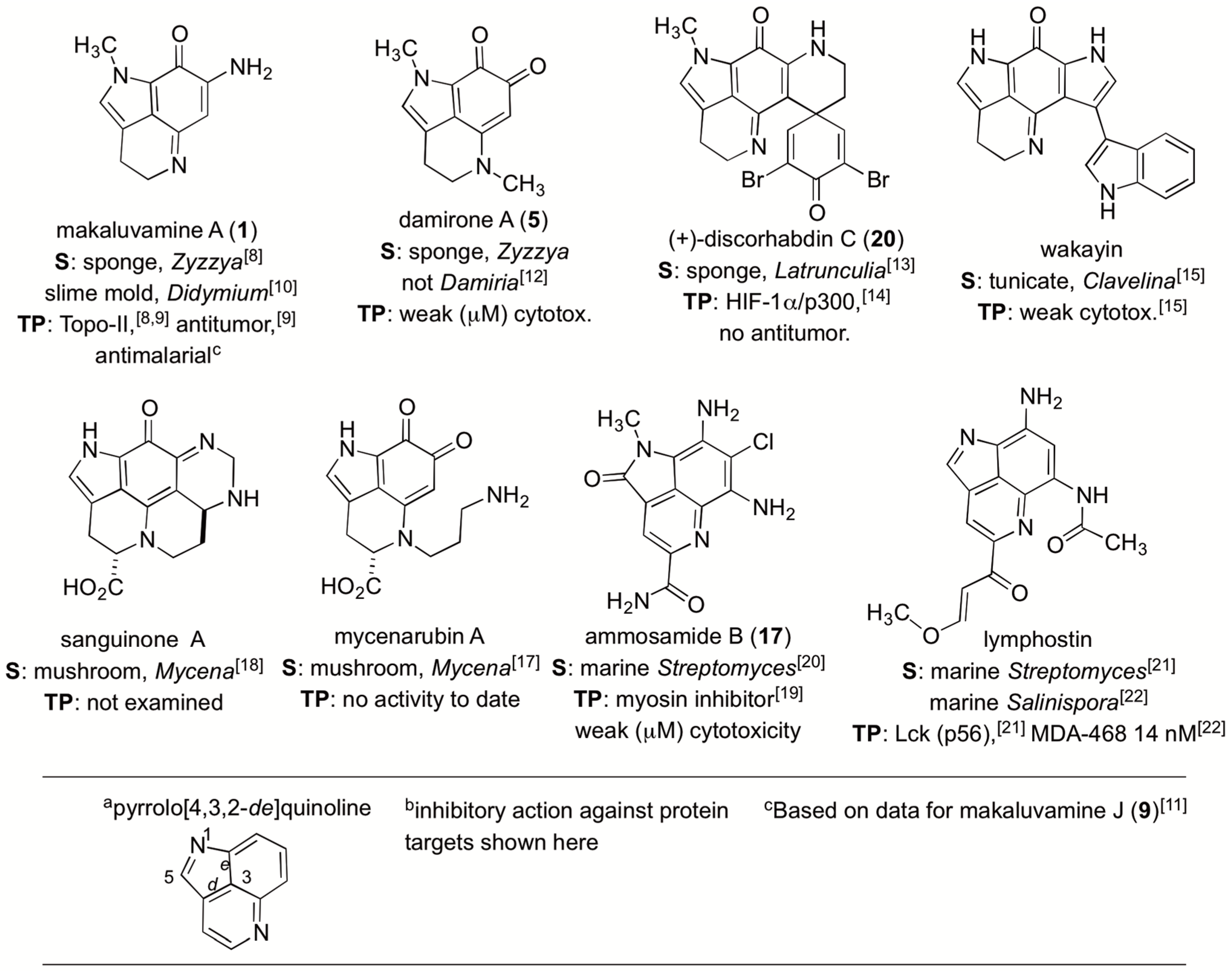
:1. Introduction
2. Results
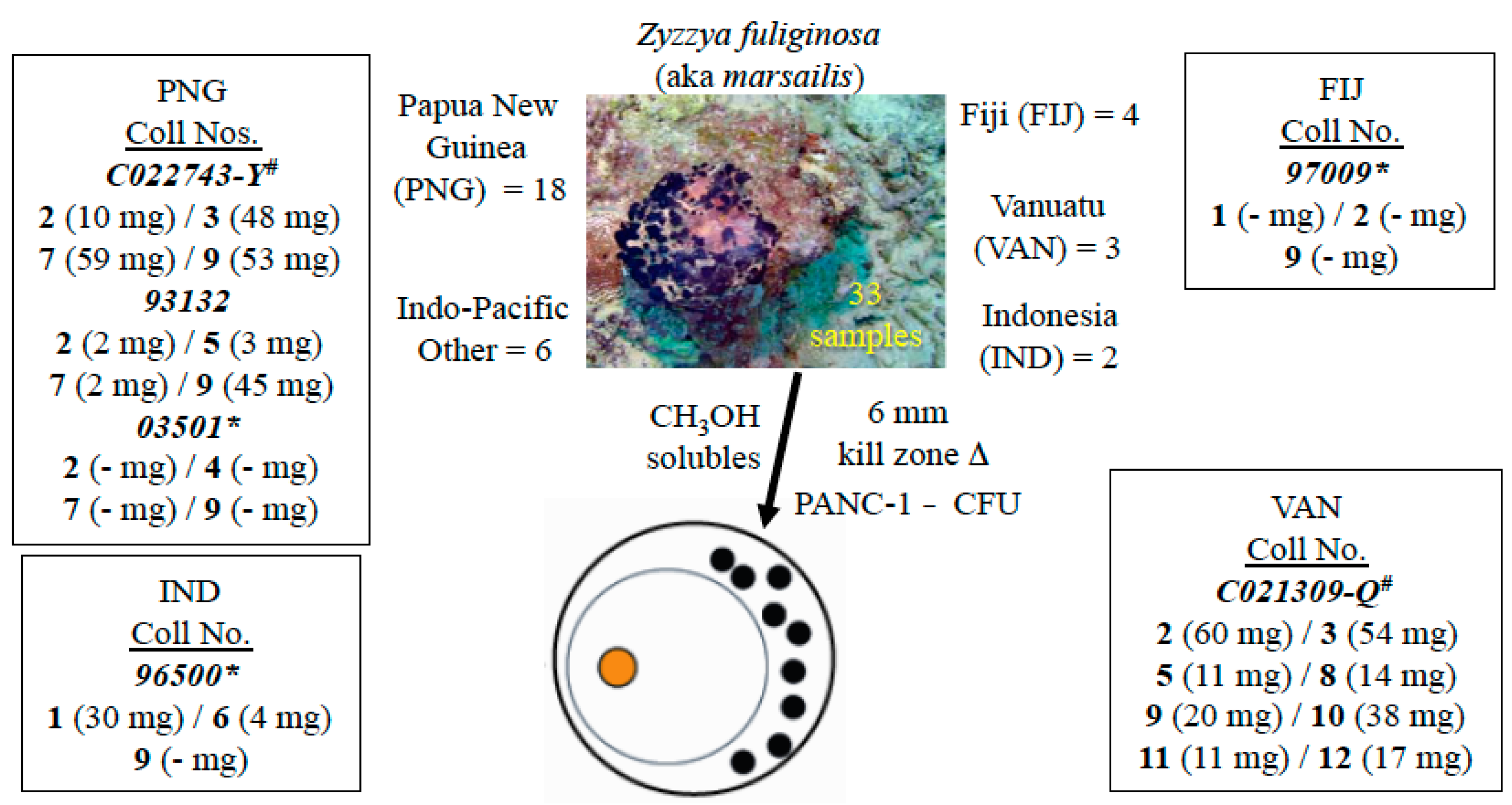
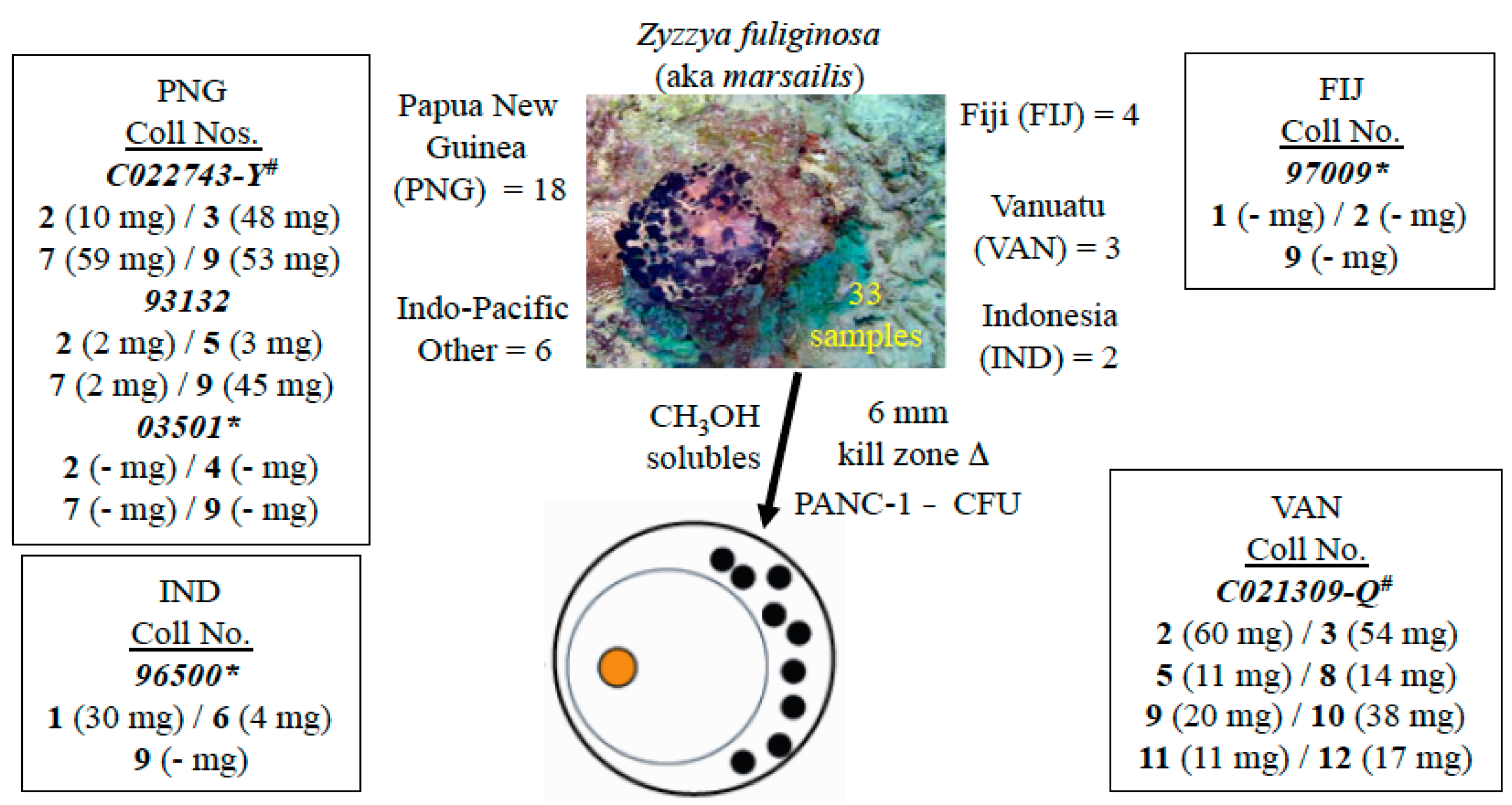
2.1. The Isolation Campaign
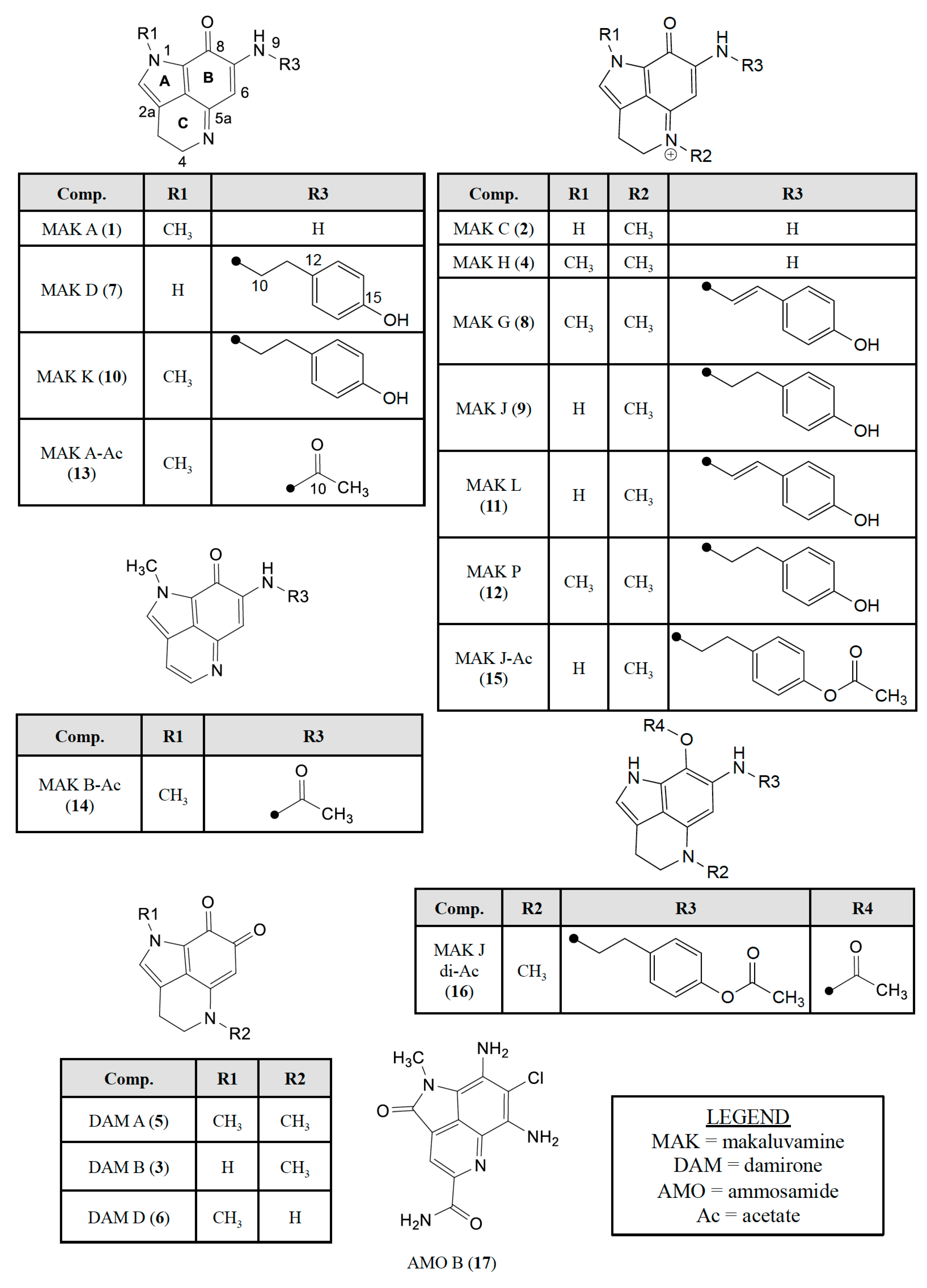
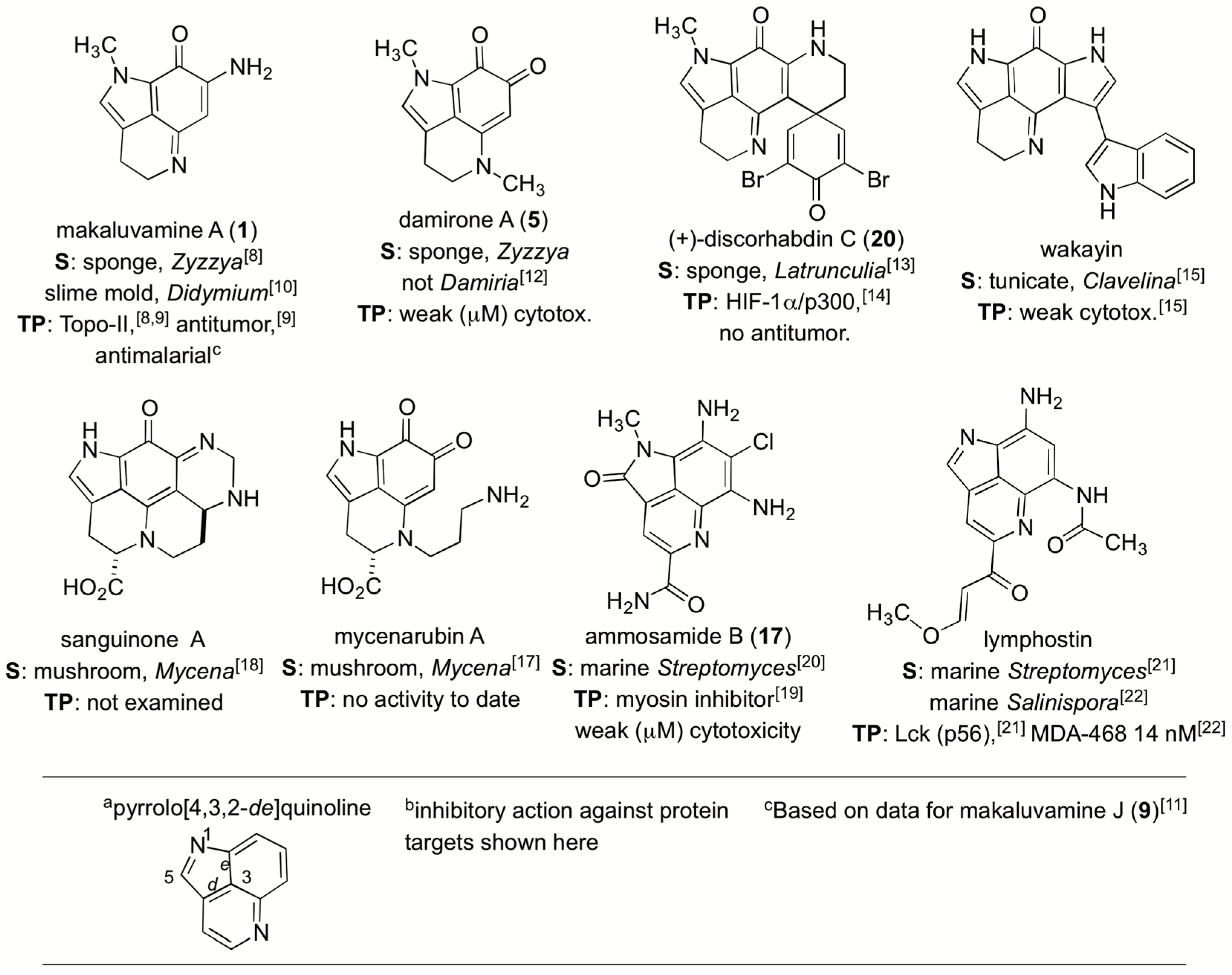
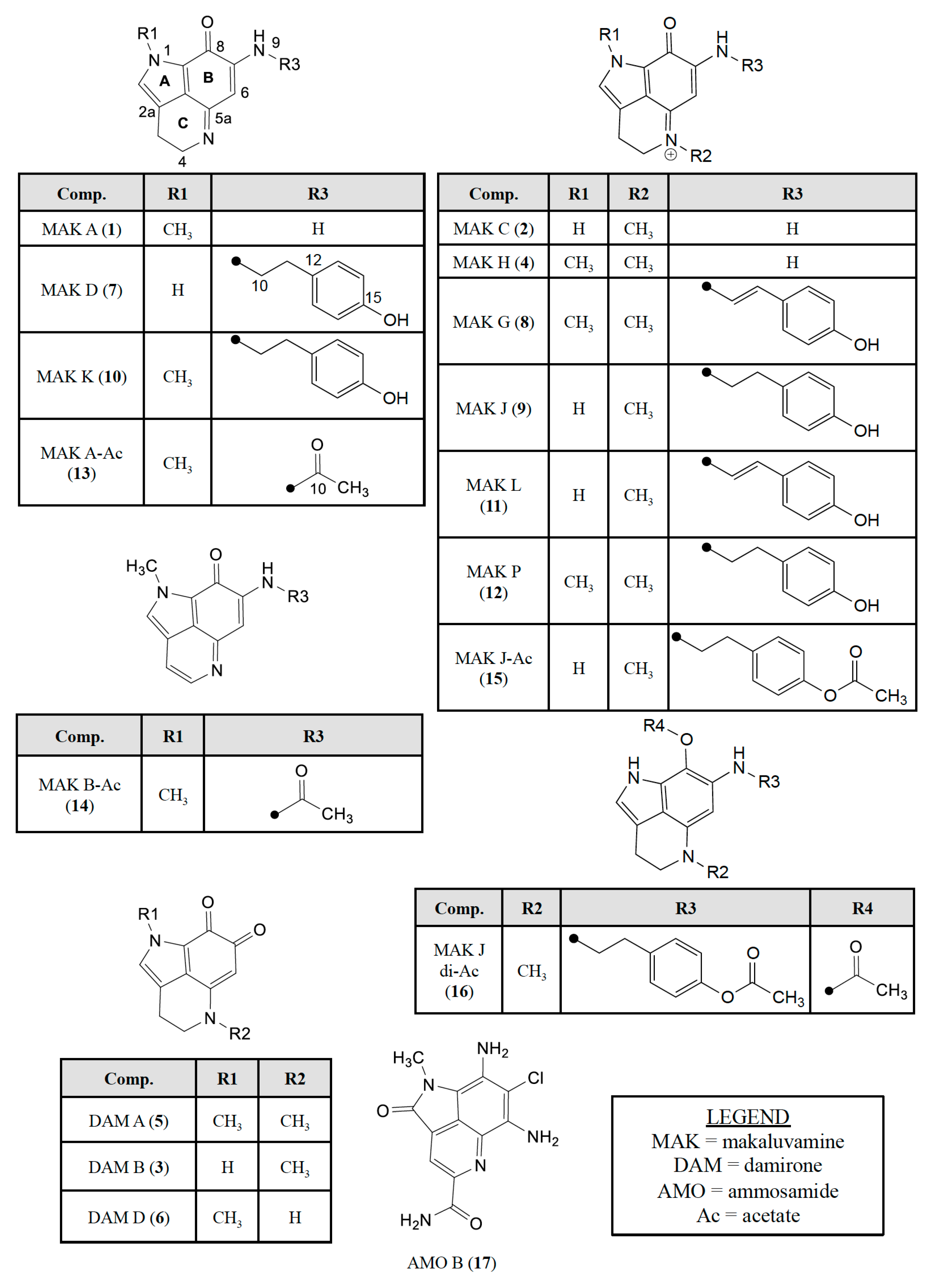
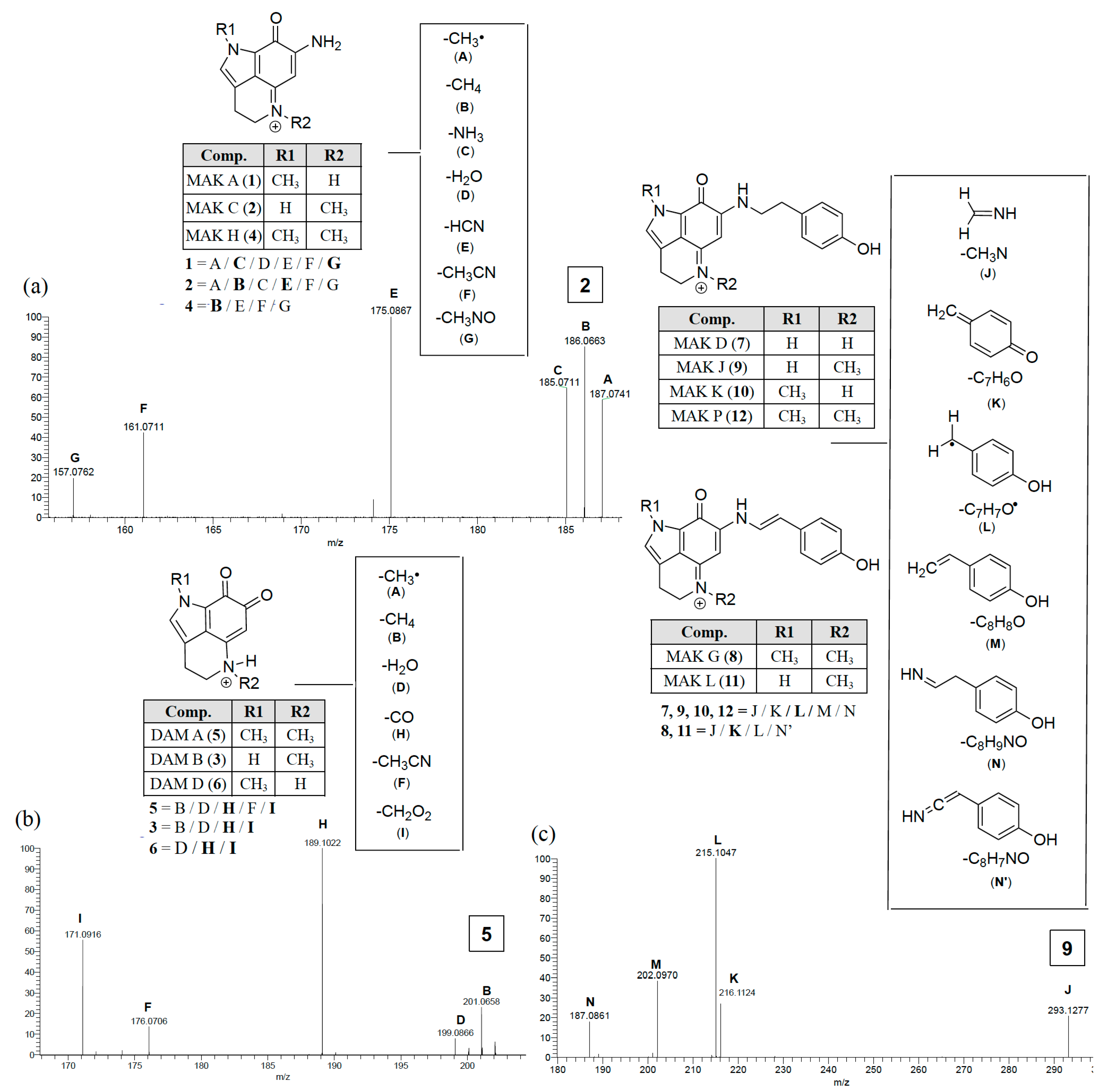
2.2. Dereplicating Pyrrolo[4,3,2-de]quinolines Using MS2 Patterns
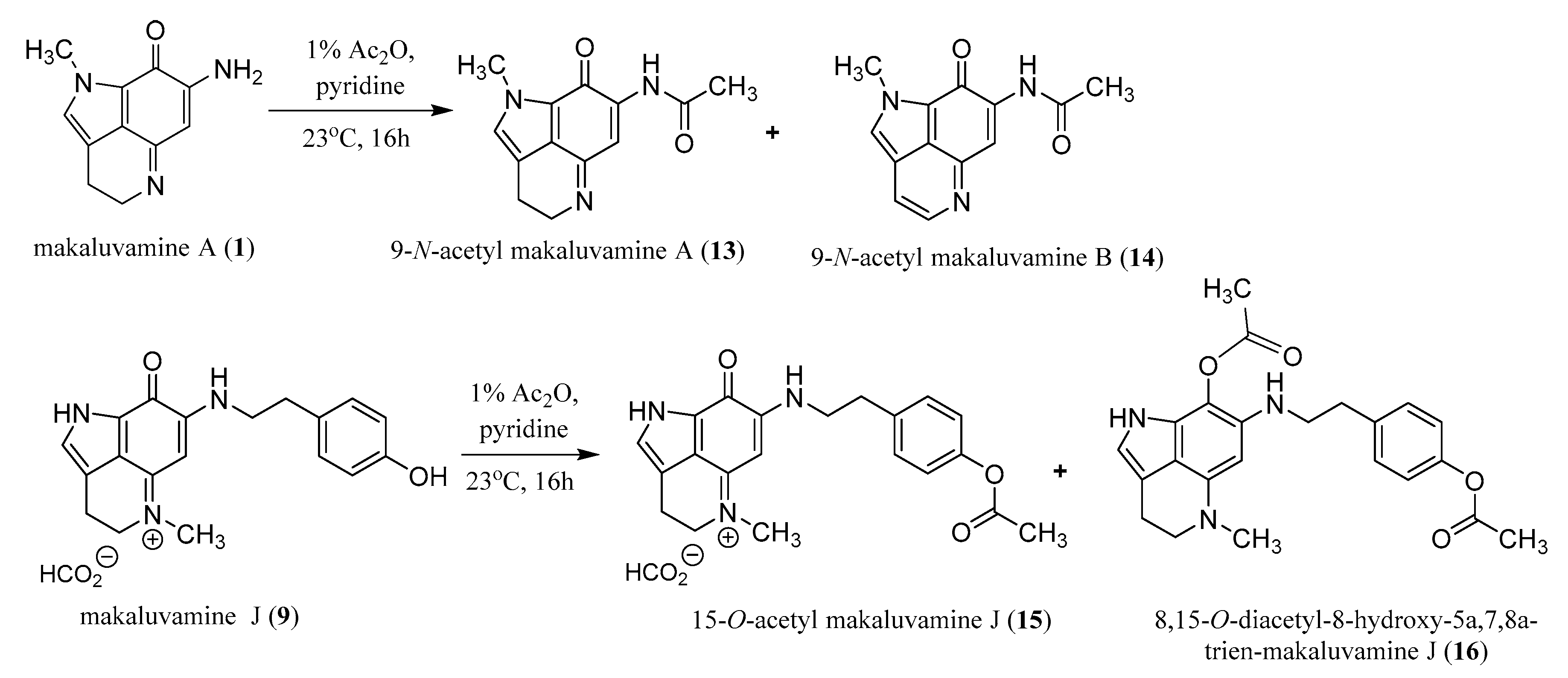
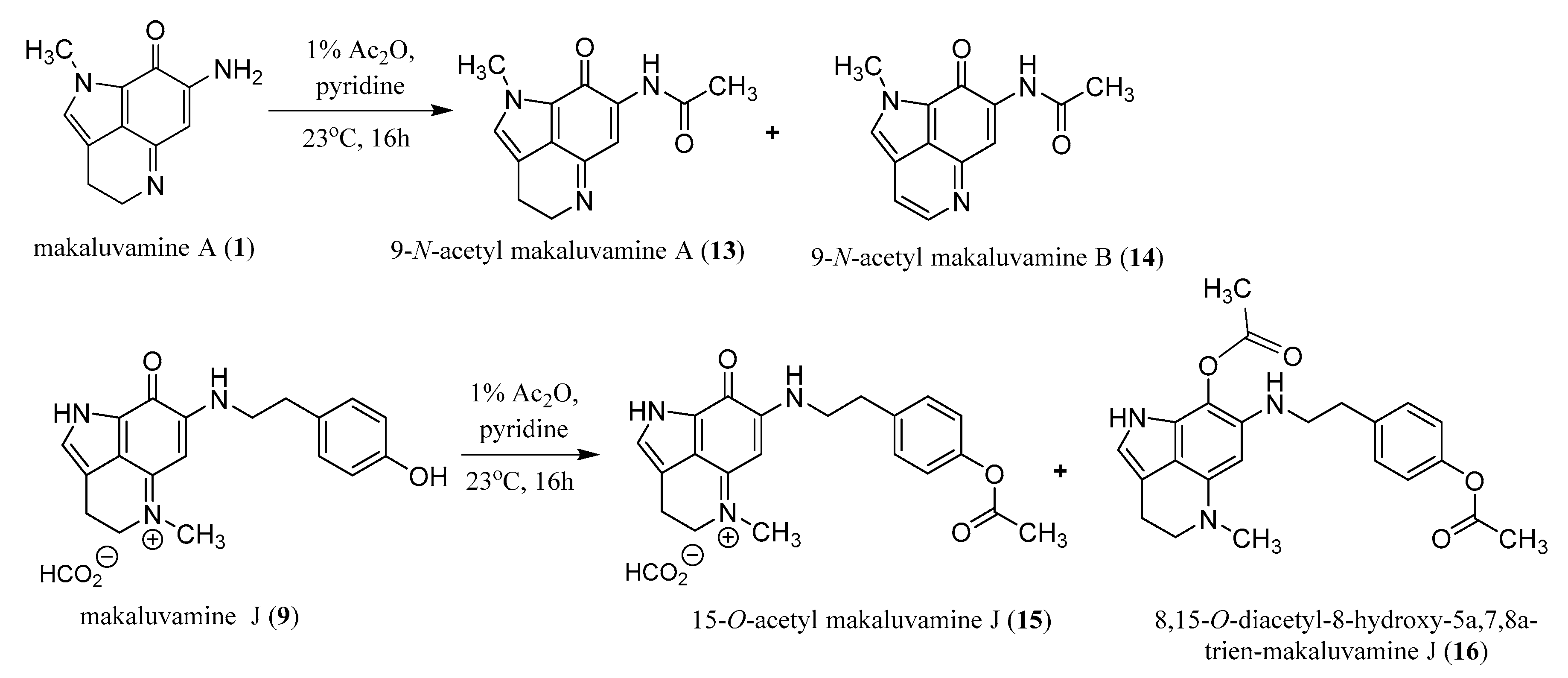
2.3. Semi-Synthesis of Acetylated Makaluvamines and Their Identification Using 1H-NMR and MS2 Data
3. Discussion
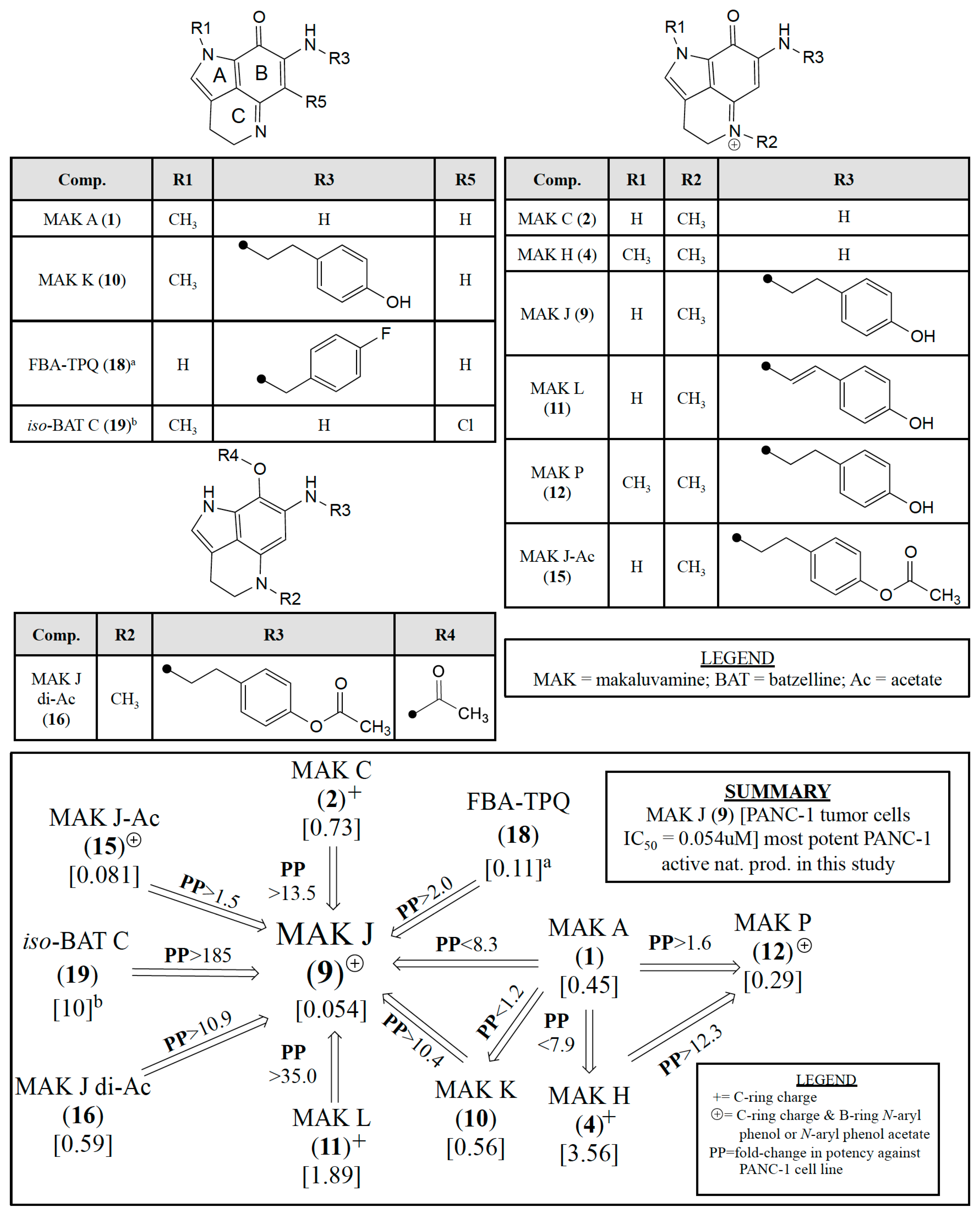
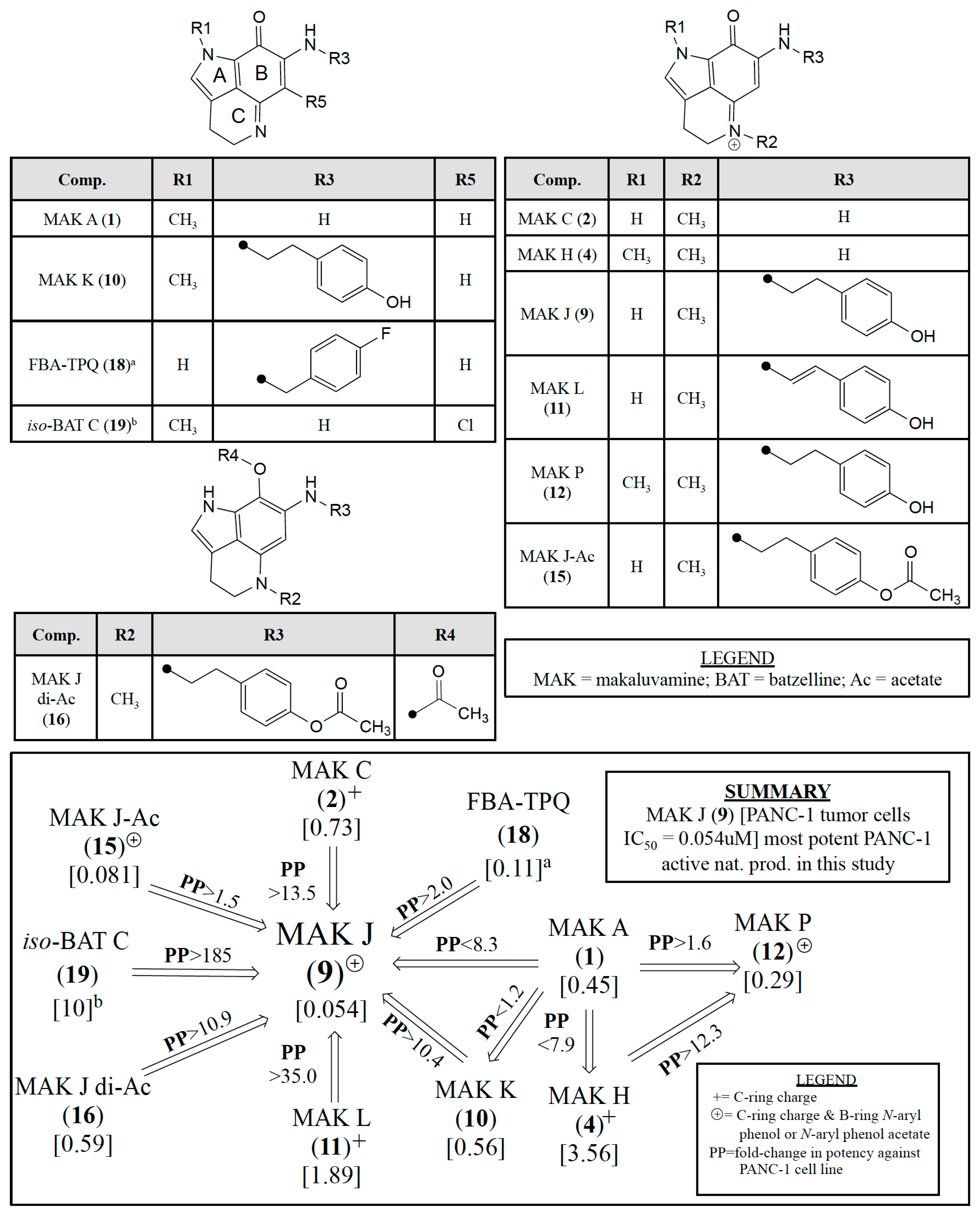
3.1. Evaluating Cytotoxicity Data for a Mini-Library of 22 Compounds
3.2. Assessing Relative PANC-1 Potencies of Pyrrolo[4,3,2-de]quinolines
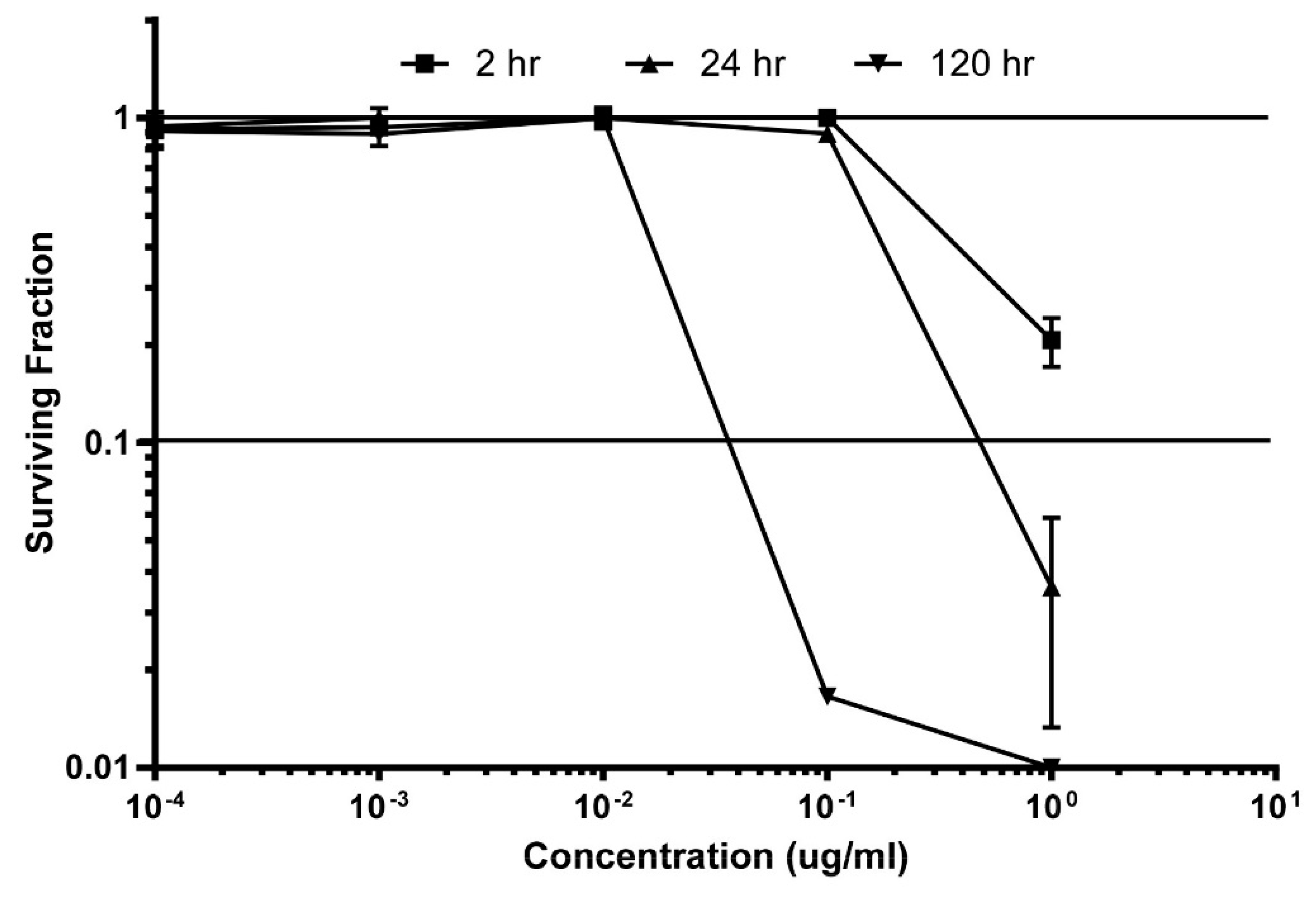
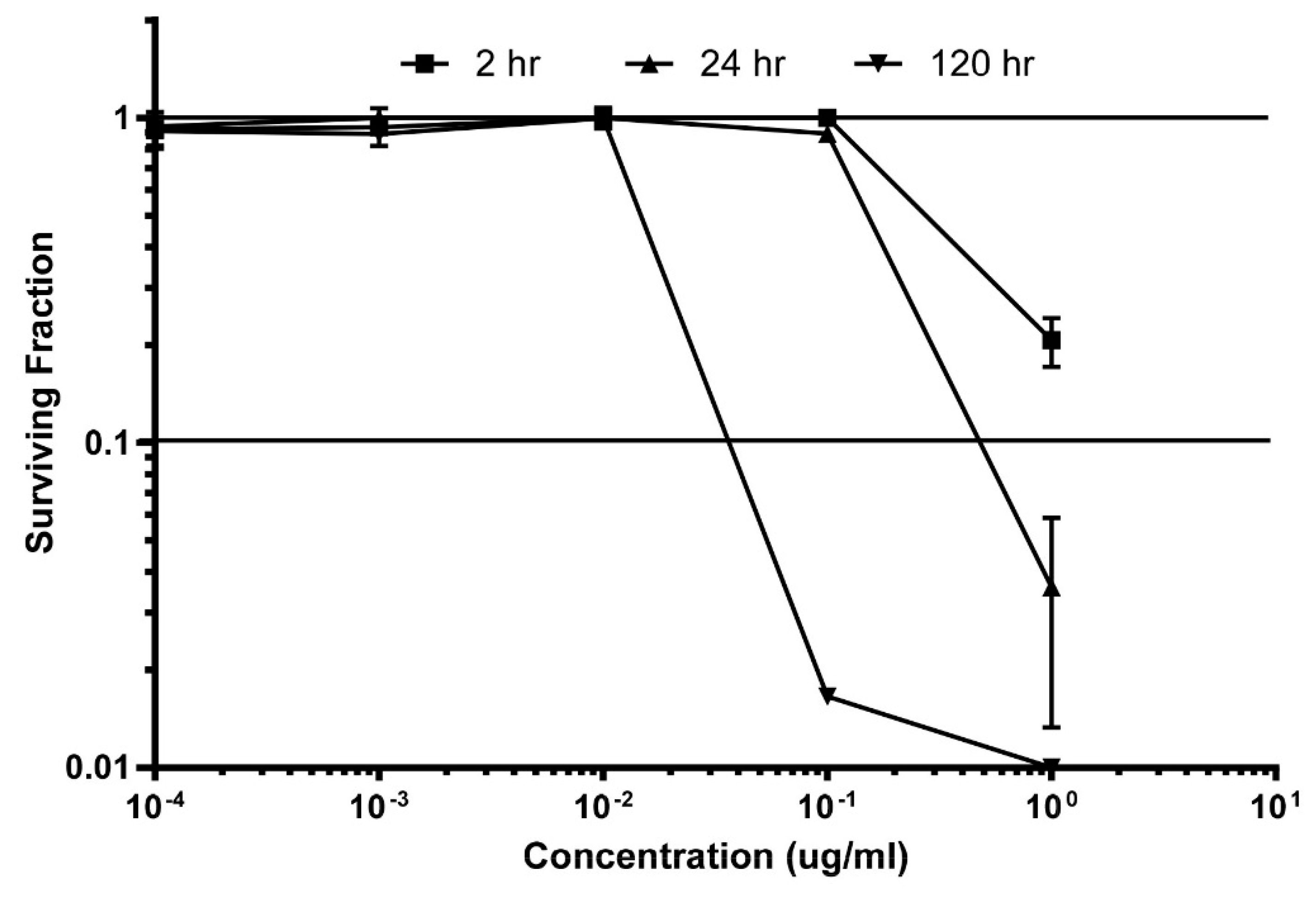
3.3. Secondary Screening
4. Material and Methods
4.1. General Experimental Procedures
4.2. Animal Material
4.3. Extraction and Isolation
4.4. Compound Properties
4.5. Mass Spectrometry
4.6. Acetylation of Makaluvamines A (1) and J (9)
4.7. Semi-Synthetic Compound Properties
4.8. Cytotoxicity Assays
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thale, Z.; Kinder, F.R.; Bair, K.W.; Bontempo, J.; Czuchta, A.M.; Versace, R.W.; Phillips, P.E.; Sanders, M.L.; Wattanasin, S.; Crews, P. Bengamides revisited: New structures and antitumor studies. J. Org. Chem. 2001, 66, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Hoffmann, H.; Zhang, J.; Debussche, L.; Haag-Richter, S.; Kurz, M.; Nardi, F.; Lukat, P.; Kochems, I.; Tietgen, H.; et al. Production of the bengamide class of marine natural products in myxobacteria: Biosynthesis and structure-activity relationships. Angew. Chem. Int. Ed. Engl. 2015, 54, 15560–15564. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Sohn, J.; Vaske, Y.M.; White, K.N.; Cohen, T.L.; Vervoort, H.C.; Tenney, K.; Valeriote, F.A.; Bjeldanes, L.F.; Crews, P. Myxobacteria versus sponge-derived alkaloids: The bengamide family identified as potent immune modulating agents by scrutiny of LC-MS/ELSD libraries. Bioorg. Med. Chem. 2012, 20, 4348–4355. [Google Scholar] [CrossRef] [PubMed]
- Furusato, A.; Kato, H.; Nehira, T.; Eguchi, K.; Kawabata, T.; Fujiwara, Y.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Takeya, M.; et al. Acanthomanzamines A-E with new manzamine frameworks from the marine sponge Acanthostrongylophora ingens. Org. Lett. 2014, 16, 3888–3891. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.L.; Peraud, O.; Kasanah, N.; Sims, J.W.; Kothalawala, N.; Anderson, M.A.; Abbas, S.H.; Rao, K.V.; Jupally, V.R.; Kelly, M.; et al. An analysis of the sponge Acanthostrongylophora ingens microbiome yields an actinomycete that produces the natural product manzamine A. Front. Mar. Sci. 2014, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sakemi, S.; Ichiba, T.; Kohmoto, S.; Saucy, G.; Higa, T. Isolation and structure elucidation of onnamide A, a new bioactive metabolite of a marine sponge, Theonella sp. J. Am. Chem. Soc. 1988, 110, 4851–4853. [Google Scholar] [CrossRef]
- Wilson, M.C.; Mori, T.; Rückert, C.; Uria, A.R.; Helf, M.J.; Takada, K.; Gernert, C.; Steffens, U.A.E.; Heycke, N.; Schmitt, S.; et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 2014, 506, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Radisky, E.S.; Barrows, L.R.; Copp, B.R.; Kramer, R.A.; Ireland, C.M. Novel cytotoxic topoisomerase II inhibiting pyrroloiminoquinones from Fijian sponges of the genus Zyzzya. J. Am. Chem. Soc. 1993, 115, 1632–1638. [Google Scholar] [CrossRef]
- Dijoux, M.-G.; Schnabel, P.C.; Hallock, Y.F.; Boswell, J.L.; Johnson, T.R.; Wilson, J.A.; Ireland, C.M.; van Soest, R.; Boyd, M.R.; Barrows, L.R.; et al. II Antitumor activity and distribution of pyrroloiminoquinones in the sponge genus Zyzzya. Bioorg. Med. Chem. 2005, 13, 6035–6044. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Iwasaki, T.; Imai, S.; Sakamoto, S.; Yamaguchi, K.; Ito, A. Laboratory culture of the Myxomycetes: Formation of fruiting bodies of Didymium bahiense and its plasmodial production of makaluvamine A. J. Nat. Prod. 2001, 64, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Buchanan, M.S.; Duffy, S.; Avery, V.M.; Charman, S.A.; Charman, W.N.; White, K.L.; Shackleford, D.M.; Edstein, M.D.; Andrews, K.T.; et al. Antimalarial activity of pyrroloiminoquinones from the Australian marine sponge Zyzzya sp. J. Med. Chem. 2012, 55, 5851–5858. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Harper, M.K.; Faulkner, D.J. Makaluvamines H-M and Damirone C from the Pohnpeian sponge Zyzzya fuliginosa. J. Nat. Prod. 1995, 58, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Perry, N.B.; Blunt, J.W.; McCombs, J.D.; Munro, M.H.G. Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem. 1986, 51, 5476–5478. [Google Scholar]
- Goey, A.K.L.; Chau, C.H.; Sissung, T.M.; Cook, K.M.; Venzon, D.J.; Castro, A.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; McMahon, J.B.; et al. Screening and biological effects of marine pyrroloiminoquinone alkaloids: Potential inhibitors of the HIF-1α/p300 interaction. J. Nat. Prod. 2016, 79, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Copp, B.R.; Ireland, C.M.; Barrows, L.R. Wakayin: A novel cytotoxic pyrroloiminoquinone alkaloid from the ascidian Clavelina species. J. Org. Chem. 1991, 56, 4596–4597. [Google Scholar] [CrossRef]
- Jordan, P.A.; Moore, B.S. Biosynthetic pathway connects cryptic ribosomally synthesized posttranslationally modified peptide genes with pyrroloquinoline alkaloids. Cell. Chem. Biol. 2016, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Spiteller, P. Mycenarubins A and B, red pyrroloquinoline alkaloids from the mushroom Mycena rosea. Eur. J. Org. Chem. 2007, 1571–1576. [Google Scholar]
- Peters, S.; Spiteller, P. Sanguinones A and B, blue pyrroloquinoline alkaloids from the fruiting bodies of the mushroom Mycena sanguinolenta. J. Nat. Prod. 2007, 70, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; MacMillan, J.B.; Gaudêncio, S.P.; Fenical, W.; La Clair, J.J. Ammosamides A and B target myosin. Angew. Chem. Int. Ed. Engl. 2009, 48, 728–732. [Google Scholar] [PubMed]
- Hughes, C.C.; MacMillan, J.B.; Gaudêncio, S.P.; Jensen, P.R.; Fenical, W. The ammosamides: Structures of cell cycle modulators from a marine-derived Streptomyces species. Angew. Chem. Int. Ed. Engl. 2009, 48, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Yano, H.; Sasaki, K.; Sato, S.; Nakanishi, S.; Takahashi, I.; Tamaoki, T. Inhibition of lymphocyte kinase Lck and phosphatidylinositol 3-kinase by a novel immunosuppressant, lymphostin. Biosci. Biotechnol. Biochem. 2002, 66, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Janso, J.E.; McDonald, L.; He, M.; Liu, H.; Barbieri, L.; Eustáquio, A.S.; Fielding, E.N.; Carter, G.T.; Jensen, P.R.; et al. Discovery and assembly-line biosynthesis of the lymphostin pyrroloquinoline alkaloid family of mTOR inhibitors in Salinispora bacteria. J. Am. Chem. Soc. 2011, 133, 13311–13313. [Google Scholar] [CrossRef] [PubMed]
- Antunes, E.M.; Copp, B.R.; Davies-Coleman, M.T.; Samaai, T. Pyrroloiminoquinone and related metabolites from marine sponges. Nat. Prod. Rep. 2005, 22, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Nadkarni, D.H.; Qin, J.-J.; Voruganti, S.; Nguyen, T.; Xu, S.; Wang, W.; Velu, S.E.; Zhang, R. Anticancer activity and molecular mechanisms of action of makaluvamines and analogues. Mol. Cell. Pharmacol. 2012, 4, 69–81. [Google Scholar]
- Zhang, X.; Xu, H.; Zhang, X.; Voruganti, S.; Murugesan, S.; Nadkarni, D.H.; Velu, S.E.; Wang, M.-H.; Wang, W.; Zhang, R. Preclinical evaluation of anticancer efficacy and pharmacological properties of FBA-TPQ, a novel synthetic makaluvamine analog. Mar. Drugs 2012, 10, 1138–1155. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.A.; Johnson, J.D.; Carrier, M.K.; Meyer, C.I.; Pitts, T.P.; Gunasekera, S.P.; Wright, A.E. Selective cytotoxic activity of the marine-derived batzelline compounds against pancreatic cancer cell lines. Anticancer Drugs 2009, 20, 149–155. [Google Scholar] [PubMed]
- Wang, W.; Nijampatnam, B.; Velu, S.E.; Zhang, R. Discovery and development of synthetic tricyclic pyrroloquinone (TPQ) alkaloid analogs for human cancer therapy. Front. Chem. Sci. Eng. 2016, 10, 1–15. [Google Scholar]
- Wang, W.; Rayburn, E.R.; Velu, S.E.; Nadkarni, D.H.; Murugesan, S.; Zhang, R. In vitro and In vivo anticancer activity of novel synthetic makaluvamine analogues. Clin. Cancer Res. 2009, 15, 3511–3518. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Li, J.N.; Zhu, J.X.; Melvin, W.S.; Bekaii-Saab, T.S.; Chen, C.S.; Muscarella, P. A structurally optimized celecoxib derivative inhibits human pancreatic cancer cell growth. J. Gastrointest. Surg. 2006, 10, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xu, T.; Guo, H.; Liu, Y.; Hu, P.; Yang, X.; Li, X.; Ge, S.; Velu, S.E.; Nadkarni, D.H.; et al. Experimental therapy of ovarian cancer with synthetic makaluvamine analog: In vitro and in vivo anticancer activity and molecular mechanisms of action. PLoS ONE 2011, 6, e20729. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.R.; Morinaka, B.I.; Arnagata, T.; Robinson, S.J.; Tenney, K.; Bray, W.M.; Gassner, N.C.; Lokey, R.S.; Media, J.; Valeriote, F.A.; et al. Biostructural features of additional jasplakinolide (Jaspamide) analogues. J. Nat. Prod. 2011, 74, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Nakeff, A.; Tenney, K.; Crews, P.; Gunatilaka, L.; Valeriote, F. A new paradigm for the development of anticancer agents from natural products. J. Exp. Ther. Oncol. 2006, 5, 195–204. [Google Scholar] [PubMed]
- Johnson, T.A.; Tenney, K.; Cichewicz, R.H.; Morinaka, B.I.; White, K.N.; Amagata, T.; Subramanian, B.; Media, J.; Mooberry, S.L.; Valeriote, F.A.; et al. Sponge-derived fijianolide polyketide class: Further evaluation of their structural and cytotoxicity properties. J. Med. Chem. 2007, 50, 3795–3803. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, R.H.; Valeriote, F.A.; Crews, P. Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the pederin family. Org. Lett. 2004, 6, 1951–1954. [Google Scholar] [CrossRef] [PubMed]
- Valeriote, F.A.; Tenney, K.; Media, J.; Pietraszkiewicz, H.; Edelstein, M.; Johnson, T.A.; Amagata, T.; Crews, P. Discovery and development of anticancer agents from marine sponges: Perspectives based on a chemistry-experimental therapeutics collaborative program. J. Exp. Ther. Oncol. 2012, 10, 119–134. [Google Scholar] [PubMed]
- Johnson, T.A.; Morgan, M.V.C.; Aratow, N.A.; Estee, S.A.; Sashidhara, K.V.; Loveridge, S.T.; Segraves, N.L.; Crews, P. Assessing pressurized liquid extraction for the high-throughput extraction of marine-sponge-derived natural products. J. Nat. Prod. 2010, 73, 359–364. [Google Scholar] [PubMed]
- McCloud, T.G. High throughput extraction of plant, marine and fungal specimens for preservation of biologically active molecules. Molecules 2010, 15, 4526–4563. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | A (1) a | A-Ac (13) a | B-Ac (14) b | J (9) a | J-Ac (15) a | J di-Ac (16) b |
|---|---|---|---|---|---|---|
| δH, mult, (J, Hz) | ||||||
| 2 | 7.27, s | 7.17, s | 8.15, s | 7.31, s | 7.13, s | 7.32, s |
| 3 | 2.81, t (7.5) | 2.70, t (7.5) | 7.75, d (6.0) | 2.92, t (7.5) | 2.95, brs | 3.04, t (7.5) |
| 4 | 3.77, t (7.5) | 3.95, t (7.5) | 8.43, d (6.0) | 3.89, t (7.5) | 3.78, brs | 3.93, t (7.5) |
| 6 | 5.61, s | 6.31, s | 8.67, s | 5.59, s | 5.42, s | 6.17, s |
| 10 | 3.60, t (7.0) | 3.63, brs | 3.76, t (7.5) | |||
| 11 | 2.83, t (7.0) | 2.85, brs | 2.98, t (7.5) | |||
| 13 | 7.05, d (8.0) | 7.05, d (8.0) | 7.03, d (8.0) | |||
| 14 | 6.69, d (8.0) | 7.29, d (8.0) | 7.31, d (8.0) | |||
| N1-Me | 3.89, s | 3.83, s | 4.39, s | |||
| N5-Me | 3.37, s | 3.28, s | 3.31, s | |||
| Ac | 2.34, s | 2.26, s | 2.25, s | 2.27, s 2.09, s | ||
| Compound | PANC-1 | OVCAR-3/-5 |
|---|---|---|
| IC50 (μM) | ||
| makaluvamine A (1) | 0.45 | |
| makaluvamine C (2) | 0.73 | 0.24 a/NT |
| damirone B (3) | 19 | |
| makaluvamine H (4) | 3.6 | 0.96 a/0.10 a |
| damirone A (5) | 160 | |
| damirone D (6) | 3.4 | |
| makaluvamine D (7) | 0.29 | |
| makaluvamine G (8) | 6.2 | |
| makaluvamine J (9) | 0.054 | NT/0.12 |
| makaluvamine K (10) | 0.56 | |
| makaluvamine L (11) | 1.9 | |
| makaluvamine P (12) | 0.3 | |
| 9-N-acetyl makaluvamine B (14) | 91 | |
| 15-O-acetyl makaluvamine J (15) | 0.081 | NT/0.0086 |
| 8,15-O-diacetyl-8-hydroxy-5a,7,8a-trien-makaluvamine J (16) | 0.59 | |
| ammosamide B (17) | 26 | |
| FBA-TBQ (18) | 0.11 b | 0.95 c/NT |
| isobatzelline C (19) | 10 d | |
| discorhabdin C (20) | NT | 0.33 e/2.6 e |
| etoposide | 0.39 | |
| teniposide | 0.041 | |
| gemcitabine | 7.2 f | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; McCauley, E.P.; Lorig-Roach, N.; Tenney, K.; Naphen, C.N.; Yang, A.-M.; Johnson, T.A.; Hernadez, T.; Rattan, R.; Valeriote, F.A.; et al. Another Look at Pyrroloiminoquinone Alkaloids—Perspectives on Their Therapeutic Potential from Known Structures and Semisynthetic Analogues. Mar. Drugs 2017, 15, 98. https://doi.org/10.3390/md15040098
Lin S, McCauley EP, Lorig-Roach N, Tenney K, Naphen CN, Yang A-M, Johnson TA, Hernadez T, Rattan R, Valeriote FA, et al. Another Look at Pyrroloiminoquinone Alkaloids—Perspectives on Their Therapeutic Potential from Known Structures and Semisynthetic Analogues. Marine Drugs. 2017; 15(4):98. https://doi.org/10.3390/md15040098
Chicago/Turabian StyleLin, Sheng, Erin P. McCauley, Nicholas Lorig-Roach, Karen Tenney, Cassandra N. Naphen, Ai-Mei Yang, Tyler A. Johnson, Thalia Hernadez, Ramandeep Rattan, Frederick A. Valeriote, and et al. 2017. "Another Look at Pyrroloiminoquinone Alkaloids—Perspectives on Their Therapeutic Potential from Known Structures and Semisynthetic Analogues" Marine Drugs 15, no. 4: 98. https://doi.org/10.3390/md15040098