
Cyanobacterial Toxins of the Laurentian Great Lakes, Their Toxicological Effects, and Numerical Limits in Drinking Water

Abstract
:1. Introduction
1.1. Cyanotoxins Overview
1.1.1. “Liver” Toxins
1.1.2. Neurotoxins
1.1.3. Dermatoxins
1.1.4. Miscellaneous
1.2. Characteristics of Bloom Forming Cyanobacteria
1.2.1. Buoyancy
1.2.2. Nutrient acquisition
1.2.3. Seasonality
1.2.4. Physical Forces Causing cyanoHABs
1.2.5. Species Dependent Effects
2. Distribution of cyanoHABs in the Great Lakes Region
3. Cyanotoxins in Drinking Water
4. Mechanisms of Toxicity
4.1. Commonly Found Cyanotoxins
4.1.1. Microcystins
4.1.2. Pathological Studies Using Pure MC Toxin
4.1.3. Repeat MC Oral Dose Studies
4.1.4. Effects on Other Tissues from Oral Exposure to MCs
4.1.5. Molecular Mechanism of MC Toxicity
4.2. Anatoxin-a
4.3. Cylindrospermopsin
4.4. Saxitoxin
5. Numerical Limits
5.1. Microcystin
5.2. Cylindrospermopsin
5.3. Anatoxin-a and Anatoxin-a(S)
5.4. Saxitoxin
5.5. Issues and Considerations in Developing Numerical Limits for Cyanotoxins
- (1)
- There have been few repeat oral dose animal studies using purified cyanotoxin. These studies have traditionally served as the basis for developing numerical limits since ingestion is the primary route of cyanotoxin exposure.
- (2)
- The contribution of cyanotoxins to chronic effects such as tumor promotion and cancer have not been considered in developing numerical limits for cyanotoxins, primarily due to a lack of data.
- (3)
- Guideline values should be matched closely with monitoring capabilities. At present it is not clear if this is the case. For example, there is currently no known method that targets TTMCs.
- (4)
- It is not clear whether the most sensitive individuals are protected at all levels of water ingestion rate and age group categories. In addition, other sensitive groups may not be protected such as those with underlying conditions that make them particularly sensitive to the effects of cyanotoxins.
6. Conclusions
Conflicts of Interest
References
- National Oceanic and Atmospheric Administration. Great Lakes Sea Surface Environmental Analysis. Available online: https://coastwatch.glerl.noaa.gov/glsea/ (accessed on 19 January 2015).
- Zamyadi, A.; MacLeod, S.L.; Fan, Y.; McQuaid, N.; Dorner, S.; Sauve, S.; Prevost, M. Toxic cyanobacterial breakthrough and accumulation in a drinking water plant: A monitoring and treatment challenge. Water Res. 2012, 46, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.Y.; Liang, S.; Lee, J. Toxin-producing cyanobacteria in freshwater: A review of the problems, impact on drinking water safety, and efforts for protecting public health. J. Microbiol. 2013, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Planavsky, N.J.; Asael, D.; Hofmann, A.; Reinhard, C.T.; Lalonde, S.V.; Knudsen, A.; Wang, X.; Ossa, F.O.; Pecoits, E.; Smith, A.J.B.; et al. Evidence for oxygenic photosynthesis half a billion years before the Great Oxidation Event. Nat. Geosci. 2014, 7, 283–286. [Google Scholar] [CrossRef]
- McFadden, G.I. Origin and evolution of plastids and photosynthesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016105. [Google Scholar] [CrossRef] [PubMed]
- Mejean, A.; Ploux, O. A genomic view of secondary metabolite production in cyanobacteria. In Advances in Botanical Research: Genomics of Cyanobacteria; Chauvat, F., Cassier-Chauvat, C., Eds.; Elsevier: San Diego, CA, USA, 2013. [Google Scholar]
- Calteau, A.; Fewer, D.P.; Latifi, A.; Coursin, T.; Laurent, T.; Jokela, J.; Kerfeld, C.A.; Sivonen, K.; Piel, J.; Gugger, M. Phylum-wide comparative genomics unravel the diversity of secondary metabolism in Cyanobacteria. BMC Genom. 2014, 15, 977. [Google Scholar] [CrossRef] [PubMed]
- Boyer, G.L. The occurrence of cyanobacterial toxins in New York lakes: Lessons from the MERHAB-Lower Great Lakes. Lake Reserv. Manag. 2007, 23, 153–160. [Google Scholar] [CrossRef]
- Rinta-Kanto, J.M.; Konopko, E.A.; DeBruyn, J.M.; Bourbonniere, R.A.; Boyer, G.L.; Wilhelm, S.W. Lake Erie Microcystis: Relationship between microcystin production, dynamics of genotypes and environmental parameters in a large lake. Harmful Algae 2009, 8, 665–673. [Google Scholar] [CrossRef]
- Vanderploeg, H.A.; Liebig, J.R.; Carmichael, W.W.; Agy, M.A.; Johengen, T.H.; Fahnenstiel, G.L.; Nalepa, T.F. Zebra mussel (Dreissena polymorpha) selective filtration promoted toxic Microcystis blooms in Saginaw Bay (Lake Huron) and Lake Erie. Can. J. Fish. Aquat. Sci. 2001, 58, 1208–1221. [Google Scholar] [CrossRef]
- Rinta-Kanto, J.M.; Ouellette, A.J.A.; Boyer, G.L.; Twiss, M.R.; Bridgeman, T.B.; Wilhelm, S.W. Quantification of toxic Microcystis spp. during the 2003 and 2004 blooms in Western Lake Erie using quantitative real-time PCR. Environ. Sci. Technol. 2005, 39, 4198–4205. [Google Scholar] [CrossRef] [PubMed]
- Sivonen, K.; Jones, G. Cyanobacterial toxins. In Toxic Cyanobacteria in Water—A Guide to Their Public Health Consequences, Monitoring and Management; E & FN Spon: London, UK, 1999; pp. 41–111. [Google Scholar]
- Milutinović, A.; Zorc-Pleskovič, R.; Živin, M.; Vovk, A.; Serša, I.; Šuput, D. Magnetic resonance imaging for rapid screening for the nephrotoxic and hepatotoxic effects of microcystins. Mar. Drugs 2013, 11, 2785–2798. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.E.; Zwiller, J.; Moore, R.; Daily, S.L.; Khatra, B.; Dukelow, M.; Boynton, A. Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. J. Biol. Chem. 1990, 265, 19401–19404. [Google Scholar] [PubMed]
- Ballot, A.; Sandvik, M.; Rundberget, T.; Botha, C.J.; Miles, C.O. Diversity of cyanobacteria and cyanotoxins in Hartbeespoort Dam, South Africa. Mar. Freshw. Res. 2014, 65, 175–189. [Google Scholar] [CrossRef]
- Briand, J.F.; Jacquet, S.; Flinois, C.; Avois-Jacquet, C.; Maisonnette, C.; Leberre, B.; Humbert, J.F. Variations in the microcystin production of Planktothrix rubescens (Cyanobacteria) assessed from a four-year survey of Lac du Bourget (France) and from laboratory experiments. Microb. Ecol. 2005, 50, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Hitzfeld, B.; Dietrich, D. Presence of Planktothrix sp. and cyanobacterial toxins in Lake Ammersee, Germany and their impact on whitefish (Coregonus lavaretus L.). Environ. Toxicol. 2001, 16, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Rinta-Kanto, J.M.; Wilhelm, S.W. Diversity of microcystin-producing cyanobacteria in spatially isolated regions of Lake Erie. Appl. Environ. Microbiol. 2006, 72, 5083–5085. [Google Scholar] [CrossRef] [PubMed]
- Sivonen, K.; Namikoshi, M.; Evans, W.R.; Carmichael, W.W.; Sun, F.; Rouhianen, L.; Luukkainen, R.; Rinehart, K.L. Isolation and characterization of a variety of Microcystins from seven strains of the cyanobacterial genus Anabaena. Appl. Environ. Microbiol. 1992, 58, 2495–2500. [Google Scholar] [PubMed]
- Carey, C.C.; Haney, J.F.; Cottingham, K.L. First report of microcystin-LR in the cyanobacterium Gloeotrichia echinulata. Environ. Toxicol. 2007, 22, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Luukkainen, R.; Sivonen, K.; Namikoshi, M.; Färdig, M.; Rinehart, K.; Niemelä, S. Isolation and identification of eight microcystins from thirteen Oscillatoria agardhii strains and structure of a new microcystin. Appl. Environ. Microbiol. 1993, 59, 2204–2209. [Google Scholar] [PubMed]
- Beattie, K.A.; Kaya, K.; Sano, T.; Codd, G.A. Three dehydrobutyrine-containing microcystins from Nostoc. Phytochemistry 1998, 47, 1289–1292. [Google Scholar] [CrossRef]
- Kaasalainen, U.; Fewer, D.P.; Jokela, J.; Wahlsten, M.; Sivonen, K.; Rikkinen, J. Cyanobacteria produce a high variety of hepatotoxic peptides in lichen symbiosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5886–5891. [Google Scholar] [CrossRef] [PubMed]
- Beattie, K.A.; Kaya, K.; Codd, G.A. The cyanobacterium Nodularia PCC 7804, of freshwater origin, produces [L-Har2]nodularin. Phytochemistry 2000, 54, 57–61. [Google Scholar] [CrossRef]
- Kleinteich, J.; Hildebrand, F.; Wood, S.A.; Ciŕs, S.; Agha, R.; Quesada, A.; Pearce, D.A.; Convey, P.; Kpper, F.C.; Dietrich, D.R. Diversity of toxin and non-toxin containing cyanobacterial mats of meltwater ponds on the Antarctic Peninsula: A pyrosequencing approach. Antarct. Sci. 2014, 26, 521–532. [Google Scholar] [CrossRef]
- Runnegar, M.T.; Xie, C.; Snider, B.B.; Wallace, G.A.; Weinreb, S.M.; Kuhlenkamp, J. In vitro hepatotoxicity of the cyanobacterial alkaloid cylindrospermopsin and related synthetic analogues. Toxicol. Sci. 2002, 67, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Humpage, A.R.; Fontaine, F.; Froscio, S.; Burcham, P.; Falconer, I.R. Cylindrospermopsin genotoxicity and cytotoxicity: Role of cytochrome P-450 and oxidative stress. J. Toxicol. Environ. Health A 2005, 68, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Steinman, A.; Biddanda, B.; Rediske, R.; Fahnenstiel, G. Occurrence of the toxin-producing cyanobacterium Cylindrospermopsis raciborskii in Mona and Muskegon Lakes, Michigan. J. Gt. Lakes Res. 2006, 32, 645–652. [Google Scholar] [CrossRef]
- Conroy, J.D.; Quinlan, E.L.; Kane, D.D.; Culver, D.A. Cylindrospermopsis in Lake Erie: Testing its association with other cyanobacterial genera and major limnological parameters. J. Gt. Lakes Res. 2007, 33, 519–535. [Google Scholar] [CrossRef]
- Devlin, J.P.; Edwards, O.E.; Gorham, P.R.; Hunter, M.R.; Pike, R.K.; Stavric, B. Anatoxin-a, a toxic alkaloid from Anabaena flos-aquae NCR-44h. Can. J. Chem. 1977, 55, 1367–1371. [Google Scholar] [CrossRef]
- Edwards, C.; Beattie, K.A.; Scrimgeour, C.M.; Codd, G.A. Identification of anatoxin-A in benthic cyanobacteria (blue-green algae) and in associated dog poisonings at Loch Insh, Scotland. Toxicon 1992, 30, 1165–1175. [Google Scholar] [CrossRef]
- Carmichael, W.W.; Biggs, D.F.; Peterson, M.A. Pharmacology of anatoxin-a, produced by the freshwater cyanophyte Anabaena flos-aquae NRC-44-1. Toxicon 1979, 17, 229–236. [Google Scholar] [CrossRef]
- Hyde, E.G.; Carmichael, W.W. Anatoxin-a(S), a naturally occurring organophosphate, is an irreversible active site-directed inhibitor of acetylcholinesterase (EC 3.1.1.7). J. Biochem. Toxicol. 1991, 6, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Stephens, M.; Wilkie, G.; Amar, M.; Lunt, G.G.; Whiting, P.; Gallagher, T.; Pereira, E.; Alkondon, M.; Albuquerque, E.X.; et al. (+)-Anatoxin-a is a potent agonist at neuronal nicotinic acetylcholine receptors. J. Neurochem. 1993, 60, 2308–2311. [Google Scholar] [CrossRef] [PubMed]
- Cook, W.O.; Beasley, V.R.; Dahlem, A.M.; Dellinger, J.A.; Harlin, K.S.; Carmichael, W.W. Comparison of effects of anatoxin-a(s) and paraoxon, physostigmine and pyridostigmine on mouse brain cholinesterase activity. Toxicon 1988, 26, 750–753. [Google Scholar] [CrossRef]
- Cook, W.O.; Dahlem, A.M.; Harlin, K.S.; Beasley, V.R.; Hooser, S.B.; Haschek, W.M.; Carmicheal, W.W. Reversal of cholinesterase inhibition and clinical signs and the postmortem findings in mice after intraperitoneal administration of anatoxin-a(s), paraoxon or pyridostigmine. Vet. Hum. Toxicol. 1991, 33, 1–4. [Google Scholar] [PubMed]
- Cook, W.O.; Dellinger, J.A.; Singh, S.S.; Dahlem, A.M.; Carmichael, W.W.; Beasley, V.R. Regional brain cholinesterase activity in rats injected intraperitoneally with anatoxin-a(s) or paraoxon. Toxicol. Lett. 1989, 49, 29–34. [Google Scholar] [CrossRef]
- Cox, P.A.; Banack, S.A.; Murch, S.J.; Rasmussen, U.; Tien, G.; Bidigare, R.R.; Metcalf, J.S.; Morrison, L.F.; Codd, G.A.; Bergman, B. Diverse taxa of cyanobacteria produce β-N-methylamino-l-alanine, a neurotoxic amino acid. Proc. Natl. Acad. Sci. USA 2005, 102, 5074–5078. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.E.; King, S.R.; Banack, S.A.; Webster, C.; Callanaupa, W.J.; Cox, P.A. Cyanobacteria (Nostoc commune) used as a dietary item in the Peruvian highlands produce the neurotoxic amino acid BMAA. J. Ethnopharmacol. 2008, 118, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.J.; Cox, P.A.; Banack, S.A.; Steele, J.C.; Sacks, O.W. Occurrence of β-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol. Scand. 2004, 110, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Pablo, J.; Banack, S.A.; Cox, P.A.; Johnson, T.E.; Papapetropoulos, S.; Bradley, W.G.; Buck, A.; Mash, D.C. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol. Scand. 2009, 120, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Field, N.C.; Metcalf, J.S.; Caller, T.A.; Banack, S.A.; Cox, P.A.; Stommel, E.W. Linking β-methylamino-l-alanine exposure to sporadic amyotrophic lateral sclerosis in Annapolis, MD. Toxicon 2013, 70, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Aigret, B.; De Borggraeve, W.M.; Spacil, Z.; Ilag, L.L. Selective LC-MS/MS method for the identification of BMAA from its isomers in biological samples. Anal. Bioanal. Chem. 2012, 403, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.; Hellenas, K.E. Determination of the neurotoxin BMAA (β-N-methylamino-l-alanine) in cycad seed and cyanobacteria by LC-MS/MS (liquid chromatography tandem mass spectrometry). Analyst 2008, 133, 1785–1789. [Google Scholar] [CrossRef] [PubMed]
- Faassen, E.J.; Gillissen, F.; Lurling, M. A comparative study on three analytical methods for the determination of the neurotoxin BMAA in cyanobacteria. PLoS ONE 2012, 7, e36667. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Tian, Z.; Li, J.; Yu, R.; Banack, S.A.; Wang, Z. Detection of the neurotoxin BMAA within cyanobacteria isolated from freshwater in China. Toxicon 2010, 55, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Arakawa, O.; Taniyama, S.; Nonaka, M.; Takatani, T.; Yamamori, K.; Fuchi, Y.; Noguchi, T. Occurrence of saxitoxins as a major toxin in the ovary of a marine puffer Arothron firmamentum. Toxicon 2004, 43, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Halme, M.; Rapinoja, M.L.; Karjalainen, M.; Vanninen, P. Verification and quantification of saxitoxin from algal samples using fast and validated hydrophilic interaction liquid chromatography-tandem mass spectrometry method. J. Chromatogr. B 2012, 880, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Kulis, D.M.; Sullivan, J.J.; Hall, S.; Lee, C. Dynamics and physiology of saxitoxin production by the dinoflagellates Alexandrium spp. Mar. Biol. 1990, 104, 511–524. [Google Scholar] [CrossRef]
- Sinha, R.; Pearson, L.A.; Davis, T.W.; Burford, M.A.; Orr, P.T.; Neilan, B.A. Increased incidence of Cylindrospermopsis raciborskii in temperate zones—Is climate change responsible? Water Res. 2012, 46, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Ballot, A.; Fastner, J.; Wiedner, C. Paralytic shellfish poisoning toxin-producing cyanobacterium Aphanizomenon gracile in northeast Germany. Appl. Environ. Microbiol. 2010, 76, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, T.B.; Penamon, W.A. Lyngbya wollei in western Lake Erie. J. Gt. Lakes Res. 2010, 36, 167–171. [Google Scholar] [CrossRef]
- Onodera, H.; Satake, M.; Oshima, Y.; Yasumoto, T.; Carmichael, W.W. New saxitoxin analogues from the freshwater filamentous cyanobacterium Lyngbya wollei. Nat. Toxins 1997, 5, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W.W.; Evans, W.R.; Yin, Q.Q.; Bell, P.; Moczydlowski, E. Evidence for paralytic shellfish poisons in the freshwater cyanobacterium Lyngbya wollei (Farlow ex Gomont) comb. nov. Appl. Environ. Microbiol. 1997, 63, 3104–3110. [Google Scholar] [PubMed]
- Kleinteich, J.; Wood, S.A.; Puddick, J.; Schleheck, D.; Küpper, F.C.; Dietrich, D. Potent toxins in Arctic environments—Presence of saxitoxins and an unusual microcystin variant in Arctic freshwater ecosystems. Chem. Biol. Interact. 2013, 206, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.; Webb, P.M.; Schluter, P.J.; Fleming, L.E.; Burns, J.W., Jr.; Gantar, M.; Backer, L.C.; Shaw, G.R. Epidemiology of recreational exposure to freshwater cyanobacteria—An international prospective cohort study. BMC Public Health 2006, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.; Robertson, I.M.; Webb, P.M.; Schluter, P.J.; Shaw, G.R. Cutaneous hypersensitivity reactions to freshwater cyanobacteria—Human volunteer studies. BMC Dermatol. 2006, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.; Schluter, P.J.; Shaw, G.R. Cyanobacterial lipopolysaccharides and human health—A review. Environ. Health 2006, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.; Seawright, A.A.; Schluter, P.J.; Shaw, G.R. Primary irritant and delayed-contact hypersensitivity reactions to the freshwater cyanobacterium Cylindrospermopsis raciborskii and its associated toxin cylindrospermopsin. BMC Dermatol. 2006, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.J.; Webb, P.M.; Shaw, G.R. The toxins of Lyngbya majuscula and their human and ecological health effects. Environ. Int. 2001, 27, 381–392. [Google Scholar] [CrossRef]
- Fujiki, H.; Mori, M.; Nakayasu, M.; Terada, M.; Sugimura, T.; Moore, R. Indole alkaloids: Dihydroteleocidin B, teleocidin, and lyngbyatoxin A as members of a new class of tumor promoters. Proc. Natl. Acad. Sci. USA 1981, 78, 3872–3876. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Suganuma, M.; Hakii, H.; Bartolini, G.; Moore, R.; Takayama, S.; Sugimura, T. A two-stage mouse skin carcinogenesis study of lyngbyatoxin A. J. Cancer Res. Clin. Oncol. 1984, 108, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Pittman, C. Springs Bring Mystery Illness. St. Petersburg Times. 2006. Available online: http://www.sptimes.com/2006/08/24/State/Springs_bring_mystery.shtml (accessed on 19 October 2014).
- Fossa, A.J.; Phlipsa, E.J.; Yilmaza, M.; Chapmanb, A. Characterization of paralytic shellfish toxins from Lyngbya wollei dominated mats collected from two Florida springs. Harmful Algae 2012, 16, 98–107. [Google Scholar] [CrossRef]
- Welker, M.; von Dohren, H. Cyanobacterial peptides—Nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Microginin, an angiotensin-converting enzyme inhibitor from the blue-green alga Microcystis aeruginosa. Tetrahedron Lett. 1993, 34, 501–504. [Google Scholar] [CrossRef]
- Sano, T.; Nohara, K.; Shiraishi, F.; Kaya, K. A method for micro-determination of total microcystin content in waterblooms of cyanobacteria (blue-green algae). Int. J. Environ. Anal. Chem. 1992, 49, 163–170. [Google Scholar] [CrossRef]
- Itou, Y.; Suzuki, S.; Ishida, K.; Murakami, M. Anabaenopeptins G and H, Potent Carboxypeptidase A inhibitors from the cyanobacterium Oscillatoria agardhii (NIES-595). Bioorg. Med. Chem. Lett. 1999, 9, 1243–1246. [Google Scholar] [CrossRef]
- Singh, R.K.; Tiwari, S.P.; Rai, A.K.; Mohapatra, T.M. Cyanobacteria: An emerging source for drug discovery. J. Antibiot. 2011, 64, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Gademann, K.; Portmann, C.; Blom, J.F.; Zeder, M.; Juttner, F. Multiple toxin production in the cyanobacterium Microcystis: Isolation of the toxic protease inhibitor cyanopeptolin 1020. J. Nat. Prod. 2010, 73, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Faltermann, S.; Zucchi, S.; Kohler, E.; Blom, J.F.; Pernthaler, J.; Fent, K. Molecular effects of the cyanobacterial toxin cyanopeptolin (CP1020) occurring in algal blooms: Global transcriptome analysis in zebrafish embryos. Aquat. Toxicol. 2014, 149, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kong, F. Effects of light and wind speed on the vertical distribution of Microcystis aeruginosa colonies of different sizes during a summer bloom. Int. Rev. Hydrobiol. 2009, 94, 258–266. [Google Scholar] [CrossRef]
- Misson, B.; Sabart, M.; Amblard, C.; Latour, D. Involvement of Microcystins and colony size in the benthic recruitment of the Cyanobacterium Microcystis (Cyanophyceae). J. Phycol. 2011, 47, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Fulton, R.S.; Paerl, H.W. Effects of colonial morphology on zooplankton utilization of algal resources during blue-green algal (Microcystis aeruginosa) blooms. Limnol. Oceanogr. 1987, 32, 634–644. [Google Scholar] [CrossRef]
- Jarvis, A.C.; Hart, R.C.; Combrink, S. Zooplankton feeding on size fractionated Microcystis colonies and Chlorella in a hypertrophic lake (Hartbeespoort Dam, South Africa): Implications to resource utilization and zooplankton succession. J. Plankton Res. 1987, 9, 1231–1249. [Google Scholar] [CrossRef]
- De Stasio, B.; Schrimpf, M.; Beranek, A.; Daniels, W. Increased Chlorophyll a, phytoplankton abundance, and cyanobacteria occurrence following invasion of Green Bay, Lake Michigan by dreissenid mussels. Aquat. Invasions 2008, 3, 21–27. [Google Scholar] [CrossRef]
- Walsby, A.E. Gas-filled structures providing buoyancy in photosynthetic organisms. Symp. Soc. Exp. Biol. 1972, 26, 233–250. [Google Scholar] [PubMed]
- Walsby, A.E. Structure and function of gas vacuoles. Bacteriol. Rev. 1972, 36, 1–32. [Google Scholar] [PubMed]
- Grant, N.G.; Walsby, A.E. The contribution of photosynthate to turgor pressure rise in the planktonic blue-green alga Anabaena flos-aquae. J. Exp. Bot. 1977, 28, 409–415. [Google Scholar] [CrossRef]
- Iblings, B.W.; Mur, L.R.; Walsby, A.E. Diurnal changes in buoyancy and vertical distribution in populations of Microcystis in two lakes. J. Plankton Res. 1991, 13, 419–436. [Google Scholar] [CrossRef]
- Fogg, G.E.; Walsby, A.E. Buoyancy regulation and the growth of planktonic blue-green algae. Mitteilungen der Internationale Vereinigung für Theoretisch und Angewandte Limnologie 1971, 19, 182–188. [Google Scholar]
- Miller, T.R.; Beversdorf, L.; Chaston, S.D.; McMahon, K.D. Spatiotemporal molecular analysis of cyanobacteria blooms reveals Microcystis-Aphanizomenon interactions. PLoS ONE 2013, 8, e74933. [Google Scholar] [CrossRef] [PubMed]
- Visser, P.M.; Ibelings, B.W.; Veer, B.V.D.; Koedood, J.; Mur, L.R. Artificial mixing prevents nuisance blooms of the cyanobacterium Microcystis in Lake Nieuwe Meer, the Netherlands. Freshw. Biol. 1996, 36, 435–450. [Google Scholar] [CrossRef]
- Bormans, M.; Sherman, B.; Webster, I. Is buoyancy regulation in cyanobacteria an adaptation to exploit separation of light and nutrients? Mar. Freshw. Res. 1999, 50, 897–906. [Google Scholar] [CrossRef]
- Raps, S.; Wyman, K.; Siegelman, H.W.; Falkowski, P.G. Adaptation of the cyanobacterium Microcystis aeruginosa to light intensity. Plant Physiol. 1983, 72, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Dokulil, M.T.; Teubner, K. Cyanobacteria dominance in lakes. Hydrobiologia 2000, 438, 1–12. [Google Scholar] [CrossRef]
- Scheffer, M.; Rinaldi, S.; Gragnani, A.; Mur, L.R.; van Nes, E.H. On the dominance of filamentous cyanobacteria in shallow, turbid lakes. Ecology 1997, 78, 272–282. [Google Scholar] [CrossRef]
- Ernst, A.; Reich, S.; Boger, P. Modification of dinitrogenase reductase in the cyanobacterium Anabaena variabilis due to C starvation and ammonia. J. Bacteriol. 1990, 172, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, M.; Hattori, A. Effect of ammonia on nitrogen fixation by the blue-green alga Anabaena cylindrica. Plant Cell Physiol. 1974, 15, 131–142. [Google Scholar]
- Beversdorf, L.J.; Miller, T.R.; McMahon, K.D. The role of nitrogen fixation in cyanobacterial bloom toxicity in a temperate, eutrophic lake. PLoS ONE 2013, 8, e56103. [Google Scholar] [CrossRef] [PubMed]
- Glibert, P.M.; Bronk, D.A. Release of dissolved organic nitrogen by marine diazotrophic cyanobacteria, Trichodesmium spp. Appl. Environ. Microbiol. 1994, 60, 3996–4000. [Google Scholar] [PubMed]
- Smith, V.H. Low nitrogen to phosphorus ratios favor dominance by blue-green algae in lake phytoplankton. Science 1983, 221, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Kromkamp, J.; van den Heuvel, A.; Mur, L.R. Phosphorus uptake and photosynthesis by phosphate-limited cultures of the cyanobacterium Microcystis aeruginosa. Br. Phycol. J. 1989, 24, 347–355. [Google Scholar] [CrossRef]
- Price, G.D.; Badger, M.R.; Woodger, F.J.; Long, B.M. Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): Functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plants. J. Exp. Bot. 2008, 59, 1441–1461. [Google Scholar] [CrossRef] [PubMed]
- Jannson, C.G. Calcifying cyanobacteria—The potential of biomineralization for carbon capture and storage. Curr. Opin. Biotechnol. 2010, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mackerras, A.H.; de Chazal, N.M.; Smith, G.D. Transient accumulations of cyanophycin in Anabaena cylindrica and Synechocystis 6308. Microbiology 1990, 136, 2057–2065. [Google Scholar] [CrossRef]
- Jacobson, L.; Halmann, M. Polyphosphate metabolism in the blue-green alga Microcystis aeru-ginosa. J. Plankton Res. 1982, 4, 481–488. [Google Scholar] [CrossRef]
- Robarts, R.D.; Zohary, T. Temperature effects on photosynthetic capacity, respiration, and growth rates of bloom-forming cyanobacteria. N. Z. J. Mar. Freshwat. Res. 1987, 21, 391–399. [Google Scholar] [CrossRef]
- Stumpf, R.P.; Wynne, T.T.; Baker, D.B.; Fahnenstiel, G.L. Interannual variability of cyanobacterial blooms in Lake Erie. PLoS ONE 2012, 7, e42444. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.; Brock, T.D. Effect of temperature on blue-green algae (Cyanobacteria) in Lake Mendota. Appl. Environ. Microbiol. 1978, 36, 572–576. [Google Scholar] [PubMed]
- Flores, E.; Herrero, A. Assimilatory nitrogen metabolism and its regulation. In The Molecular Biology of Cyanobacteria; Bryant, D., Ed.; Springer: Dordrecht, The Netherlands, 2004; Volume 1, pp. 487–517. [Google Scholar]
- Paerl, H.W.; Otten, T.G. Duelling ‘CyanoHABs’: Unravelling the environmental drivers controlling dominance and succession among diazotrophic and non-N2-fixing harmful cyanobacteria. Environ. Microbiol. 2016, 18, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Spiller, H.; Latorre, C.; Hassan, M.E.; Shanmugam, K.T. Isolation and characterization of nitrogenase-derepressed mutant strains of cyanobacterium Anabaena variabilis. J. Bacteriol. 1986, 165, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Gobler, C.J.; Davis, T.W.; Coyne, K.J.; Boyer, G.L. Interactive influences of nutrient loading, zooplankton grazing, and microcystin synthetase gene expression on cyanobacterial bloom dynamics in a eutrophic New York lake. Harmful Algae 2007, 6, 119–133. [Google Scholar] [CrossRef]
- Ernst, B.; Hoeger, S.J.; O’Brien, E.; Dietrich, D.R. Abundance and toxicity of Planktothrix rubescens in the pre-alpine Lake Ammersee, Germany. Harmful Algae 2009, 8, 329–342. [Google Scholar] [CrossRef]
- Padisak, J.; Scheffler, W.; Kasprzak, P.; Koschel, R.; Krienitz, L. Interannual variability in the phytoplankton composition of Lake Stechlin (1994–2000). Arch. Hydrobiol. Spec. Issues 2003, 58, 101–133. [Google Scholar]
- Lepisto, L.; Rapala, J.; Lyra, C.; Berg, K.A.; Erkomaa, K.; Johanna, I. Occurrence and toxicity of cyanobacterial blooms dominated by Anabaena lemmermannii P. Richter and Aphanizomenon spp. in boreal lakes in 2003. Algol. Stud. 2005, 117, 315–328. [Google Scholar] [CrossRef]
- Rantala, A.; Rajaniemi-Wacklin, P.; Lyra, C.; Lepistö, L.; Rintala, J.; Mankiewicz-Boczek, J.; Sivonen, K. Detection of microcystin-producing cyanobacteria in Finnish lakes with genus-specific microcystin synthetase gene E (mcyE) PCR and associations with environmental factors. Appl. Environ. Microbiol. 2006, 72, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Kanoshina, I.; Lips, U.; Leppänen, J.-M. The influence of weather conditions (temperature and wind) on cyanobacterial bloom development in the Gulf of Finland (Baltic Sea). Harmful Algae 2003, 2, 29–41. [Google Scholar] [CrossRef]
- Wallace, B.B.; Hamilton, D.P. Simulation of water-bloom formation in the cyanobacterium Microcystis aeruginosa. J. Plankton Res. 2000, 22, 1127–1138. [Google Scholar] [CrossRef]
- Lei, L.; Li, C.; Peng, L.; Han, B.P. Competition between toxic and non-toxic Microcystis aeruginosa and its ecological implication. Ecotoxicology 2015, 24, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, S.; Suda, S.; Li, R.; Matsumoto, S.; Watanabe, M.M. Morphological variability of colonies of Microcystis morphospecies in culture. J. Gen. Appl. Microbiol. 2000, 46, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Nkrumah, P.N.; Peng, Q. Different tolerances to chemical contaminants between unicellular and colonial morph of Microcystis aeruginosa: Excluding the differences among different strains. J. Hazard. Mater. 2015, 285, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Huang, W.M.; Li, D.H.; Liu, Y.D. Effects of iron on growth, pigment content, photosystem II efficiency, and siderophores production of Microcystis aeruginosa and Microcystis wesenbergii. Curr. Microbiol. 2007, 55, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.-H.; Ha, K.; Joo, G.-J.; Takamura, N. Toxin production of cyanobacteria is increased by exposure to zooplankton. Freshw. Biol. 2003, 48, 1540–1550. [Google Scholar] [CrossRef]
- Huisman, J.; Sharples, J.; Stroom, J.M.; Visser, P.M.; Kardinaal, W.E.A.; Verspagen, J.M.H.; Sommeijer, B. Changes in turbulent mixing shift competition for light between phytoplankton species. Ecology 2004, 85, 2960–2970. [Google Scholar] [CrossRef]
- Reynolds, C.S.; Oliver, R.L.; Walsby, A.E. Cyanobacterial dominance: The role of buoyancy regulation in dynamic lake environments. N. Z. J. Mar. Freshw. Res. 1987, 21, 379–390. [Google Scholar] [CrossRef]
- Bridgeman, T.B.; Chaffin, J.D.; Kane, D.D.; Conroy, J.D.; Panek, S.E.; Armenio, P.M. From river to lake: Phosphorus partitioning and algal community compositional changes in Western Lake Erie. J. Gt. Lakes Res. 2012, 38, 90–97. [Google Scholar] [CrossRef]
- Makarewicz, J.C.; Boyer, G.L.; Lewis, T.W.; Guenther, W.; Atkinson, J.; Arnold, M. Spatial and temporal distribution of the cyanotoxin microcystin-LR in the Lake Ontario ecosystem: Coastal embayments, rivers, nearshore and offshore, and upland lakes. J. Gt. Lakes Res. 2009, 35, 83–89. [Google Scholar] [CrossRef]
- Rediske, R.R.; O’Keefe, J.; Rieger, K.; Rediske, J.D. Assessment of E. coli and Microcystins in Cladophora Mats in the Nearshore Waters of Grand Traverse Bay. Available online: https://www.gvsu.edu/wri/envchem/assessment-of-e-coli-and-microcystins-in-cladophora-17.htm (accessed on 28 December 2015).
- Todd, M.; University of Wisconsin-Milwaukee, Milwaukee, WI, USA. Spatial Analysis of microcystins in Green Bay (unpublished work). 2014. [Google Scholar]
- Beversdorf, L.J.; Chaston, S.D.; Miller, T.R.; McMahon, K.D. Microcystin mcyA and mcyE gene abundances are not appropriate indicators of microcystin concentrations in lakes. PLoS ONE 2015, 10, e0125353. [Google Scholar] [CrossRef] [PubMed]
- Hotto, A.M.; Satchwell, M.F.; Berry, D.L.; Gobler, C.J.; Boyer, G.L. Spatial and temporal diversity of microcystins and microcystin-producing genotypes in Oneida Lake, NY. Harmful Algae 2008, 7, 671–681. [Google Scholar] [CrossRef]
- Pawlik-Skowrońska, B.; Pirszel, J.; Kornijów, R. Spatial and temporal variation in microcystin concentrations during perennial bloom of Planktothrix agardhii in a hypertrophic lake. Ann. Limnol. 2008, 44, 145–150. [Google Scholar] [CrossRef]
- Boyer, G.L. Cyanobacterial toxins in New York and the lower Great Lakes ecosystems. In Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs; Springer: New York, NY, USA, 2008; pp. 153–165. [Google Scholar]
- Munawar, M.; Munawar, I.F. A lakewide study of phytoplankton biomass and its species composition in Lake Erie, April–December 1970. J. Fish. Board Can. 1976, 33, 581–600. [Google Scholar] [CrossRef]
- Rinta-Kanto, J.M.; Saxton, M.A.; DeBruyn, J.M.; Smith, J.L.; Marvin, C.H.; Krieger, K.A.; Sayler, G.S.; Boyer, G.L.; Wilhelm, S.W. The diversity and distribution of toxigenic Microcystis spp. in present day and archived pelagic and sediment samples from Lake Erie. Harmful Algae 2008, 8, 385–394. [Google Scholar] [CrossRef]
- Ohio Environmental Protection Agency. Recreational and Beach Monitoring Data. Available online: http://www.epa.ohio.gov/ddagw/hab.aspx (accessed on 28 December 2015).
- Millie, D.; Fahnenstiel, G.; Dyble Bressie, J.; Pigg, R.; Rediske, R.; Klarer, D.; Tester, P.; Litaker, R.W. Late-summer phytoplankton in western Lake Erie (Laurentian Great Lakes): Bloom distributions, toxicity, and environmental influences. Aquat. Ecol. 2009, 43, 915–934. [Google Scholar] [CrossRef]
- Dyble, J.; Fahnenstiel, G.L.; Litaker, R.W.; Millie, D.F.; Tester, P.A. Microcystin concentrations and genetic diversity of Microcystis in the lower Great Lakes. Environ. Toxicol. 2008, 23, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Rea, C.; Yu, Z.; Lee, J. Relative importance of Microcystis abundance and diversity in determining microcystin dynamics in Lake Erie coastal wetland and downstream beach water. J. Appl. Microbiol. 2016, 120, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gruden, C.L.; Bridgeman, T.B.; Chaffin, J.D. Detection and quantification of Microcystis spp. and microcystin-LR in Western Lake Erie during the summer of 2007. Water Sci. Technol. 2009, 60, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Francy, D.S.; Graham, J.L.; Stelzer, E.A.; Ecker, C.D.; Brady, A.M.G.; Struffolino, P.; Loftin, K.A. Water Quality, Cyanobacteria, and Environmental Factors and Their Relations to Microcystin Concentrations for Use in Predictive Models at Ohio Lake Erie and Inland Lake Recreational Sites, 2013–2014; United States Geological Survey: Reston, VA, USA, 2015. [Google Scholar]
- Hitzfeld, B.C.; Hoger, S.J.; Dietrich, D.R. Cyanobacterial toxins: Removal during drinking water treatment, and human risk assessment. Environ. Health Perspect. 2000, 108 (Suppl. 1), 113–122. [Google Scholar] [CrossRef] [PubMed]
- Westrick, J.A.; Szlag, D.C.; Southwell, B.J.; Sinclair, J. A review of cyanobacteria and cyanotoxins removal/inactivation in drinking water treatment. Anal. Bioanal. Chem. 2010, 397, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Hoeger, S.J.; Hitzfeld, B.C.; Dietrich, D.R. Occurrence and elimination of cyanobacterial toxins in drinking water treatment plants. Toxicol. Appl. Pharmacol. 2005, 203, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W.W. Assessment of Blue-Green Algal Toxins in Raw and Finished Drinking Water; AWWA Research Foundation and American Water Works Association: Denver, CO, USA, 2000. [Google Scholar]
- Francis, G. Poisonous Australian lake. Nature 1878, 18, 11–12. [Google Scholar] [CrossRef]
- Fitch, C.P.; Bishop, L.M.; Boyd, W.L. “Water bloom” as a cause of poisoning in domestic animals. J. Ser. Minn. Agric. Exp. Stn. 1934, 1248, 30–39. [Google Scholar]
- Gorham, P.R. Laboratory studies on the toxins produced by waterblooms of blue-green algae. Am. J. Public Health 1962, 52, 2100–2105. [Google Scholar] [CrossRef]
- Hughes, E.O.; Gorham, P.R.; Zehnder, A. Toxicity of unialgal culture of Microcystis aeruginosa. Can. J. Microbiol. 1958, 4, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, C.T.; Mason, M.F. Observations on the pathological changes produced by a toxic substance present in blue-green algae (Microcystis aeruginosa). Am. J. Pathol. 1946, 22, 369–383. [Google Scholar] [PubMed]
- Wheeler, R.E.; Lackey, J.B.; Schott, S. A contribution on the toxicity of algae. Public Health Rep. 1942, 57, 1695–1701. [Google Scholar] [CrossRef]
- Bishop, C.T.; Anet, E.F.L.J.; Gorham, P.R. Isolation and identification of the fast-death factor in Microcystis aeruginosa NRC-1. Can. J. Biochem. Physiol. 1959, 37, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Konst, H.; McKercher, P.D.; Gorham, P.R.; Robertson, A.; Howell, J. Symptoms and pathology produced by toxic Microcystis aeruginosa NRC-1 in laboratory and domestic animals. Can. J. Comp. Med. Vet. Sci. 1965, 29, 221–228. [Google Scholar] [PubMed]
- Botes, D.P.; Kruger, H.; Viljoen, C.C. Isolation and characterization of four toxins from the blue-green alga, Microcystis aeruginosa. Toxicon 1982, 20, 945–954. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Harada, K.; Namikoshi, M.; Chen, C.; Harvis, C.A.; Munro, M.H.G.; Blunt, J.W.; Mulligan, P.E.; Beasley, V.R.; Dahlem, A.M.; et al. Nodularin, microcystin, and the configuration of Adda. J. Am. Chem. Soc. 1988, 110, 8557–8558. [Google Scholar] [CrossRef]
- Carmichael, W.W.; Gorham, P.R. An improved method for obtaining axenic clones of planktonic blue-green algae. J. Phycol. 1974, 10, 238–240. [Google Scholar]
- Dierstein, R.; Kaiser, I.; Weckesser, J. Rapid determination of Microcystis sp. toxins by reversed-phase liquid chromatography. FEMS Microbiol. Lett. 1988, 49, 143–147. [Google Scholar] [CrossRef]
- Harada, K.-I.; Matsuura, K.; Suzuki, M.; Oka, H.; Watanabe, M.F.; Oishi, S.; Dahlem, A.M.; Beasley, V.R.; Carmichael, W.W. Analysis and purification of toxic peptides from cyanobacteria by reversed-phase high-performance liquid chromatography. J. Chromatogr. 1988, 448, 275–283. [Google Scholar] [CrossRef]
- Hooser, S.B.; Beasley, V.R.; Lovell, R.A.; Carmichael, W.W.; Haschek, W.M. Toxicity of microcystin LR, a cyclic heptapeptide hepatotoxin from Microcystis aeruginosa, to rats and mice. Vet. Pathol. 1989, 26, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Runnegar, M.T.; Kong, S.; Berndt, N. Protein phosphatase inhibition and in vivo hepatotoxicity of microcystins. Am. J. Physiol. 1993, 265, G224–G230. [Google Scholar] [PubMed]
- Lovell, R.A.; Schaeffer, D.J.; Hooser, S.B.; Haschek, W.M.; Dahlem, A.M.; Carmichael, W.W.; Beasley, V.R. Toxicity of intraperitoneal doses of microcystin-LR in two strains of male mice. J. Environ. Pathol. Toxicol. Oncol. 1989, 9, 221–237. [Google Scholar] [PubMed]
- Fawell, J.K.; Mitchell, R.E.; Everett, D.J.; Hill, R.E. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Falconer, I.R.; Burch, M.D.; Steffensen, D.A.; Choice, M.; Coverdale, O.R. Toxicity of the blue-green alga (cyanobacterium) Microcystis aeruginosa in drinking water to growing pigs, as an animal model for human injury and risk assessment. Environ. Toxicol. Water Qual. 1994, 9, 131–139. [Google Scholar] [CrossRef]
- Heinze, R. Toxicity of the cyanobacterial toxin microcystin-LR to rats after 28 days intake with the drinking water. Environ. Toxicol. 1999, 14, 57–60. [Google Scholar] [CrossRef]
- Zhou, Y.; Yuan, J.; Wu, J.; Han, X. The toxic effects of microcystin-LR on rat spermatogonia in vitro. Toxicol. Lett. 2012, 212, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xiang, Z.; Li, D.; Han, X. Regulation of microcystin-LR-induced toxicity in mouse spermatogonia by miR-96. Environ. Sci. Technol. 2014, 48, 6383–6390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, L.; Yan, Y. Microcystin-LR impairs zebrafish reproduction by affecting oogenesis and endocrine system. Chemosphere 2015, 120, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xie, P.; Li, G.; Jun, C.; Cai, Y.; Xiong, Q.; Zhao, Y. The proteomic study on cellular responses of the testes of zebrafish (Danio rerio) exposed to microcystin-RR. Proteomics 2012, 12, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Z.; Zhang, F.-Q.; Li, C.-F.; Yi, D.; Fu, X.-L.; Cui, L.-X. A Cyanobacterial toxin, microcystin-LR, induces apoptosis of sertoli cells by changing the expression levels of apoptosis-related proteins. Tohoku J. Exp. Med. 2011, 224, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, C.; Wu, Y.; Ye, B.; Han, L.; Shou, X.; Wang, M.; Wang, J.; Jia, X. Toxic effects of microcystin-LR on the reproductive system of male Rana nigromaculata in vitro. Aquat. Toxicol. 2013, 126, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Kist, L.W.; Rosemberg, D.B.; Pereira, T.C.; de Azevedo, M.B.; Richetti, S.K.; de Castro Leao, J.; Yunes, J.S.; Bonan, C.D.; Bogo, M.R. Microcystin-LR acute exposure increases AChE activity via transcriptional ache activation in zebrafish (Danio rerio) brain. Comp. Biochem. Physiol. C 2012, 155, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yan, W.; Cai, F.; Li, C.; Chen, N.; Wang, J. Spatial learning and memory impairment and pathological change in rats induced by acute exposure to microcystin-LR. Environ. Toxicol. 2014, 29, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Maidana, M.; Carlis, V.; Galhardi, F.G.; Yunes, J.S.; Geracitano, L.A.; Monserrat, J.M.; Barros, D.M. Effects of microcystins over short- and long-term memory and oxidative stress generation in hippocampus of rats. Chem. Biol. Interact. 2006, 159, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, F.; Cai, F.; Yan, W.; Zhou, Q.; Xie, L. Microcystin-LR inhibited hippocampal long-term potential via regulation of the glycogen synthase kinase-3β pathway. Chemosphere 2013, 93, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Ju, J.; Li, Y.; Yin, L.; Pu, Y. Alterations in neurobehaviors and inflammation in hippocampus of rats induced by oral administration of microcystin-LR. Environ. Sci. Pollut. Res. Int. 2014, 21, 12419–12425. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.; Ekambaram, P. Involvement of nitric oxide in learning & memory processes. Indian J. Med. Res. 2011, 133, 471–478. [Google Scholar] [PubMed]
- Li, X.; Zhang, X.; Ju, J.; Li, Y.; Yin, L.; Pu, Y. Maternal repeated oral exposure to microcystin-LR affects neurobehaviors in developing rats. Environ. Toxicol. Chem. 2015, 34, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Li, Y.; Han, X. Decline of sperm quality and testicular function in male mice during chronic low-dose exposure to microcystin-LR. Reprod. Toxicol. 2011, 31, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, X. Microcystin-LR causes cytotoxicity effects in rat testicular Sertoli cells. Environ. Toxicol. Pharmacol. 2012, 33, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Xie, P.; Li, H.; Hao, L.; Li, G.; Qiu, T.; Liu, Y. Involvement of Fas/FasL system in apoptotic signaling in testicular germ cells of male Wistar rats injected i.v. with microcystins. Toxicon 2009, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of Ser/Thr protein phosphatases: Specificity, use and common forms of abuse. Methods Mol. Biol. 2007, 365, 23–38. [Google Scholar] [PubMed]
- Armi, Z.; Turki, S.; Trabelsi, E.; Ceredi, A.; Riccardi, E.; Milandri, A. Occurrence of diarrhetic shellfish poisoning (DSP) toxins in clams (Ruditapes decussatus) from Tunis north lagoon. Environ. Monit. Assess. 2012, 184, 5085–5095. [Google Scholar] [CrossRef] [PubMed]
- van den Top, H.J.; Gerssen, A.; McCarron, P.; van Egmond, H.P. Quantitative determination of marine lipophilic toxins in mussels, oysters and cockles using liquid chromatography-mass spectrometry: Inter-laboratory validation study. Food Addit. Contam. 2011, 28, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.; Pan, J.; Liang, J.; Zhou, Y.; Wu, J. Identification of the okadaic acid-based toxin profile of a marine dinoflagellate strain Prorocentrum lima by LC-MS/MS and NMR spectroscopic data. J. Sep. Sci. 2012, 35, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, S.; Matsushima, R.; Watanabe, M.F.; Harada, K.; Ichihara, A.; Carmichael, W.W.; Fujiki, H. Inhibition of protein phosphatases by microcystins and nodularin associated with hepatotoxicity. J. Cancer Res. Clin. Oncol. 1990, 116, 609–614. [Google Scholar] [CrossRef] [PubMed]
- MacKintosh, R.W.; Dalby, K.N.; Campbell, D.G.; Cohen, P.T.; Cohen, P.; MacKintosh, C. The cyanobacterial toxin microcystin binds covalently to cysteine-273 on protein phosphatase 1. FEBS Lett. 1995, 371, 236–240. [Google Scholar] [PubMed]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev. 2008, 27, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, 229–238. [Google Scholar] [CrossRef]
- Wang, S.S.; Esplin, E.D.; Li, J.L.; Huang, L.; Gazdar, A.; Minna, J.; Evans, G.A. Alterations of the PPP2R1B gene in human lung and colon cancer. Science 1998, 282, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Futamura, M.; Yamaguchi, K.; Aoki, S.; Takahashi, T.; Saji, S. Alterations of the PPP2R1B gene located at 11q23 in human colorectal cancers. Gut 2000, 47, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Loukola, A.; Monni, O.; Kuokkanen, K.; Franssila, K.; Elonen, E.; Vilpo, J.; Joensuu, H.; Kere, J.; Aaltonen, L.; Knuutila, S. PPP2R1B gene in chronic lymphocytic leukemias and mantle cell lymphomas. Leuk. Lymphoma 2001, 41, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Guergnon, J.; Godet, A.N.; Galioot, A.; Falanga, P.B.; Colle, J.H.; Cayla, X.; Garcia, A. PP2A targeting by viral proteins: A widespread biological strategy from DNA/RNA tumor viruses to HIV-1. Biochim. Biophys. Acta 2011, 1812, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, Y. The role of PP2A-associated proteins and signal pathways in microcystin-LR toxicity. Toxicol. Lett. 2015, 236, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Agudo, A.; Canto, K.P.; Chan, P.C.; Chorus, I.; Falconer, I.R.; Fan, A.; Fujiki, H.; Karagas, M. Ingested nitrate and nitrite, and cyanobacterial peptide toxins. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2006. [Google Scholar]
- Falconer, I.R.; Yeung, D.S. Cytoskeletal changes in hepatocytes induced by Microcystis toxins and their relation to hyperphosphorylation of cell proteins. Chem. Biol. Interact. 1992, 81, 181–196. [Google Scholar] [CrossRef]
- Zhou, M.; Tu, W.W.; Xu, J. Mechanisms of microcystin-LR-induced cytoskeletal disruption in animal cells. Toxicon 2015, 101, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, L.; Liu, W.; Qiao, Q.; Wu, K.; Wen, J.; Huang, C.; Tang, R.; Zhang, X. Involvement of oxidative stress and cytoskeletal disruption in microcystin-induced apoptosis in CIK cells. Aquat. Toxicol. 2015, 165, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Come, C.; Laine, A.; Chanrion, M.; Edgren, H.; Mattila, E.; Liu, X.; Jonkers, J.; Ivaska, J.; Isola, J.; Darbon, J.M.; et al. CIP2A is associated with human breast cancer aggressivity. Clin. Cancer. Res. 2009, 15, 5092–5100. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; Copland, M.; Slupsky, J.R.; Clark, R.E. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood 2011, 117, 6660–6668. [Google Scholar] [CrossRef] [PubMed]
- Rincon, R.; Cristobal, I.; Zazo, S.; Arpi, O.; Menendez, S.; Manso, R.; Lluch, A.; Eroles, P.; Rovira, A.; Albanell, J.; et al. PP2A inhibition determines poor outcome and doxorubicin resistance in early breast cancer and its activation shows promising therapeutic effects. Oncotarget 2015, 6, 4299–4314. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Puustinen, P.; Niemela, M.; Ahola, R.; Arnold, H.; Bottzauw, T.; Ala-aho, R.; Nielsen, C.; Ivaska, J.; Taya, Y.; et al. CIP2A inhibits PP2A in human malignancies. Cell 2007, 130, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.M. Microcystin-LR and okadaic acid-induced cellular effects: a dualistic response. FEBS Lett. 2004, 557, 1–8. [Google Scholar] [CrossRef]
- Fischer, W.J.; Altheimer, S.; Cattori, V.; Meier, P.J.; Dietrich, D.R.; Hagenbuch, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol. Appl. Pharmacol. 2005, 203, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.T.; Mudge, L.M. Protein phosphatase activity in cyanobacteria: Consequences for microcystin toxicity analysis. Toxicon 1993, 31, 1179–1186. [Google Scholar] [CrossRef]
- Feurstein, D.; Holst, K.; Fischer, A.; Dietrich, D.R. Oatp-associated uptake and toxicity of microcystins in primary murine whole brain cells. Toxicol. Appl. Pharmacol. 2009, 234, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, T.; Kagalou, I.; Bacopoulos, V.; Leonardos, I.D. Accumulation of microcystins in water and fish tissues: An estimation of risks associated with microcystins in most of the Greek Lakes. Environ. Toxicol. 2010, 25, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Meriluoto, J.A.; Nygard, S.E.; Dahlem, A.M.; Eriksson, J.E. Synthesis, organotropism and hepatocellular uptake of two tritium-labeled epimers of dihydromicrocystin-LR, a cyanobacterial peptide toxin analog. Toxicon 1990, 28, 1439–1446. [Google Scholar] [CrossRef]
- Fischer, W.J.; Dietrich, D.R. Pathological and biochemical characterization of microcystin-induced hepatopancreas and kidney damage in carp (Cyprinus carpio). Toxicol. Appl. Pharmacol. 2000, 164, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Cazenave, J.; Wunderlin, D.A.; de Los Angeles Bistoni, M.; Ame, M.V.; Krause, E.; Pflugmacher, S.; Wiegand, C. Uptake, tissue distribution and accumulation of microcystin-RR in Corydoras paleatus, Jenynsia multidentata and Odontesthes bonariensis. A field and laboratory study. Aquat. Toxicol. 2005, 75, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Pouria, S.; de Andrade, A.; Barbosa, J.; Cavalcanti, R.L.; Barreto, V.T.; Ward, C.J.; Preiser, W.; Poon, G.K.; Neild, G.H.; Codd, G.A. Fatal microcystin intoxication in haemodialysis unit in Caruaru, Brazil. Lancet 1998, 352, 21–26. [Google Scholar] [CrossRef]
- Meier-Abt, F.; Hammann-Hanni, A.; Stieger, B.; Ballatori, N.; Boyer, J.L. The organic anion transport polypeptide 1D1 (OATP1D1) mediates hepatocellular uptake of phalloidin and microcystin into skate liver. Toxicol. Appl. Pharmacol. 2007, 218, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar] [PubMed]
- Ose, A.; Kusuhara, H.; Endo, C.; Tohyama, K.; Miyajima, M.; Kitamura, S.; Sugiyama, Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab. Dispos. 2010, 38, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yuan, J.; Li, Z.; Wang, Z.; Cheng, D.; Du, Y.; Li, W.; Kan, Q.; Zhang, W. Genetic polymorphisms and function of the organic anion-transporting polypeptide 1A2 and its clinical relevance in drug disposition. Pharmacology 2015, 95, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lee, A.C.; Krafczyk, K.; Zhu, L.; Murray, M. Protein kinase C regulates the internalization and function of the human organic anion transporting polypeptide 1A2. Br. J. Pharmacol. 2011, 162, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.M.; Scherkenbach, L.A.; Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics 2009, 10, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, H.; Bailey, D.G.; Dresser, G.K.; Gregor, J.C.; Schwarz, U.I.; McGrath, J.S.; Jolicoeur, E.; Lee, W.; Leake, B.F.; Tirona, R.G.; et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin. Pharmacol. Ther. 2007, 81, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Zeller, P.; Clement, M.; Fessard, V. Similar uptake profiles of microcystin-LR and -RR in an in vitro human intestinal model. Toxicology 2011, 290, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B.; Gui, C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica 2008, 38, 778–801. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Backman, J.T.; Kajosaari, L.I.; Leathart, J.B.; Neuvonen, M.; Daly, A.K.; Eichelbaum, M.; Kivisto, K.T.; Neuvonen, P.J. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin. Pharmacol. Ther. 2005, 77, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Kivisto, K.T.; Diczfalusy, U.; Bodin, K.; Bertilsson, L.; Fromm, M.F.; Eichelbaum, M. Effect of SLCO1B1 polymorphism on induction of CYP3A4 by rifampicin. Pharmacogenet. Genom. 2006, 16, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Takane, H.; Miyata, M.; Burioka, N.; Kurai, J.; Fukuoka, Y.; Suyama, H.; Shigeoka, Y.; Otsubo, K.; Ieiri, I.; Shimizu, E. Severe toxicities after irinotecan-based chemotherapy in a patient with lung cancer: A homozygote for the SLCO1B1*15 allele. Ther. Drug Monit. 2007, 29, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Iwai, M.; Suzuki, H.; Ieiri, I.; Otsubo, K.; Sugiyama, Y. Functional analysis of single nucleotide polymorphisms of hepatic organic anion transporter OATP1B1 (OATP-C). Pharmacogenetics 2004, 14, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Saito, S.; Sekine, A.; Mishima, C.; Kondo, K.; Kitamura, Y.; Harigae, S.; Osawa, S.; Nakamura, Y. Catalog of 258 single-nucleotide polymorphisms (SNPs) in genes encoding three organic anion transporters, three organic anion-transporting polypeptides, and three NADH: Ubiquinone oxidoreductase flavoproteins. J. Hum. Genet. 2001, 46, 668–683. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gorham, P.R. Toxic Waterblooms of blue-green algae. Can. Vet. J. 1960, 1, 235–245. [Google Scholar] [PubMed]
- Gorham, P.R. Toxic algae as a public health hazard. J. Am. Water Works Assoc. 1964, 56, 1481–1488. [Google Scholar]
- Carmichael, W.W.; Biggs, D.F.; Gorham, P.R. Toxicology and pharmacological action of Anabaena flos-aquae toxin. Science 1975, 187, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Aronstam, R.S.; Witkop, B. Anatoxin-a interactions with cholinergic synaptic molecules. Proc. Natl. Acad. Sci. USA 1981, 78, 4639–4643. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.S. The crystal structure and absolute configuration of 2,9-diacetyl-9-azabicyclo[4,2,1]non-2,3-ene. Acta Crystallogr. Sect. B 1972, B28, 2577. [Google Scholar] [CrossRef]
- Gampbell, H.F.; Edwards, O.E.; Kolt, R. Synthesis of nor-anatoxin-a and anatoxin-a. Can. J. Chem. 1977, 55, 1372–1379. [Google Scholar] [CrossRef]
- Sharples, C.G.; Kaiser, S.; Soliakov, L.; Marks, M.J.; Collins, A.C.; Washburn, M.; Wright, E.; Spencer, J.A.; Gallagher, T.; Whiteaker, P.; et al. UB-165: A novel nicotinic agonist with subtype selectivity implicates the α4β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J. Neurosci. 2000, 20, 2783–2791. [Google Scholar] [PubMed]
- Biggs, D.F.; Dryden, W.F. Action of anatoxin 1 at the neuromuscular junction. Proc. West. Pharmacol. Soc. 1977, 20, 461. [Google Scholar] [PubMed]
- Adeyemo, O.M.; Siren, A.L. Cardio-respiratory changes and mortality in the conscious rat induced by (+)- and (+/−)-anatoxin-a. Toxicon 1992, 30, 899–905. [Google Scholar] [CrossRef]
- Kofuji, P.; Aracava, Y.; Swanson, K.L.; Aronstam, R.S.; Rapoport, H.; Albuquerque, E.X. Activation and blockade of the acetylcholine receptor-ion channel by the agonists (+)-anatoxin-a, the N-methyl derivative and the enantiomer. J. Pharmacol. Exp. Ther. 1990, 252, 517–525. [Google Scholar] [PubMed]
- Fawell, J.K.; Mitchell, R.E.; Hill, R.E.; Everett, D.J. The toxicity of cyanobacterial toxins in the mouse: II anatoxin-a. Hum. Exp. Toxicol. 1999, 18, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.K.; Krieger, R.I. Effect of route of exposure and repeated doses on the acute toxicity in mice of the cyanobacterial nicotinic alkaloid anatoxin-a. Toxicon 1991, 29, 134–138. [Google Scholar] [CrossRef]
- Fitzgeorge, R.B.; Clark, S.A.; Keevil, C.W. Routes of intoxication. In Detection Methods for Cyanobacterial Toxins; Codd, G.A., Jefferies, T.M., Keevil, C.W., Potter, E., Eds.; The Royal Society of Chemistry: London, UK, 1994; pp. 69–74. [Google Scholar]
- Astrachan, N.B.; Archer, B.G.; Hilbelink, D.R. Evaluation of the subacute toxicity and teratogenicity of anatoxin-a. Toxicon 1980, 18, 684–688. [Google Scholar] [CrossRef]
- Wonnacott, S.; Swanson, K.L.; Albuquerque, E.X.; Huby, N.J.; Thompson, P.; Gallagher, T. Homoanatoxin: A potent analogue of anatoxin-A. Biochem. Pharmacol. 1992, 43, 419–423. [Google Scholar] [CrossRef]
- Skulberg, O.M.; Carmichael, W.W.; Andersen, R.A.; Mtsunaga, S.; Moore, R.E.; Skulberg, R. Investigations of a neurotoxic oscillatorialean strain (cyanophyceae) and its toxin. Isolation and characterization of homoanatoxin-a. Environ. Toxicol. Chem. 1992, 11, 321–329. [Google Scholar] [CrossRef]
- Moustaka-Gouni, M.; Kormas, K.A.; Vardaka, E.; Katsiapi, M.; Gkelis, S. Raphidiopsis mediterranea Skuja represents non-heterocytous life-cycle stages of Cylindrospermopsis raciborskii (Woloszynska) Seenayya et Subba Raju in Lake Kastoria (Greece), its type locality: Evidence by morphological and phylogenetic analysis. Harmful Algae 2009, 8, 864–872. [Google Scholar] [CrossRef]
- Namikoshi, M.; Murakami, T.; Fujiwara, T.; Nagai, H.; Niki, T.; Harigaya, E.; Watanabe, M.F.; Oda, T.; Yamada, J.; Tsujimura, S. Biosynthesis and transformation of homoanatoxin-a in the cyanobacterium Raphidiopsis mediterranea Skuja and structures of three new homologues. Chem. Res. Toxicol. 2004, 17, 1692–1696. [Google Scholar] [CrossRef] [PubMed]
- Araoz, R.; Molgo, J.; Tandeau de Marsac, N. Neurotoxic cyanobacterial toxins. Toxicon 2010, 56, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.A.; Carmichael, W.W. The pharmacology of anatoxin-a(s), a neurotoxin produced by the freshwater cyanobacterium Anabaena flos-aquae NRC 525-17. Toxicon 1986, 24, 425–434. [Google Scholar] [CrossRef]
- Mahmood, N.A.; Carmichael, W.W. Anatoxin-a(s), an anticholinesterase from the cyanobacterium Anabaena flos-aquae NRC-525-17. Toxicon 1987, 25, 1221–1227. [Google Scholar] [CrossRef]
- Cook, W.O.; Iwamoto, G.A.; Schaeffer, D.J.; Beasley, V.R. Effect of anatoxin-a(s) from Anabaena flos-aquae NRC-525-17 on blood pressure, heart rate, respiratory rate, tidal volume, minute volume, and phrenic nerve activity in rats. J. Environ. Pathol. Toxicol. Oncol. 1989, 9, 393–400. [Google Scholar] [PubMed]
- Blumberg, S.; Silman, I. Inactivation of electric eel acetylcholinesterase by acylation with N-hydroxysuccinimide esters of amino acid derivatives. Biochemistry 1978, 17, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.A.; Carmichael, W.W.; Pfahler, D. Anticholinesterase poisonings in dogs from a cyanobacterial (blue-green algae) bloom dominated by Anabaena flos-aquae. Am. J. Vet. Res. 1988, 49, 500–503. [Google Scholar] [PubMed]
- Cook, W.O.; Iwamoto, G.A.; Schaeffer, D.J.; Carmichael, W.W.; Beasley, V.R. Pathophysiologic effects of anatoxin-a(s) in anaesthetized rats: The influence of atropine and artificial respiration. Pharmacol. Toxicol. 1990, 67, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Rymuszka, A.; Sieroslawska, A. Effects of neurotoxin—Anatoxin-a on common carp (Cyprinus carpio L.) innate immune cells in vitro. Neuro Endocrinol. Lett. 2011, 32, 84–88. [Google Scholar] [PubMed]
- Astrachan, N.B.; Archer, B.G. Simplified monitoring of anatoxin-a by reverse-phase high performance liquid chromatography and the sub-acute effects of anatoxin-a in rats. In The Water Environment; Carmichael, W., Ed.; Springer: New York, NY, USA, 1981; pp. 437–446. [Google Scholar]
- Carmichael, W.W.; Azevedo, S.M.; An, J.S.; Molica, R.J.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Byth, S. Palm Island mystery disease. Med. J. Aust. 1980, 2, 40–42. [Google Scholar] [PubMed]
- Griffiths, D.J.; Saker, M.L. The Palm Island mystery disease 20 years on: A review of research on the cyanotoxin cylindrospermopsin. Environ. Toxicol. 2003, 18, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.R.; Griffiths, D.J. Chemical and Biological Monitoring of Water Quality at Solomon Dam, Palm Island: Final Report; Department of Botany, School of Biological Sciences, James Cook University of North Queensland: Townsville, Australia, 1983. [Google Scholar]
- Hawkins, P.R.; Runnegar, M.T.; Jackson, A.R.; Falconer, I.R. Severe hepatotoxicity caused by the tropical cyanobacterium (blue-green alga) Cylindrospermopsis raciborskii (Woloszynska) Seenaya and Subba Raju isolated from a domestic water supply reservoir. Appl. Environ. Microbiol. 1985, 50, 1292–1295. [Google Scholar] [PubMed]
- Ohtani, I.; Moore, R.E.; Runnegar, M.T.C. Cylindrospermopsin: A potent hepatotoxin from the blue-green alga Cylindrospermopsis raciborskii. J. Am. Chem. Soc. 1992, 114, 7941–7942. [Google Scholar] [CrossRef]
- Harada, K.I.; Ohtani, I.; Iwamoto, K.; Suzuki, M.; Watanabe, M.F.; Watanabe, M.; Terao, K. Isolation of cylindrospermopsin from a cyanobacterium Umezakia natans and its screening method. Toxicon 1994, 32, 73–84. [Google Scholar] [CrossRef]
- Neumann, C.; Bain, P.; Shaw, G. Studies of the comparative in vitro toxicology of the cyanobacterial metabolite deoxycylindrospermopsin. J. Toxicol. Environ. Health A 2007, 70, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Banker, R.; Teltsch, B.; Sukenik, A.; Carmeli, S. 7-Epicylindrospermopsin, a toxic minor metabolite of the cyanobacterium Aphanizomenon ovalisporum from lake Kinneret, Israel. J. Nat. Prod. 2000, 63, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Looper, R.E.; Williams, R.M. A concise asymmetric synthesis of the marine hepatotoxin 7-epicylindrospermopsin. Angew. Chem. Int. Ed. 2004, 43, 2930–2933. [Google Scholar] [CrossRef] [PubMed]
- Terao, K.; Ohmori, S.; Igarashi, K.; Ohtani, I.; Watanabe, M.F.; Harada, K.I.; Ito, E.; Watanabe, M. Electron microscopic studies on experimental poisoning in mice induced by cylindrospermopsin isolated from blue-green alga Umezakia natans. Toxicon 1994, 32, 833–843. [Google Scholar] [CrossRef]
- Runnegar, M.T.; Kong, S.M.; Zhong, Y.Z.; Ge, J.L.; Lu, S.C. The role of glutathione in the toxicity of a novel cyanobacterial alkaloid cylindrospermopsin in cultured rat hepatocytes. Biochem. Biophys. Res. Commun. 1994, 201, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.L.; Seawright, A.A.; Shaw, G.R.; Senogles, P.; Eaglesham, G.K.; Smith, M.J.; Chiswell, R.K.; Moore, M.R. Hepatic xenobiotic metabolism of cylindrospermopsin in vivo in the mouse. Toxicon 2002, 40, 471–476. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Humpage, A.R.; Falconer, I.R. Oral toxicity of the cyanobacterial toxin cylindrospermopsin in male Swiss albino mice: Determination of no observed adverse effect level for deriving a drinking water guideline value. Environ. Toxicol. 2003, 18, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Froscio, S.M.; Humpage, A.R.; Wickramasinghe, W.; Shaw, G.; Falconer, I.R. Interaction of the cyanobacterial toxin cylindrospermopsin with the eukaryotic protein synthesis system. Toxicon 2008, 51, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Falconer, I.R.; Humpage, A.R. Preliminary evidence for in vivo tumour initiation by oral administration of extracts of the blue-green alga cylindrospermopsis raciborskii containing the toxin cylindrospermopsin. Environ. Toxicol. 2001, 16, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Humpage, A.R.; Fenech, M.; Thomas, P.; Falconer, I.R. Micronucleus induction and chromosome loss in transformed human white cells indicate clastogenic and aneugenic action of the cyanobacterial toxin, cylindrospermopsin. Mutat. Res. 2000, 472, 155–161. [Google Scholar] [CrossRef]
- Straser, A.; Filipic, M.; Zegura, B. Genotoxic effects of the cyanobacterial hepatotoxin cylindrospermopsin in the HepG2 cell line. Arch. Toxicol. 2011, 85, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Broude, E.V.; Demidenko, Z.N.; Vivo, C.; Swift, M.E.; Davis, B.M.; Blagosklonny, M.V.; Roninson, I.B. p21 (CDKN1A) is a negative regulator of p53 stability. Cell Cycle 2007, 6, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Colman, M.S.; Afshari, C.A.; Barrett, J.C. Regulation of p53 stability and activity in response to genotoxic stress. Mutat. Res. 2000, 462, 179–188. [Google Scholar] [CrossRef]
- Humpage, A.R.; Falconer, I.R. Oral Toxicity of Cylindrospermopsin: No Observed Adverse Effect Level Determination in Male Swiss Albino Mice; Cooperative Research Centre for Water Quality and Treatment: Salisbury, Australia, 2002. [Google Scholar]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fingerman, M.; Forester, R.H.; Stover, J.H., Jr. Action of shellfish poison on peripheral nerve and skeletal muscle. Proc. Soc. Exp. Biol. Med. 1953, 84, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.H. Paralytic effects of “paralytic shellfish poison” on frog nerve and muscle. Br. J. Pharmacol. Chemother. 1964, 22, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Nishiyama, A. Actions of saxitoxin on peripheral neuromuscular systems. J. Physiol. 1965, 180, 50–66. [Google Scholar] [PubMed]
- Wang, J.; Salata, J.J.; Bennett, P.B. Saxitoxin is a gating modifier of HERG K+ channels. J. Gen. Physiol. 2003, 121, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Sheets, M.; Ishida, H.; Li, F.; Barry, W.H. Saxitoxin blocks L-type ICa. J. Pharmacol. Exp. Ther. 2004, 308, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef] [PubMed]
- Sommer, H.; Meyer, K. Mussel poisoning. Calif. West. Med. 1935, 42, 423–426. [Google Scholar]
- Sommer, H.; Whedon, W.; Kofoid, C.; Stohler, R. Relation of paralytic shellfish poison to certain plankton organisms of the genus gonyaulax. Arch. Pathol. 1937, 24, 537–559. [Google Scholar]
- Stüken, A.; Orr, R.J.S.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of Nuclear-Encoded Genes for the Neurotoxin Saxitoxin in Dinoflagellates. PLoS ONE 2011, 6, e20096. [Google Scholar] [CrossRef] [PubMed]
- Al-Tebrineh, J.; Mihali, T.K.; Pomati, F.; Neilan, B.A. Detection of saxitoxin-producing cyanobacteria and Anabaena circinalis in environmental water blooms by quantitative PCR. Appl. Environ. Microbiol. 2010, 76, 7836–7842. [Google Scholar] [CrossRef] [PubMed]
- Deeds, J.R.; Landsberg, J.H.; Etheridge, S.M.; Pitcher, G.C.; Longan, S.W. Non-traditional vectors for paralytic shellfish poisoning. Mar. Drugs 2008, 6, 308–348. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, L.E.; Moczydlowski, E.G. Characterization of saxitoxin binding to saxiphilin, a relative of the transferrin family that displays pH-dependent ligand binding. Biochemistry 1994, 33, 12312–12322. [Google Scholar] [CrossRef] [PubMed]
- Yotsu-Yamashita, M.; Okoshi, N.; Watanabe, K.; Araki, N.; Yamaki, H.; Shoji, Y.; Terakawa, T. Localization of pufferfish saxitoxin and tetrodotoxin binding protein (PSTBP) in the tissues of the pufferfish, Takifugu pardalis, analyzed by immunohistochemical staining. Toxicon 2013, 72, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Noguchia, T.; Ebesub, J.S.M. Puffer poisoning: Epidemiology and treatment. J. Toxicol. Toxin Rev. 2001, 20, 1–10. [Google Scholar] [CrossRef]
- Wiberg, G.; Stephenson, N. Toxicologic studies on paralytic shellfish poison. Toxicol. Appl. Pharmacol. 1960, 2, 607–615. [Google Scholar] [CrossRef]
- Alexander, J.; Benford, D.; Cockburn, A.; Cravedi, J.-P.; Dogliotti, E.; Domenico, A.D.; Fernández-Cruz, M.L.; Fink-Gremmels, J.; Fürst, P.; Galli, C.; et al. Marine biotoxins in shellfish—Saxitoxin group. EFSA J. 2009, 1019, 1–76. [Google Scholar]
- Smith, E.A.; Grant, F.; Ferguson, C.M.; Gallacher, S. Biotransformations of paralytic shellfish toxins by bacteria isolated from bivalve molluscs. Appl. Environ. Microbiol. 2001, 67, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Kotaki, Y.; Oshima, Y.; Yasumoto, T. Bacterial transformation of paralytic shellfish toxins in coral reef crabs and a marine snail. Nippon Suisan Gakkaishi 1985, 51, 1009–1013. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Li, D.; Shen, Y.; Li, G.; Liu, Y. First report of aphantoxins in China—Waterblooms of toxigenic Aphanizomenon flos-aquae in Lake Dianchi. Ecotoxicol. Environ. Saf. 2006, 65, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.W.; Bullerjahn, G.S.; Tuttle, T.; McKay, R.M.; Watson, S.B. Effects of increasing nitrogen and phosphorus concentrations on phytoplankton community growth and toxicity during Planktothrix Blooms in Sandusky Bay, Lake Erie. Environ. Sci. Technol. 2015, 49, 7197–7207. [Google Scholar] [CrossRef] [PubMed]
- Chorus, I. Current Approaches to Cyanotoxin Risk Assessment, Risk Management and Regulations in Different Countries; Federal Environment Agency (Umweltbundesamt): Dessau-Roßlau, Germany, 2012. [Google Scholar]
- Stewart, I.; Webb, P.M.; Schluter, P.J.; Shaw, G.R. Recreational and occupational field exposure to freshwater cyanobacteria—A review of anecdotal and case reports, epidemiological studies and the challenges for epidemiologic assessment. Environ. Health 2006, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Hilborn, E.D.; Roberts, V.A.; Backer, L.; DeConno, E.; Egan, J.S.; Hyde, J.B.; Nicholas, D.C.; Wiegert, E.J.; Billing, L.M.; DiOrio, M.; et al. Algal bloom-associated disease outbreaks among users of freshwater lakes—United States, 2009–2010. Morb. Mortal. Wkly. Rep. 2014, 63, 11–15. [Google Scholar]
- Weirich, C.A.; Miller, T.R. Freshwater harmful algal blooms: Toxins and children’s health. Curr. Probl. Pediatr. Adolesc. Health Care 2014, 44, 2–24. [Google Scholar] [CrossRef] [PubMed]
- D’Anglada, L.V.; Donohue, J.M.; Strong, J. Health Effects Support Document for the Cyanobacterial Toxin Microcystins. Available online: https://www.epa.gov/nutrient-policy-data/health-effects-support-documents (accessed on 28 December 2015).
- D’Anglada, L.V.; Donohue, J.M.; Strong, J. Health Effects Support Document for the Cyanobacterial Toxin Cylindrospermopsin. Available online: https://www.epa.gov/nutrient-policy-data/health-effects-support-documents (accessed on 28 December 2015).
- D’Anglada, L.V.; Donohue, J.M.; Strong, J. Health Effects Support Document for the Cyanobacterial Toxin Anatoxin-a. Available online: https://www.epa.gov/nutrient-policy-data/health-effects-support-documents (accessed on 28 December 2015).
- D’Anglada, L.V.; Strong, J. Drinking Water Health Advisory for the Cyanobacterial Microcystin Toxins. Available online: https://www.epa.gov/nutrient-policy-data/drinking-water-health-advisory-documents-cyanobacterial-toxins (accessed on 28 December 2015).
- Kahn, H.D.; Stralka, K. Estimated daily average per capita water ingestion by child and adult age categories based on USDA’s 1994–1996 and 1998 continuing survey of food intakes by individuals. J. Expos. Sci. Environ. Epidemiol. 2008, 19, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey Data. Available online: https://www.cdc.gov/nchs/nhanes/ (accessed on 28 December 2015).
- Zhang, X.; Xie, P.; Wang, W.; Li, D.; Li, L.; Tang, R.; Lei, H.; Shi, Z. Dose-dependent effects of extracted microcystins on embryonic development, larval growth and histopathological changes of southern catfish (Silurus meridionalis). Toxicon 2008, 51, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ying, F.; Chen, Y.; Han, X. Microcystin (-LR) affects hormones level of male mice by damaging hypothalamic-pituitary system. Toxicon 2012, 59, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Stern, S.; Soderholm, S.C.; Cox, C.; Sharma, A.; Inglis, G.B.; Preston, R.; Balys, M.; Reuhl, K.R.; Gelein, R. Developmental neurotoxicity of methanol exposure by inhalation in rats. Res. Rep. 1996, 73, 1–64. [Google Scholar]
- Murray, T.G.; Burton, T.C.; Rajani, C.; Lewandowski, M.F.; Burke, J.M.; Eells, J.T. Methanol poisoning. A rodent model with structural and functional evidence for retinal involvement. Arch. Ophthalmol. 1991, 109, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Kraut, J.A.; Kurtz, I. Toxic Alcohol Ingestions: Clinical Features, Diagnosis, and Management. Clin. J. Am. Soc. Nephrol. 2008, 3, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Ufelmann, H.; Krüger, T.; Luckas, B.; Schrenk, D. Human and rat hepatocyte toxicity and protein phosphatase 1 and 2A inhibitory activity of naturally occurring desmethyl-microcystins and nodularins. Toxicology 2012, 293, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, I.; Jokela, J.; Fewer, D.P.; Wahlsten, M.; Rikkinen, J.; Sivonen, K. Discovery of rare and highly toxic microcystins from lichen-associated cyanobacterium Nostoc sp. strain IO-102-I. Appl. Environ. Microbiol. 2004, 70, 5756–5763. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Sano, T.; Kubota, R.; Kobayashi, N.; Tahara, M.; Obama, T.; Sugimoto, N.; Nishimura, T.; Ikarashi, Y. Effects of the amino acid constituents of microcystin variants on cytotoxicity to primary cultured rat hepatocytes. Toxins 2013, 6, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Pant, S.C.; Vijayaraghavan, R.; Rao, P.V.L. Comparative toxicity evaluation of cyanobacterial cyclic peptide toxin microcystin variants (LR, RR, YR) in mice. Toxicology 2003, 188, 285–296. [Google Scholar] [CrossRef]
- Niedermeyer, T.H.J.; Daily, A.; Swiatecka-Hagenbruch, M.; Moscow, J.A. Selectivity and potency of microcystin congeners against OATP1B1 and OATP1B3 expressing cancer cells. PLoS ONE 2014, 9, e91476. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O.; Sandvik, M.; Nonga, H.E.; Rundberget, T.; Wilkins, A.L.; Rise, F.; Ballot, A. Thiol derivatization for LC-MS identification of microcystins in complex matrices. Environ. Sci. Technol. 2012, 46, 8937–8944. [Google Scholar] [CrossRef] [PubMed]
- Burch, M.D. Effective doses, guidelines & regulations. Adv. Exp. Med. Biol. 2008, 619, 831–853. [Google Scholar] [PubMed]
- D’Anglada, L.V.; Strong, J. Drinking Water Health Advisory for the Cyanobacterial Toxin Cylindrospermopsin. Available online: https://www.epa.gov/nutrient-policy-data/drinking-water-health-advisory-documents-cyanobacterial-toxins (accessed on 28 December 2015).
- Munday, R.; Reeve, J. Risk assessment of shellfish toxins. Toxins 2013, 5, 2109–2137. [Google Scholar] [CrossRef] [PubMed]
- Mons, M.N.; Egmond, H.P.V.; Speijers, G.J.A. Paralytic Shelffish Poisoning: A Review; National Institute Public Health and Environment: Bilthoven, The Netherlands, 1998. [Google Scholar]
- Egmond, H.P.; Apeldoorn, M.E.V.; Speijers, G.J.A. Marine Biotoxins. Food Agric. Organ. U. N. 2004, 80, 1–278. [Google Scholar]
- Garcı́a, C.; del Carmen Bravo, M.A.; Lagos, M.; Lagos, N. Paralytic shellfish poisoning: Post-mortem analysis of tissue and body fluid samples from human victims in the Patagonia fjords. Toxicon 2004, 43, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Gessner, B.D.; Bell, P.; Doucette, G.J.; Moczydlowski, E.; Poli, M.A.; Van Dolah, F.; Hall, S. Hypertension and identification of toxin in human urine and serum following a cluster of mussel-associated paralytic shellfish poisoning outbreaks. Toxicon 1997, 35, 711–722. [Google Scholar] [CrossRef]
- Schantz, E.J. Chemistry and biology of saxitoxin and related toxins. Ann. N. Y. Acad. Sci. 1986, 479, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K. Food poisoning. N. Engl. J. Med. 1953, 249, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Tennant, A.D.; Naubert, J.; Corbeil, H.E. An outbreak of paralytic shellfish poisoning. Can. Med. Assoc. J. 1955, 72, 436–439. [Google Scholar] [PubMed]
- Beversdorf, L.J.; Weirich, C.A.; Bartlett, S.L.; Miller, T.R. Variable cyanobacterial toxin and metabolite profiles across six eutrophic lakes of differing physiochemical characteristics. Toxins 2017, 9, 62. [Google Scholar] [CrossRef] [PubMed]
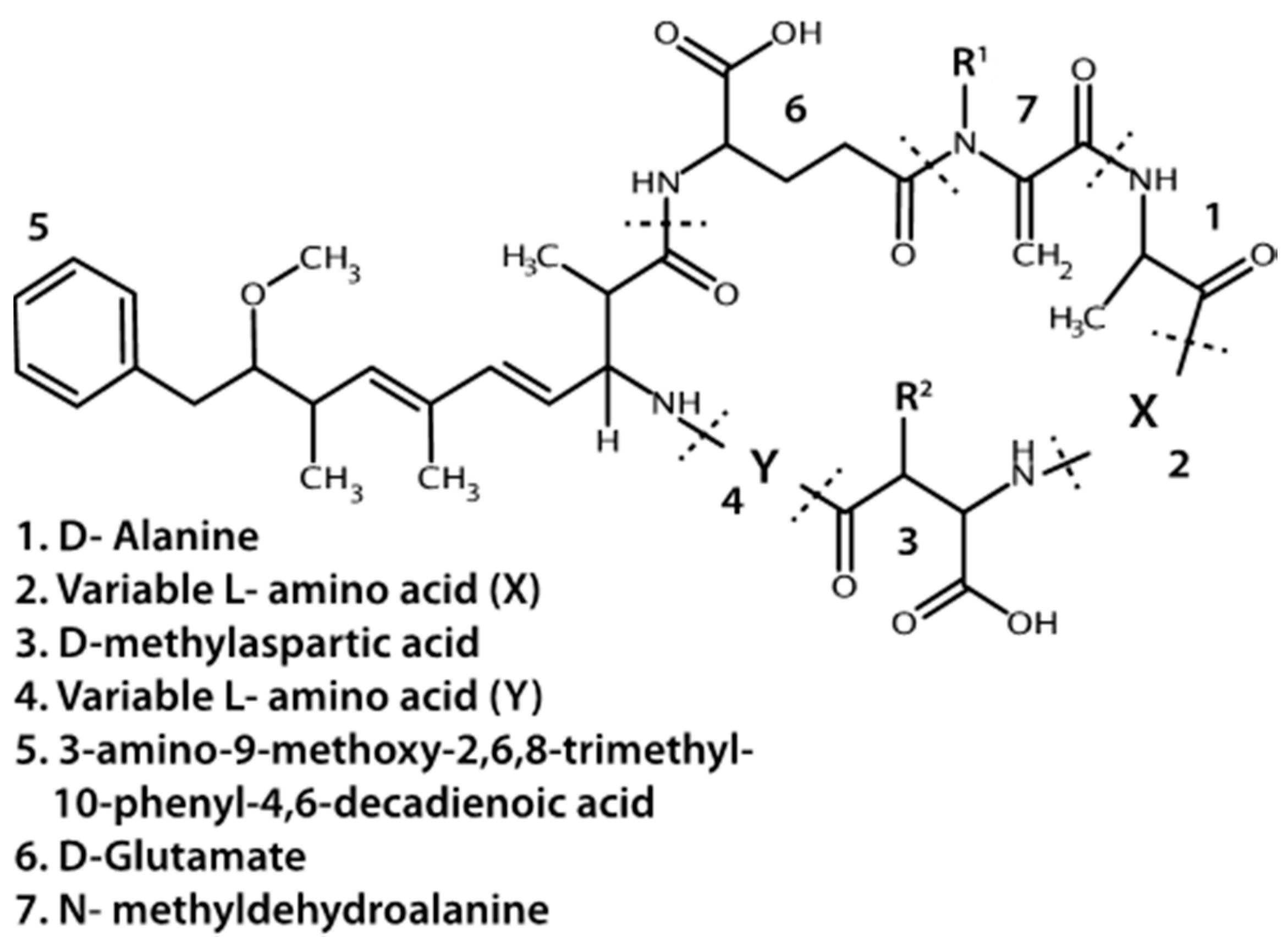
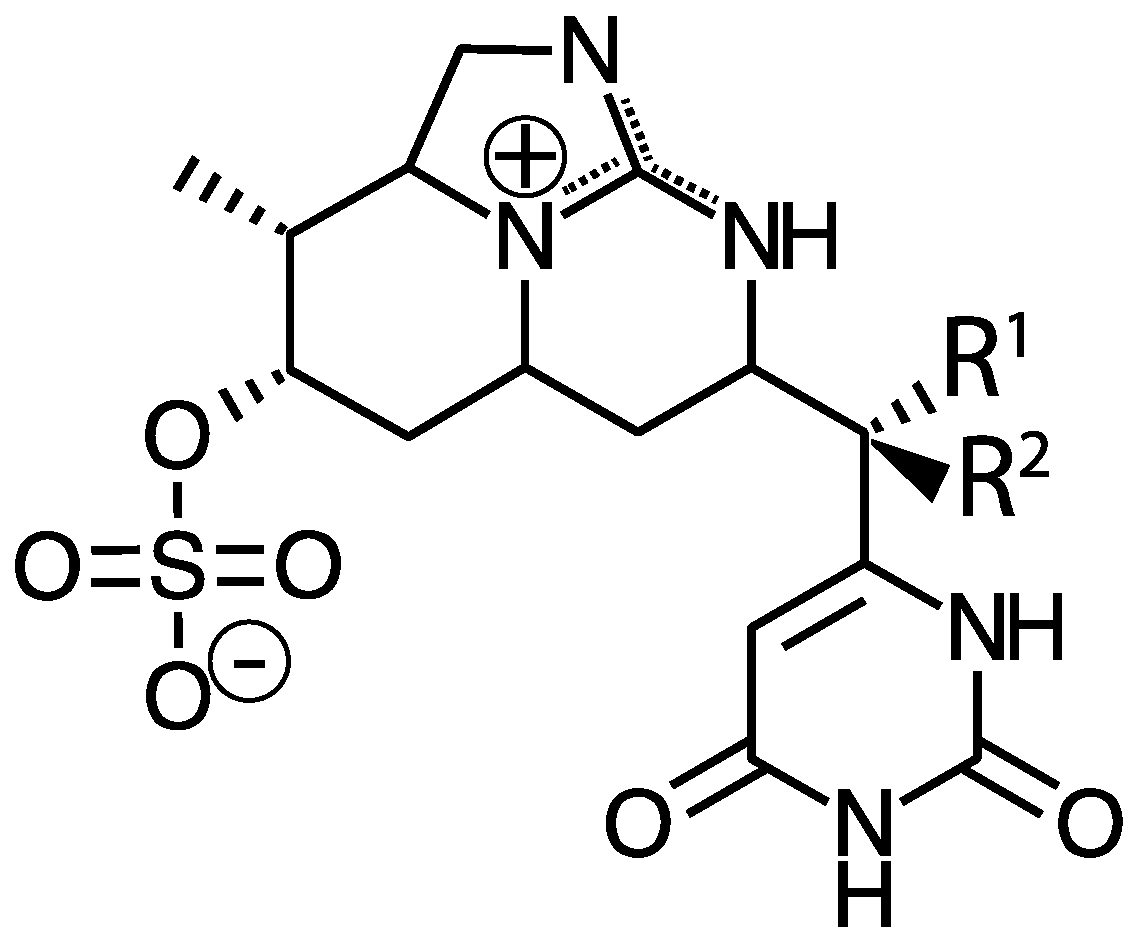
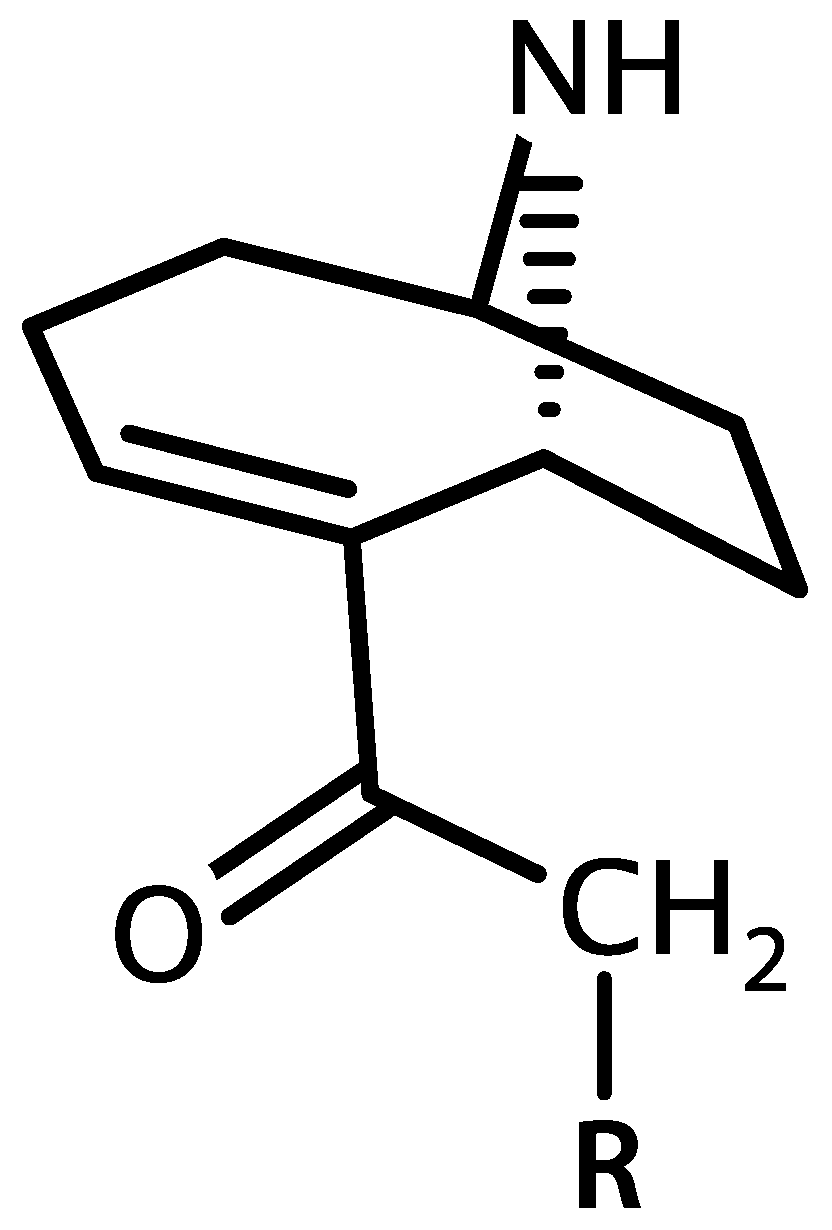
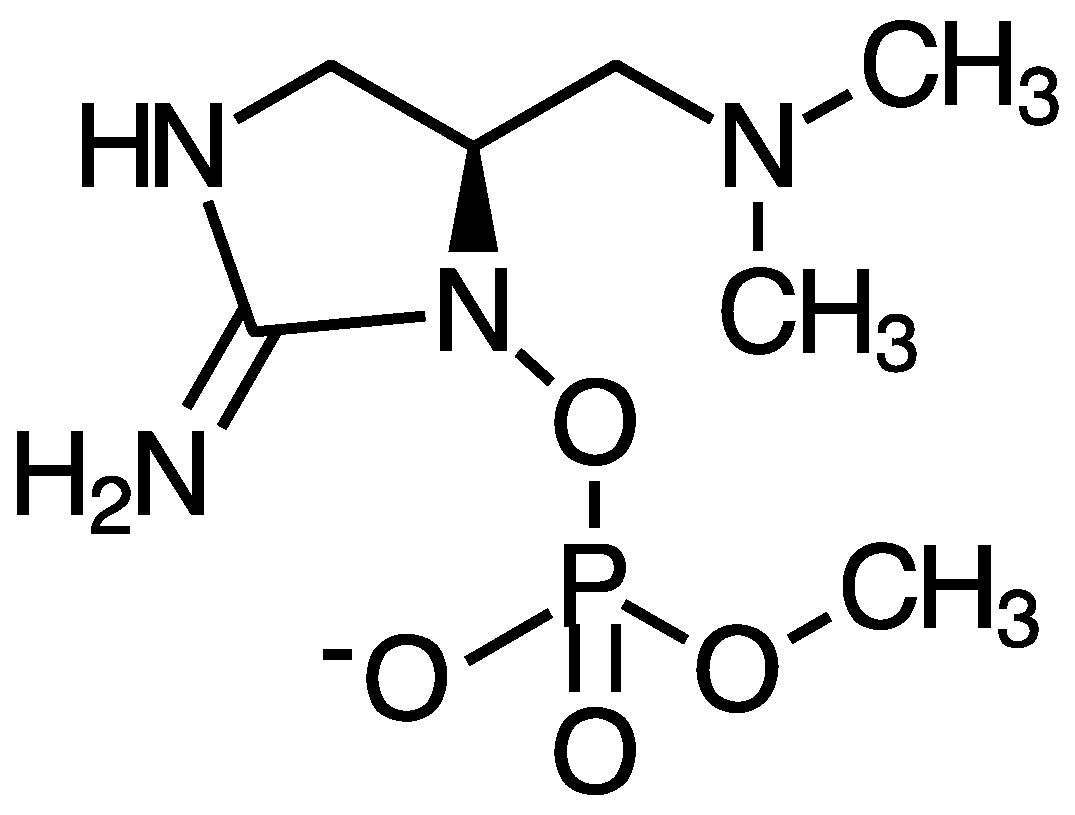

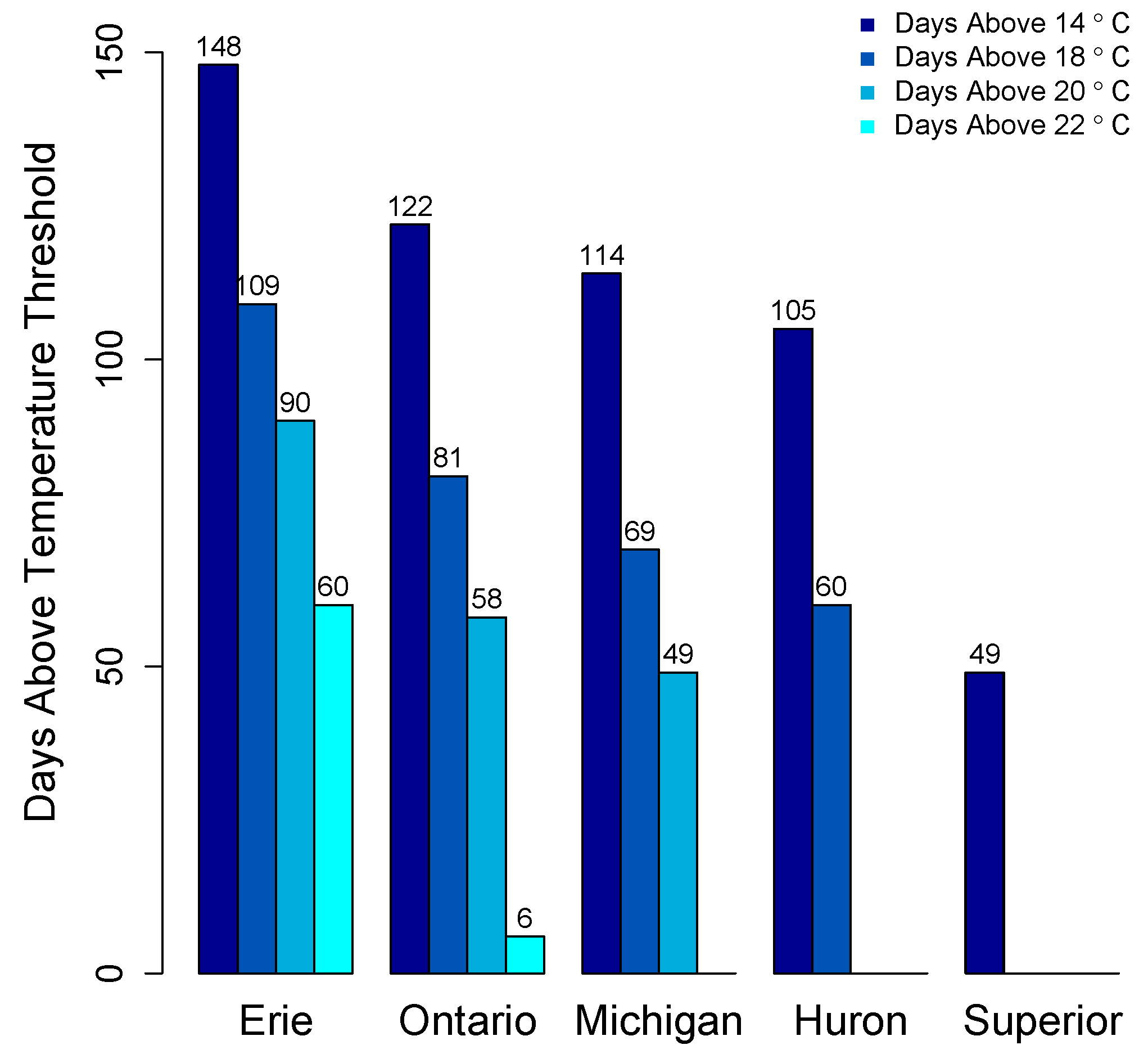
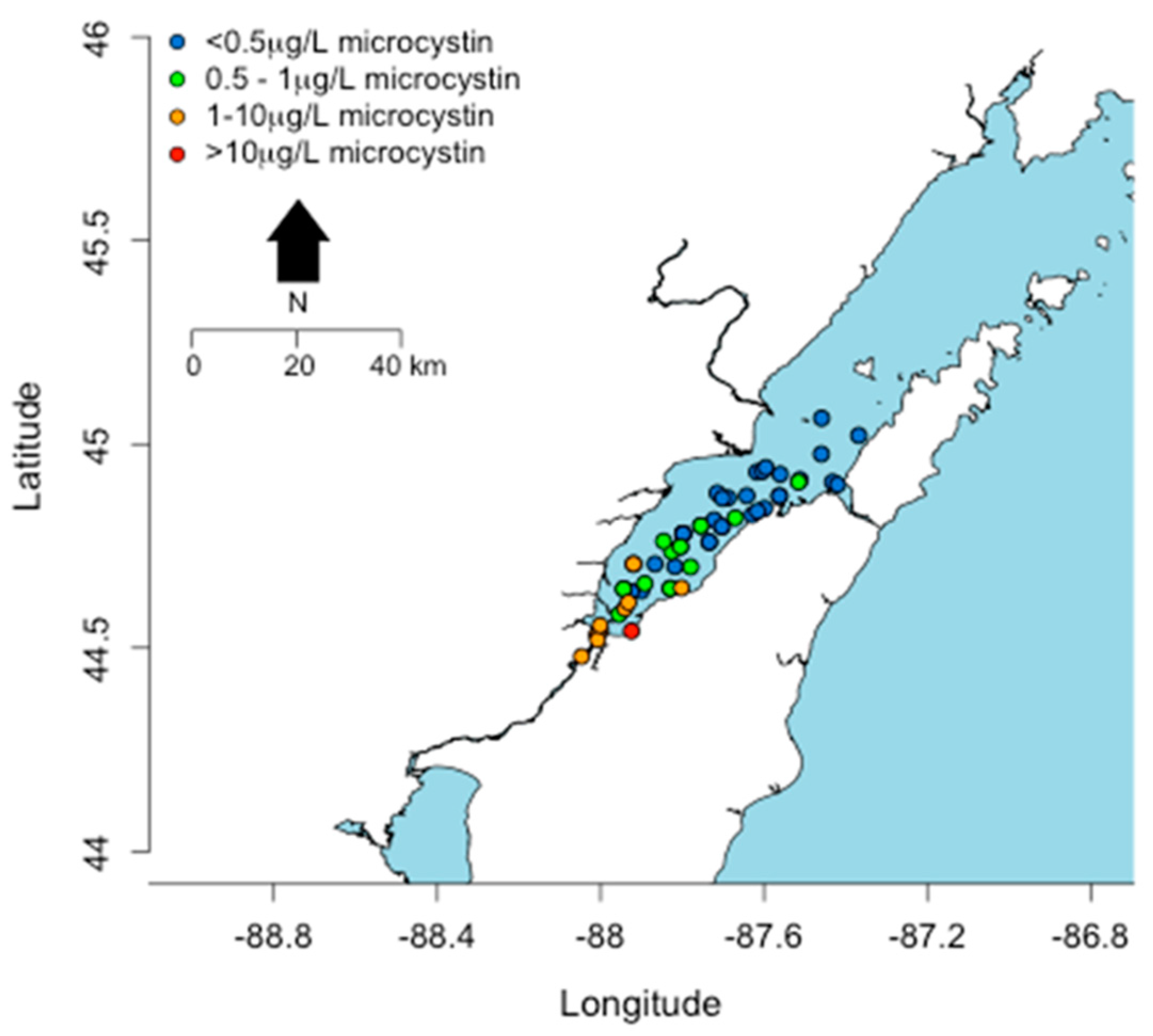
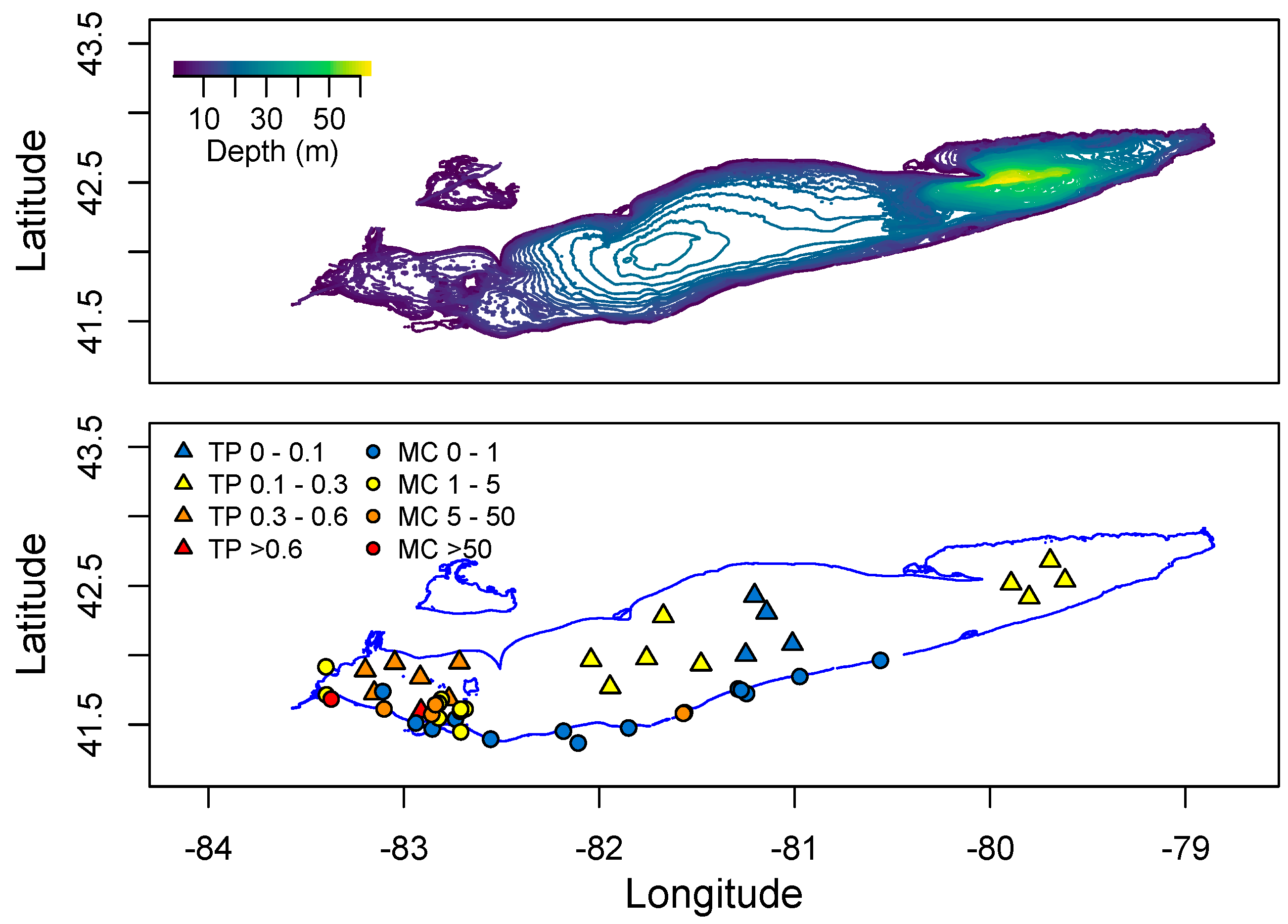
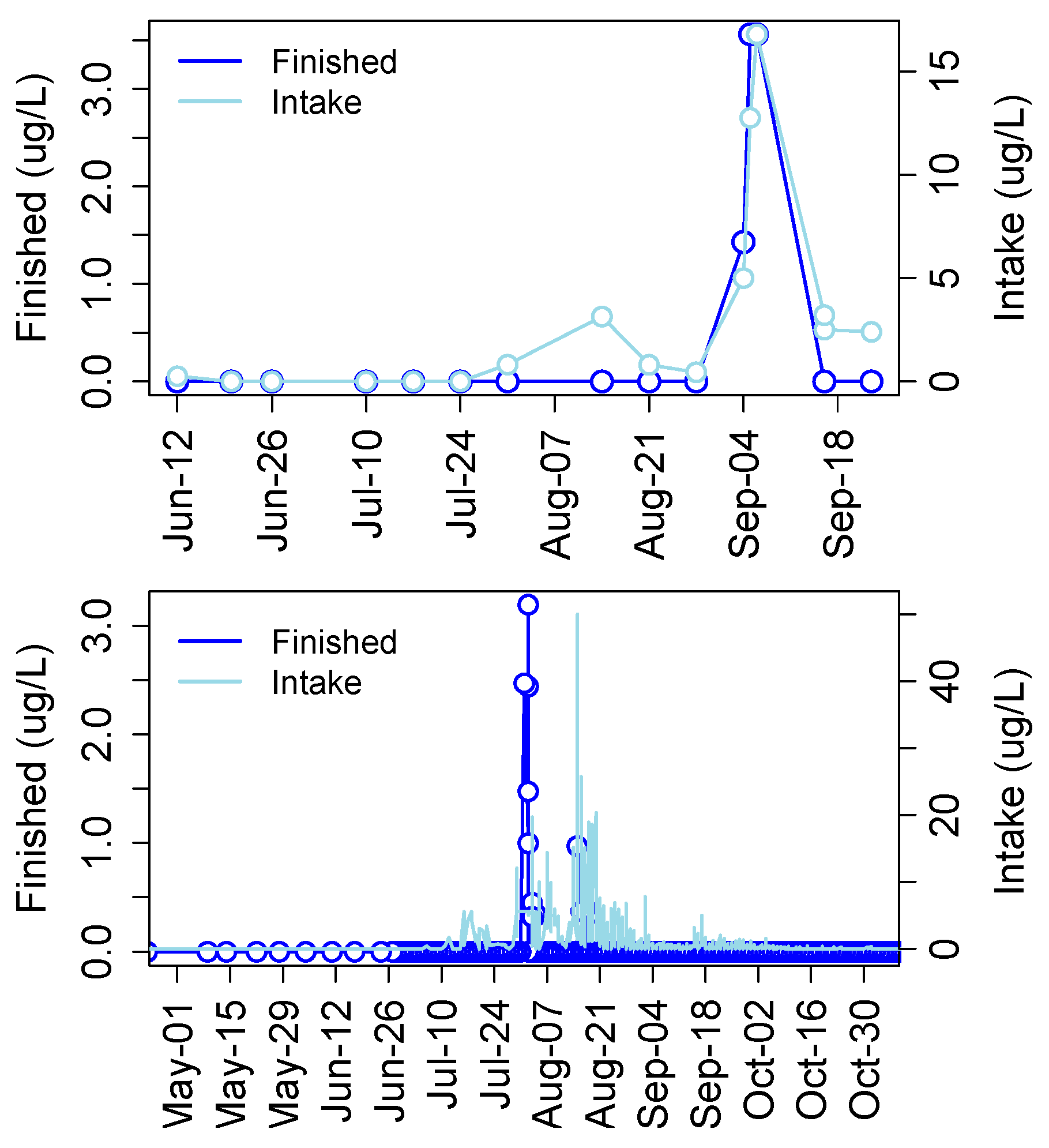
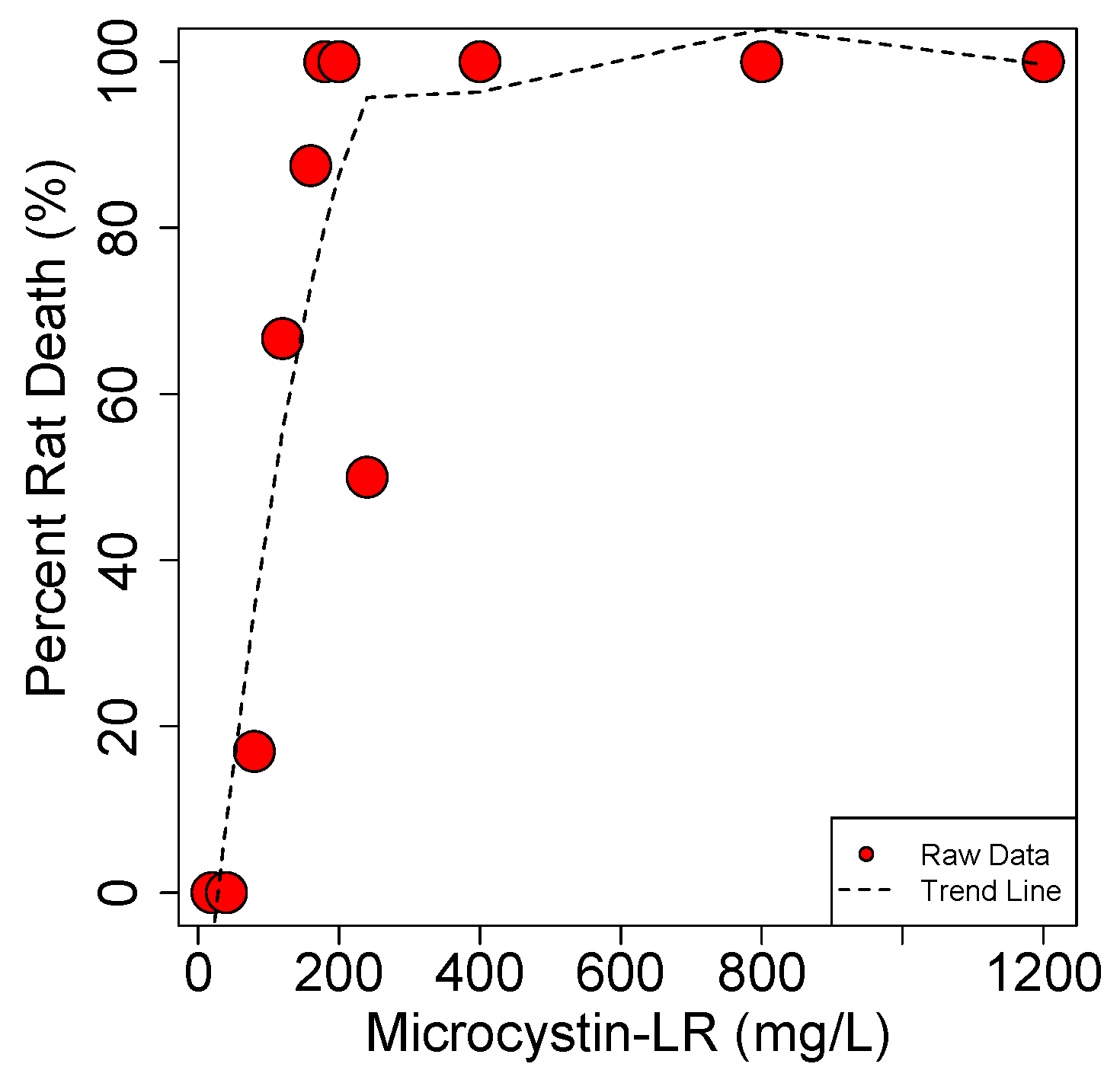
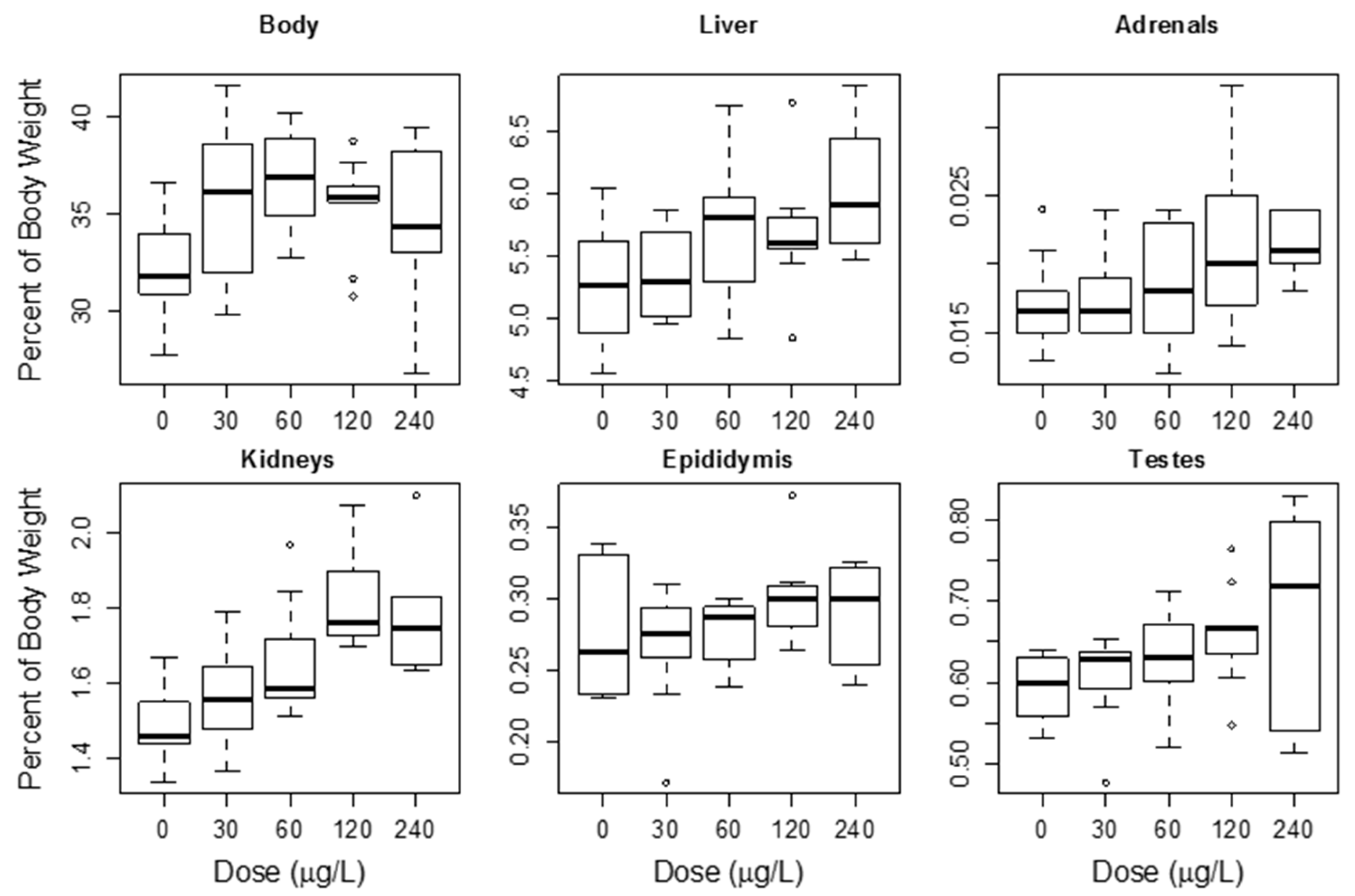
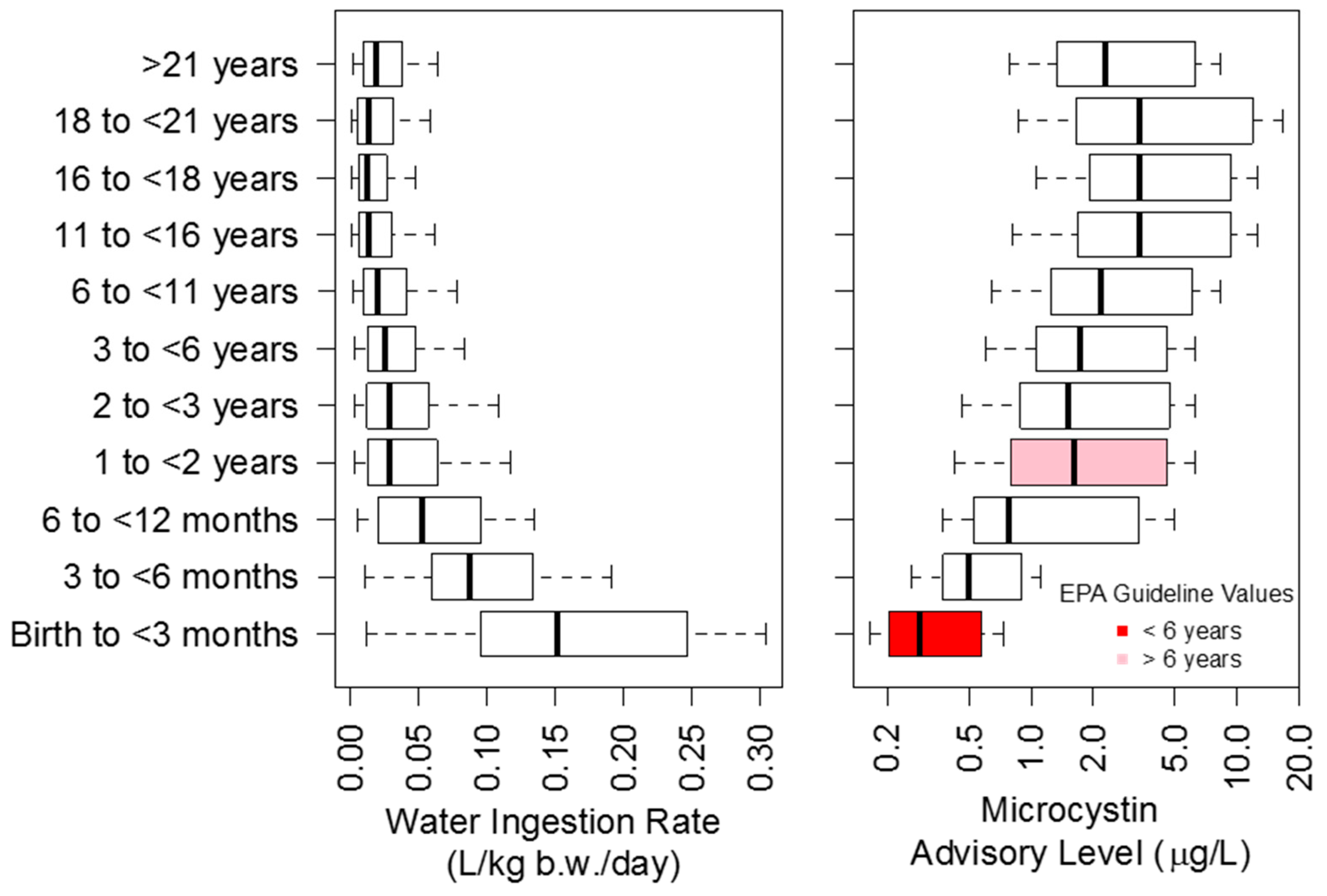
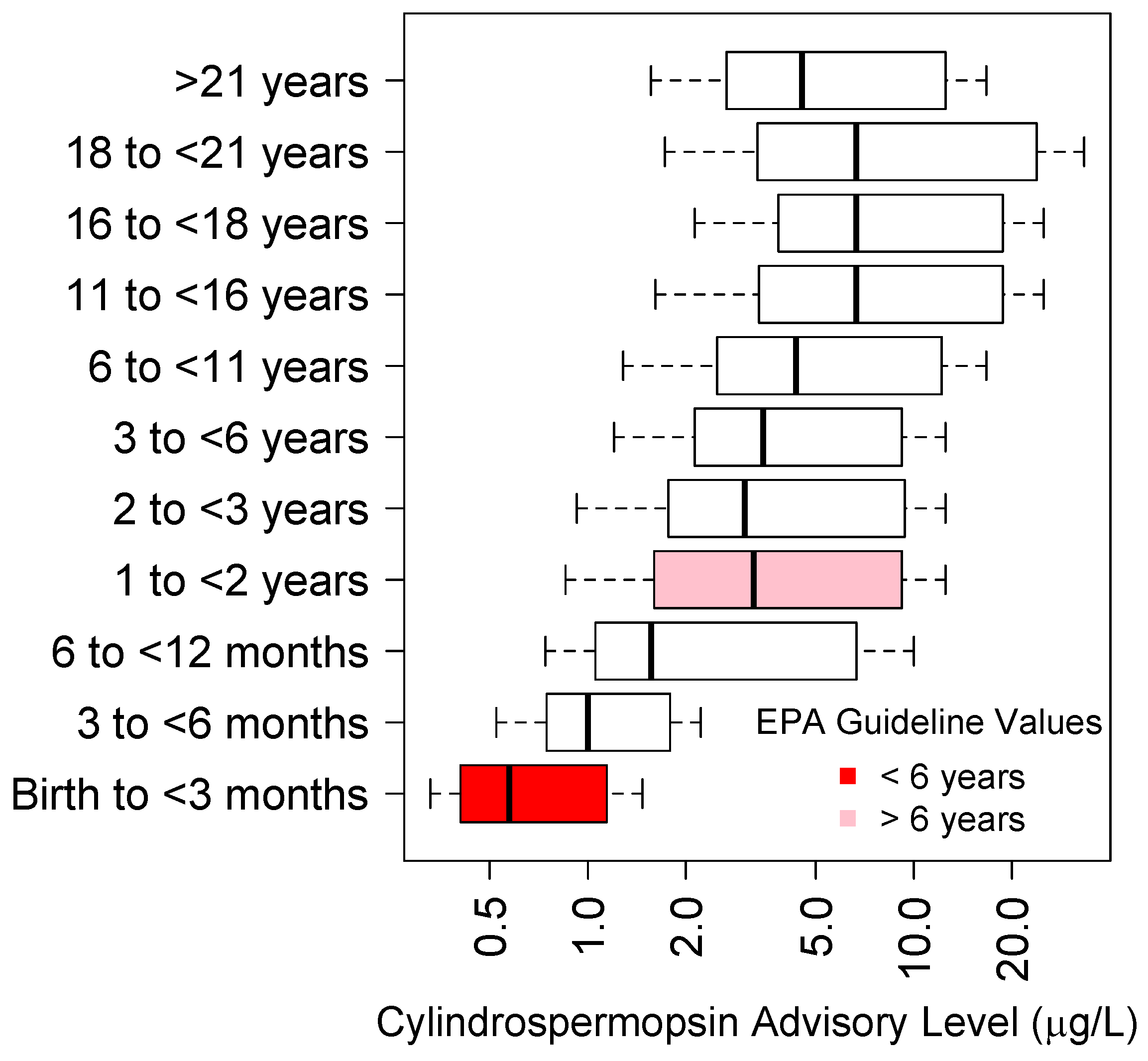
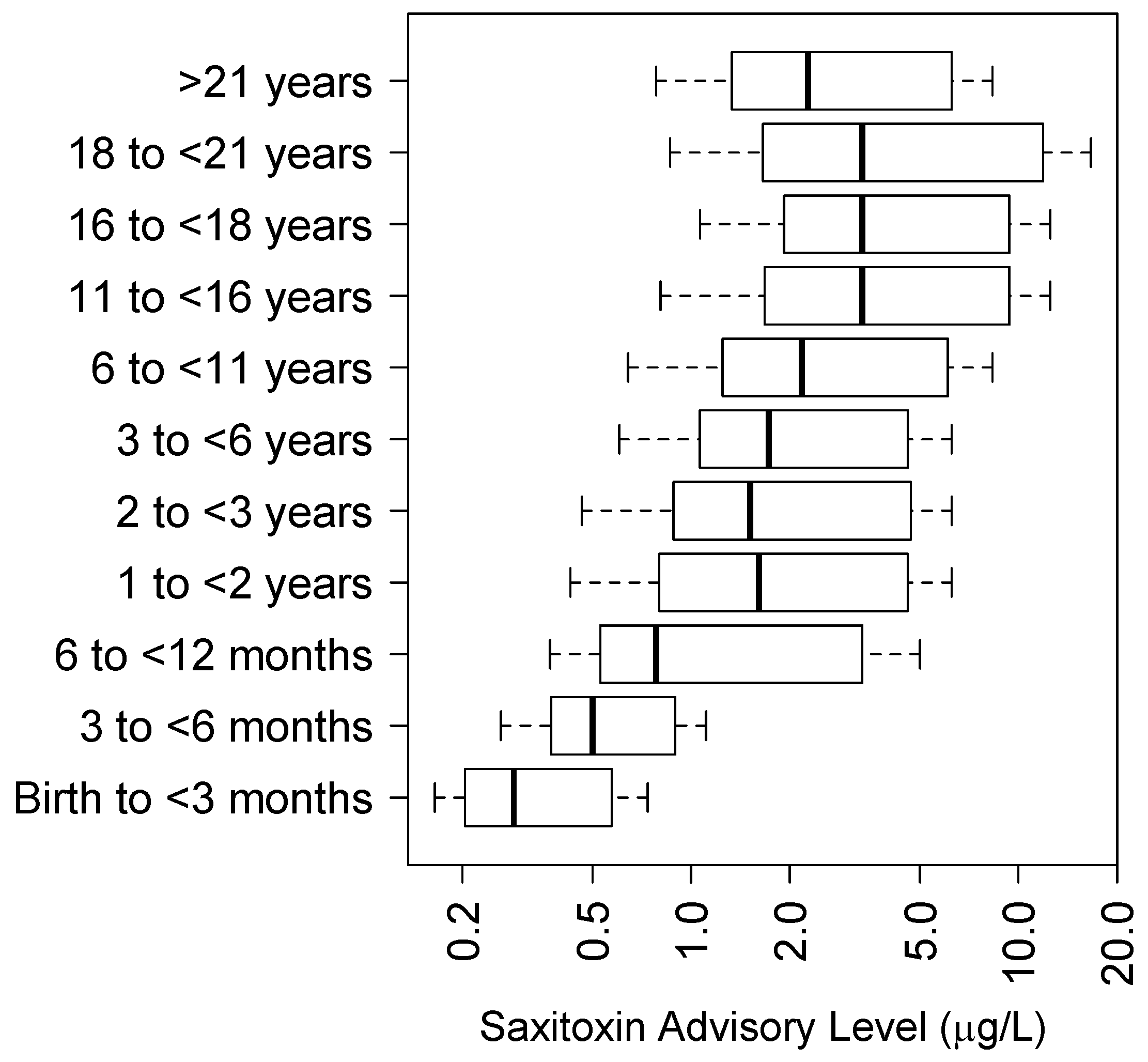

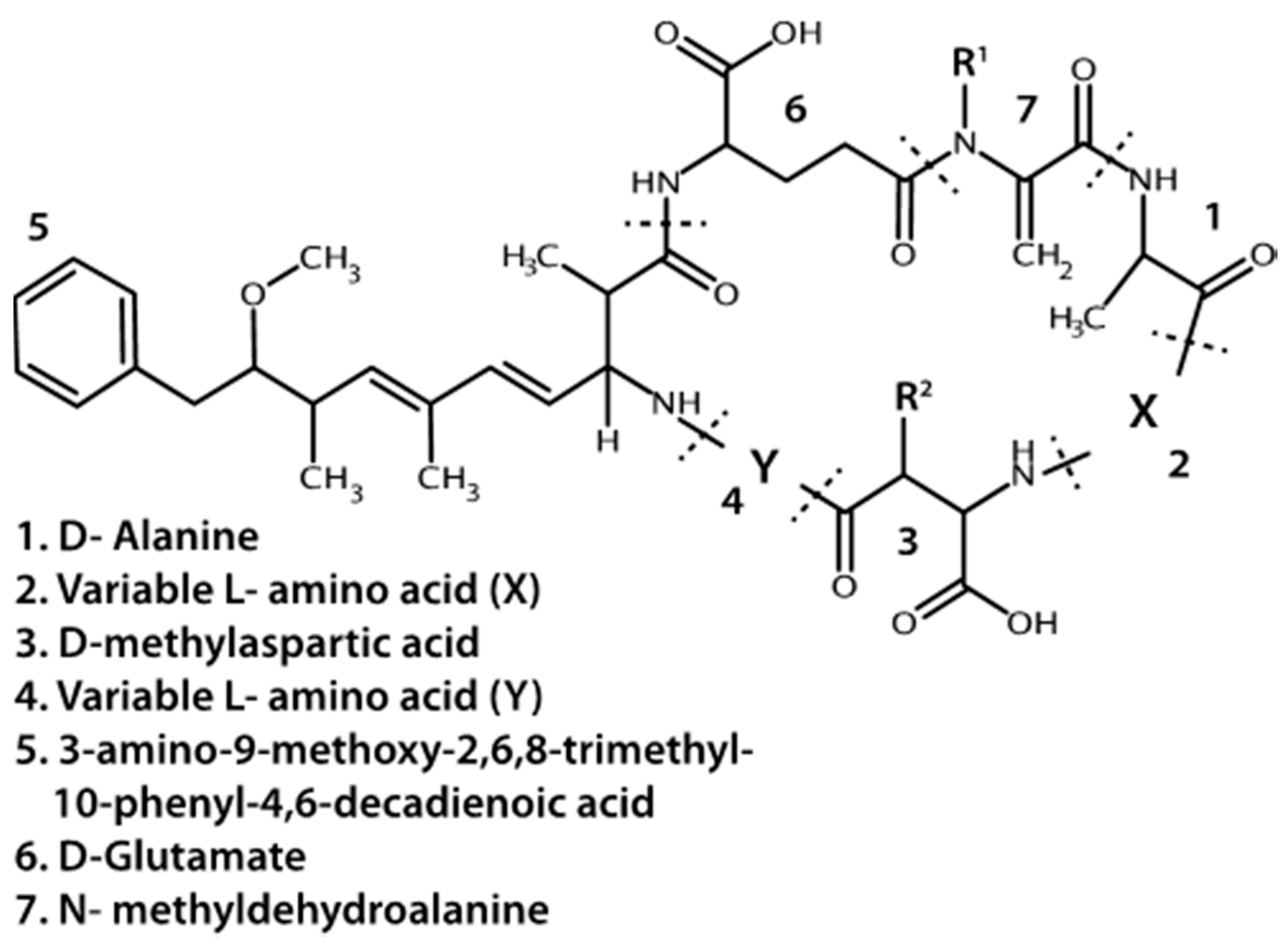{kind=link}
{kind=link}
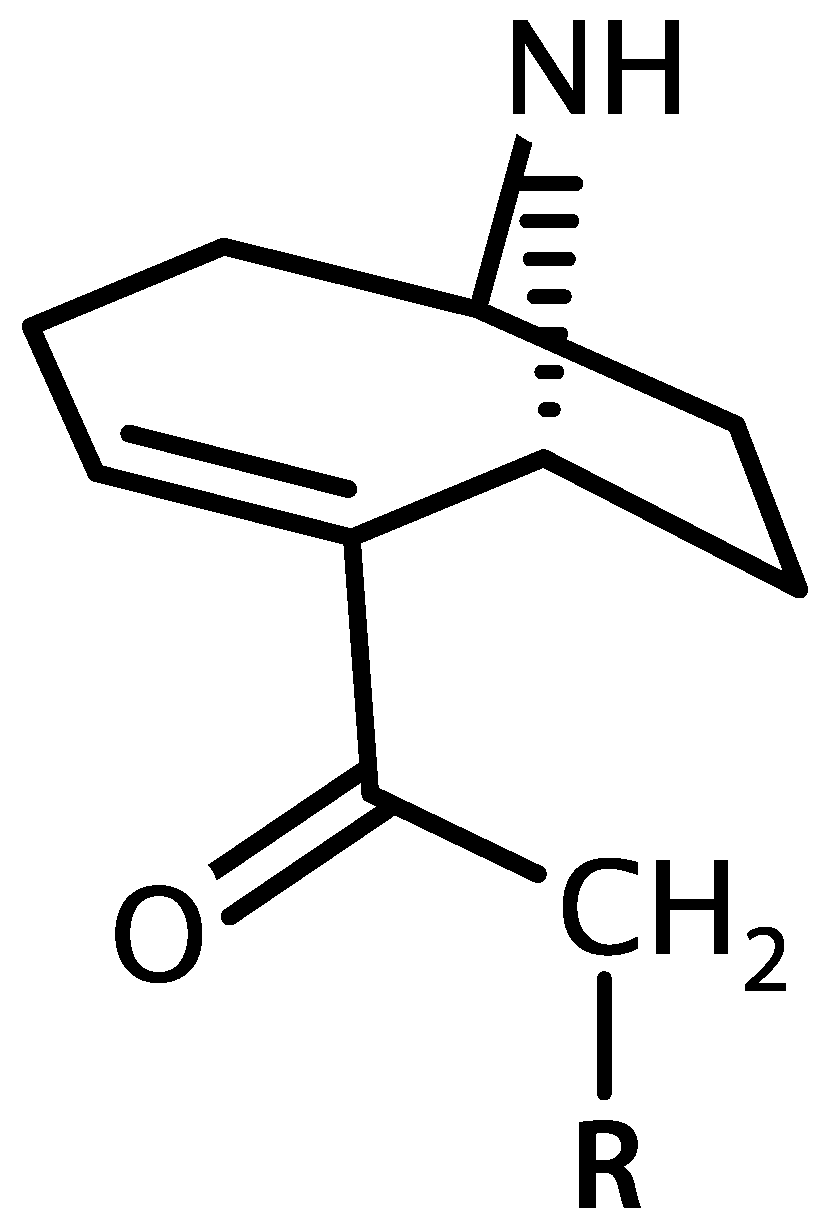{kind=link}
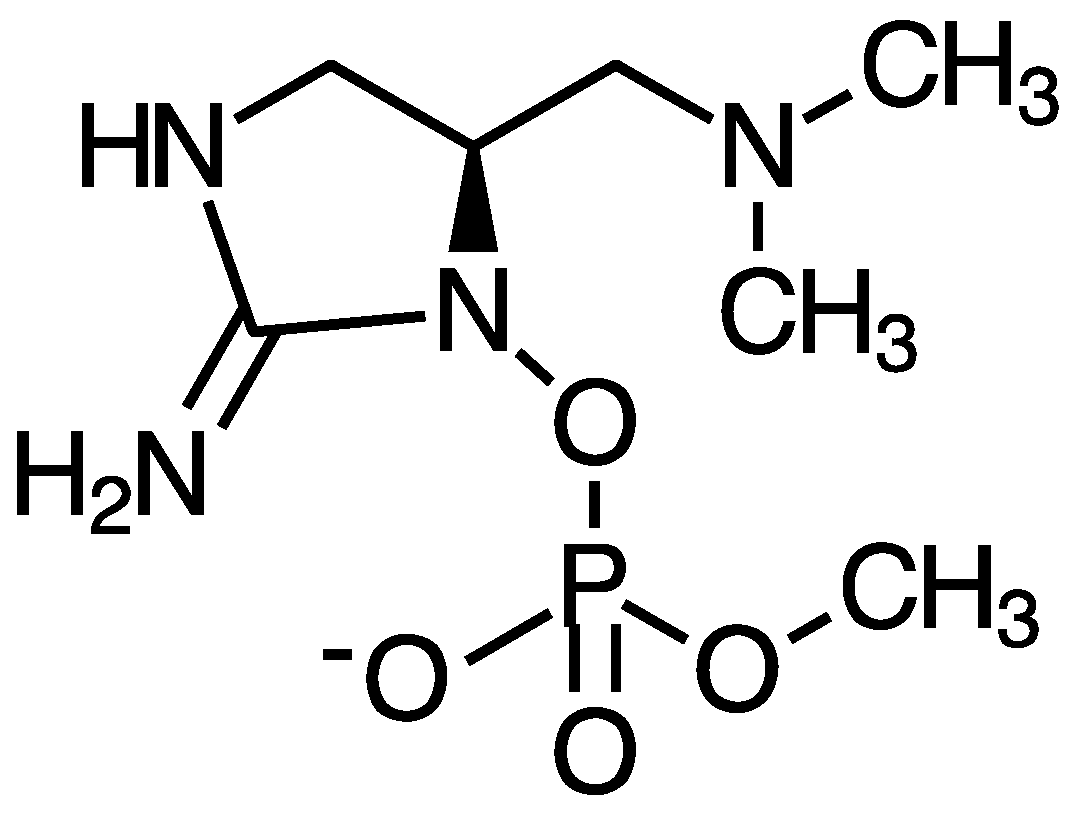{kind=link}
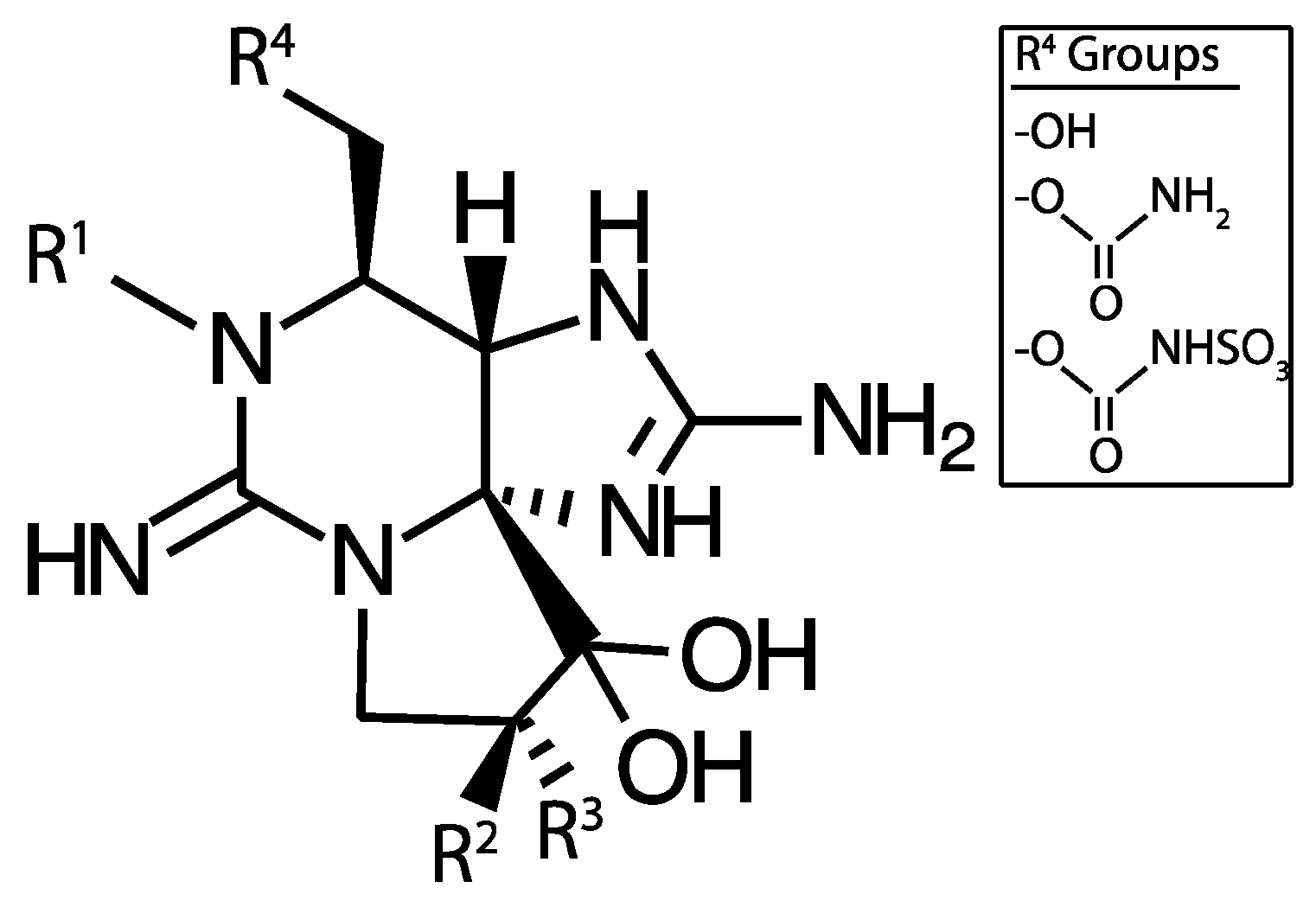{kind=link}
{kind=link}
{kind=link}
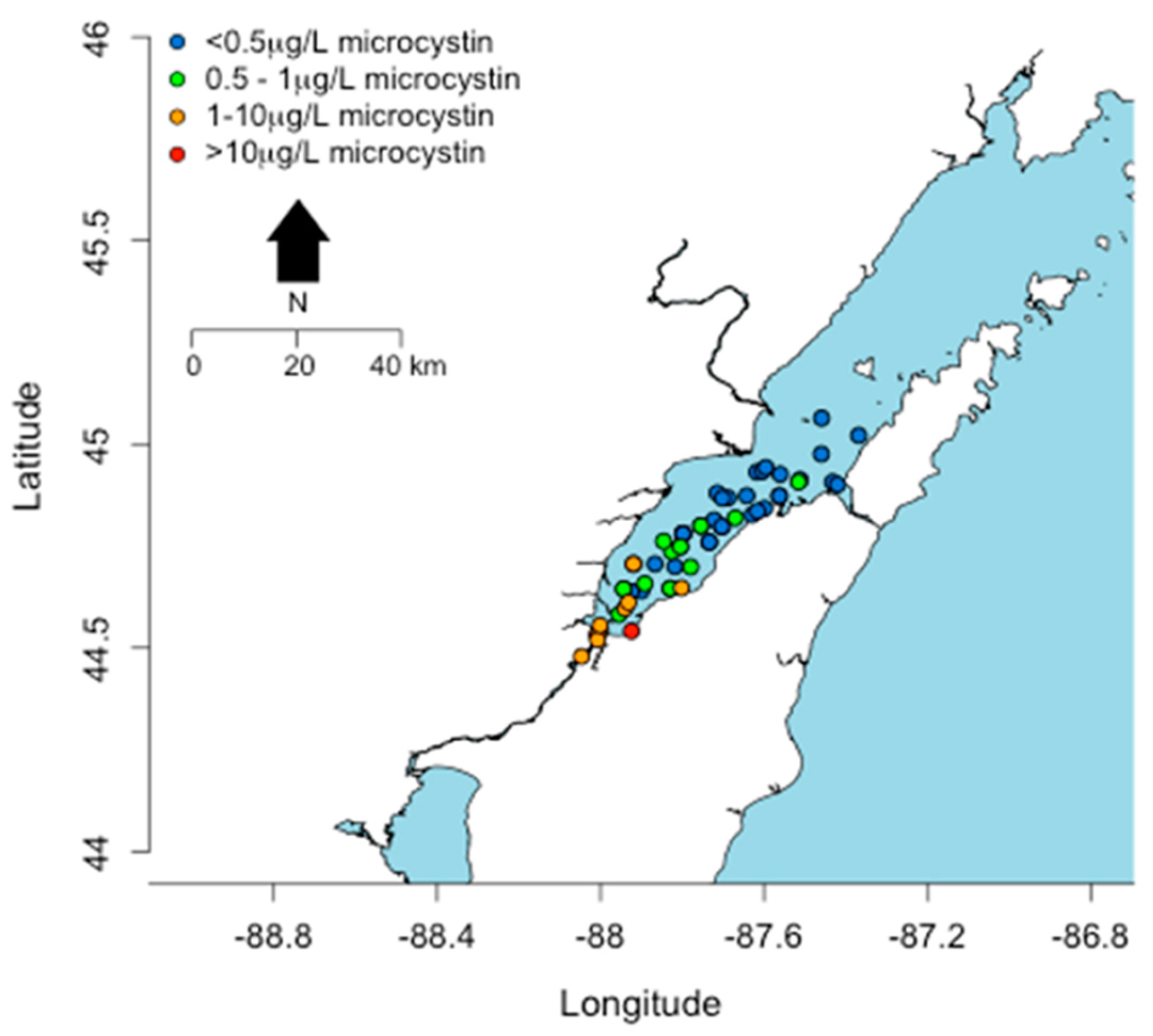{kind=link}
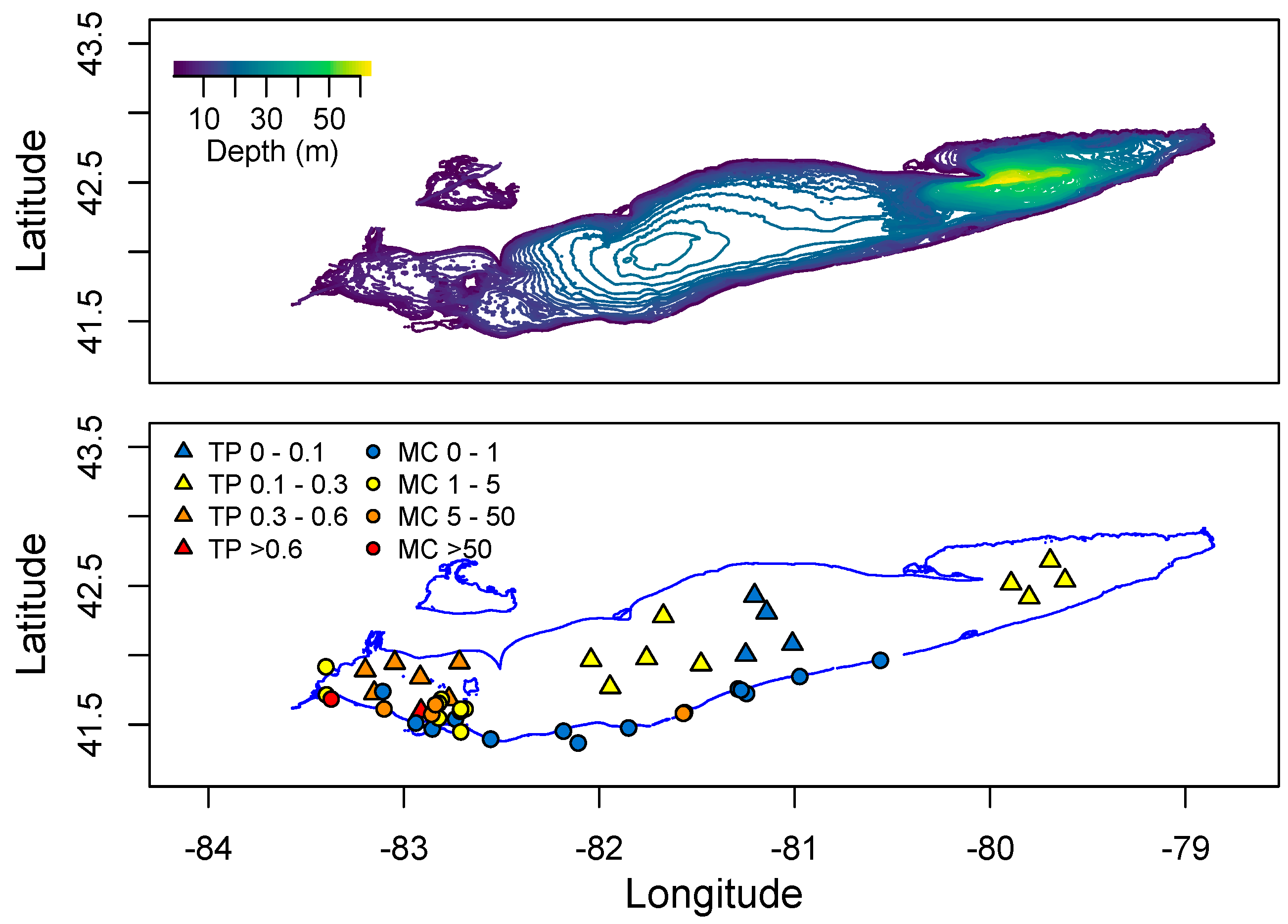{kind=link}
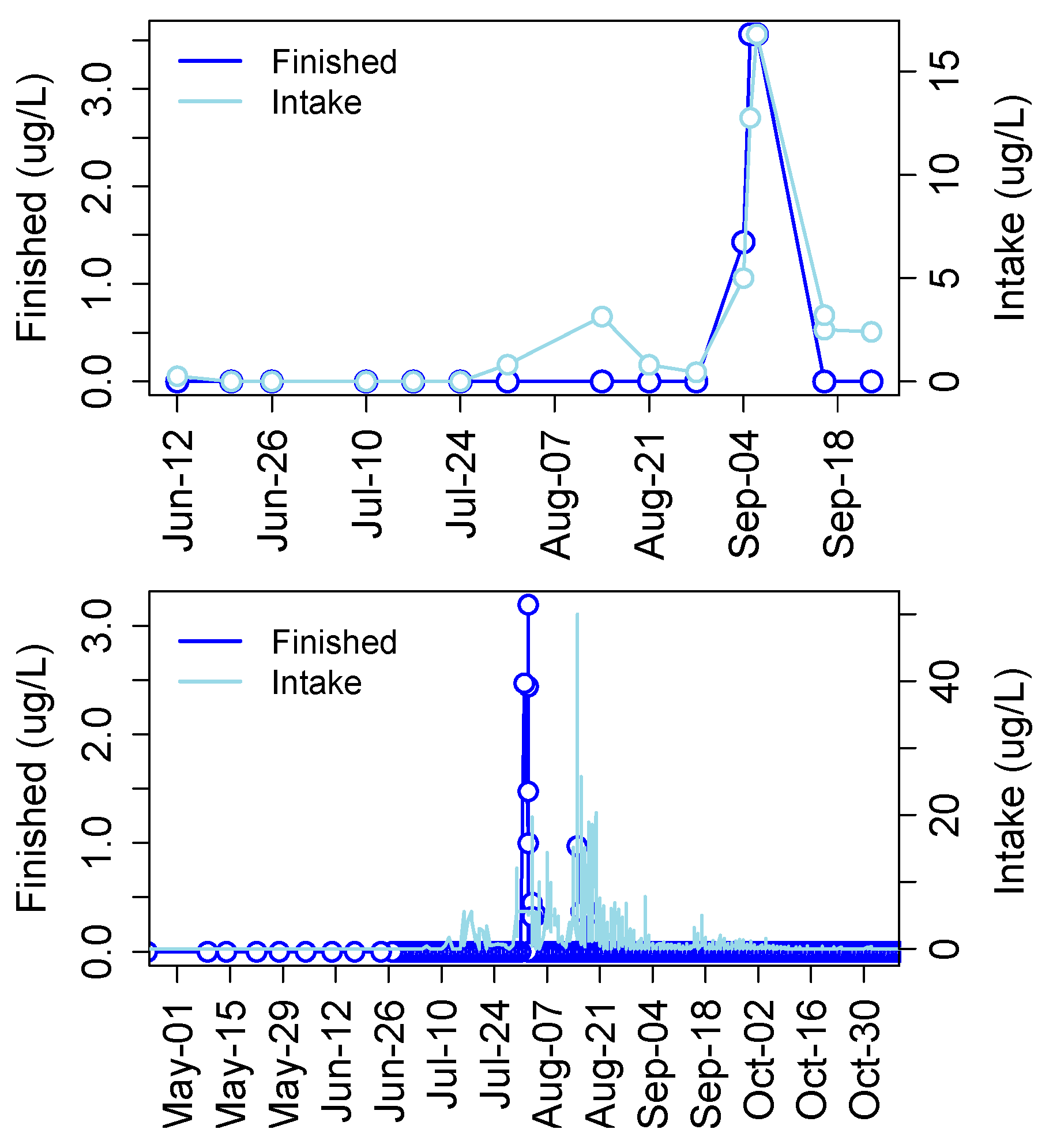{kind=link}
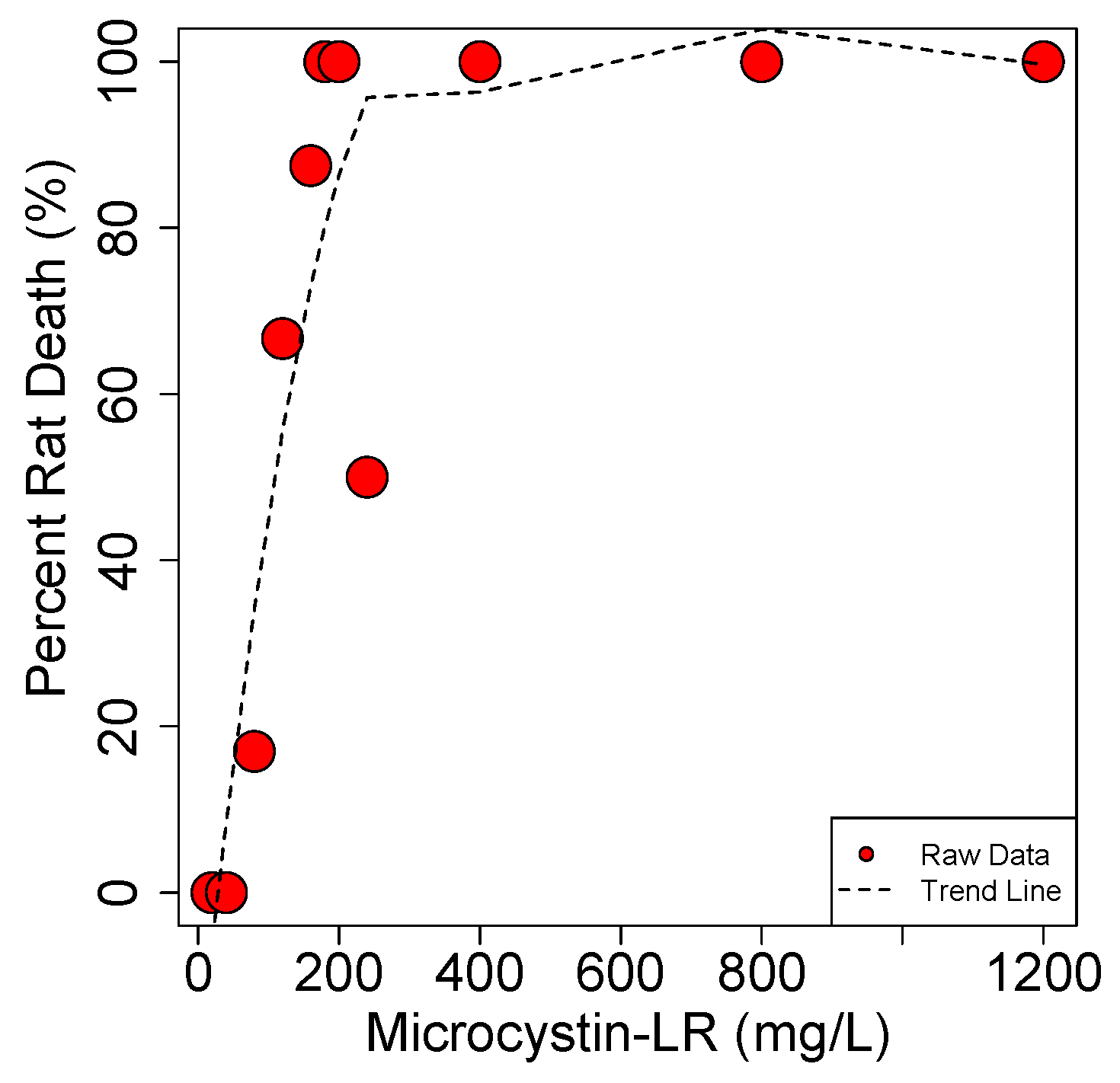{kind=link}
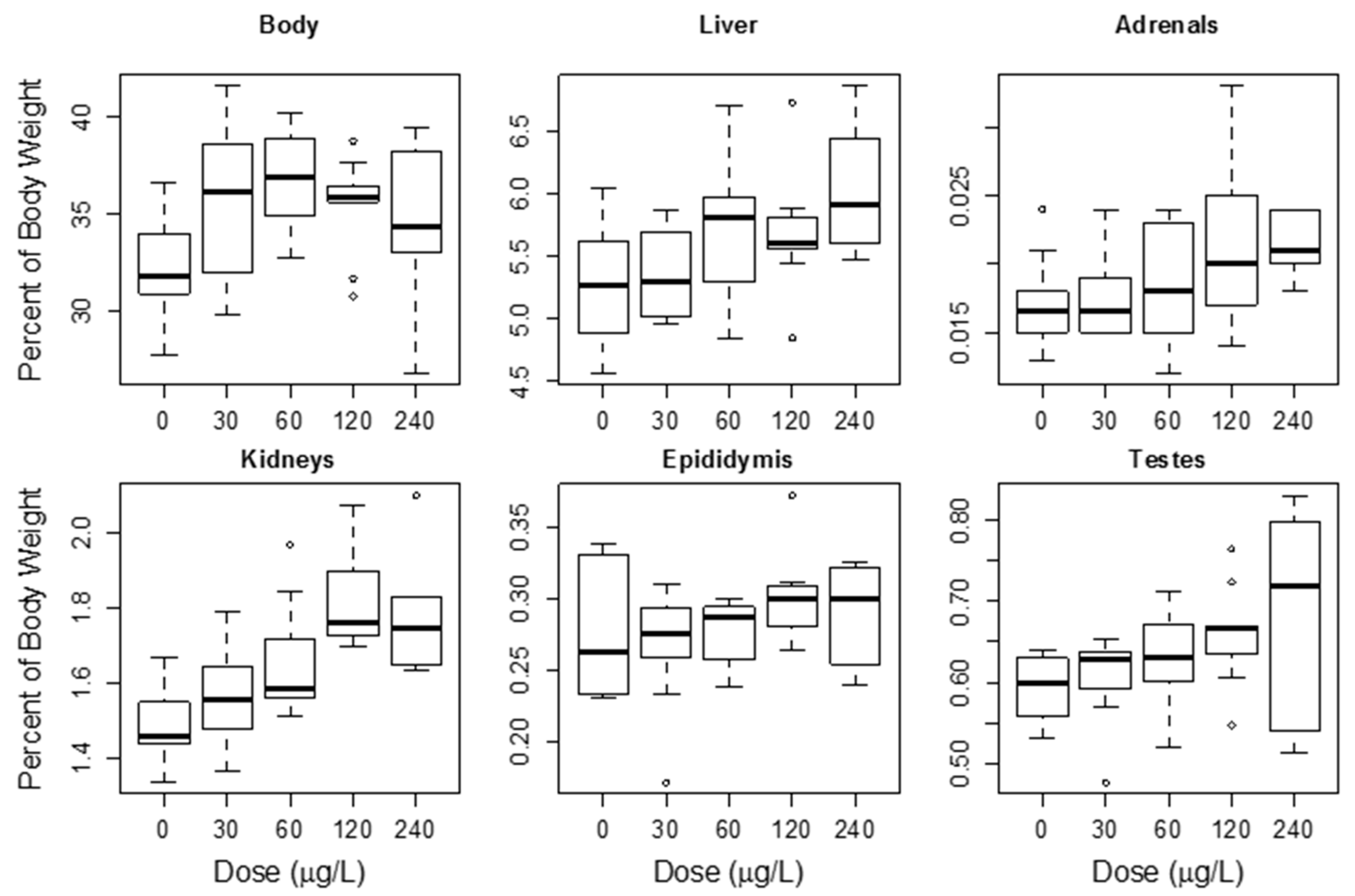{kind=link}
{kind=link}
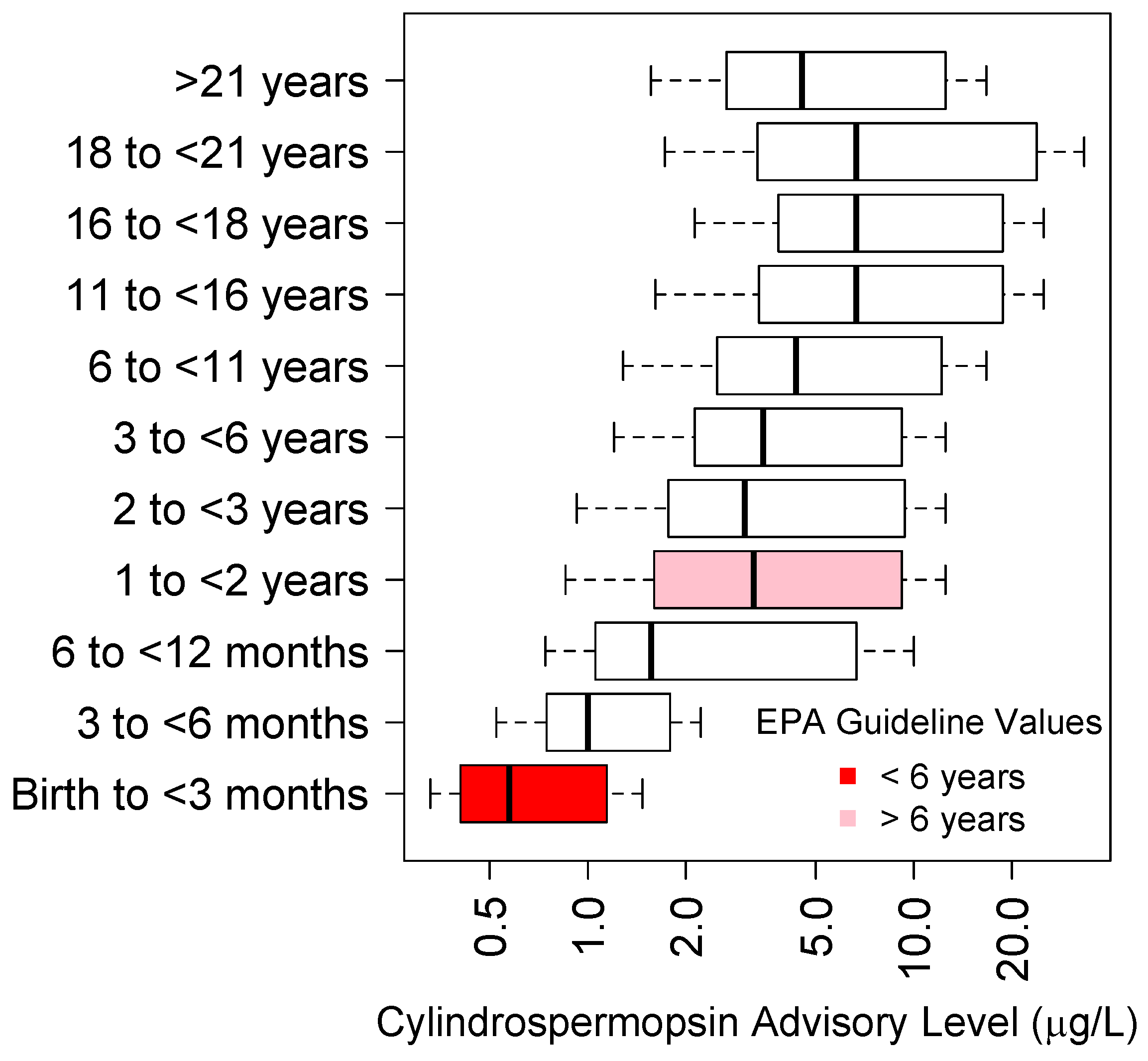{kind=link}
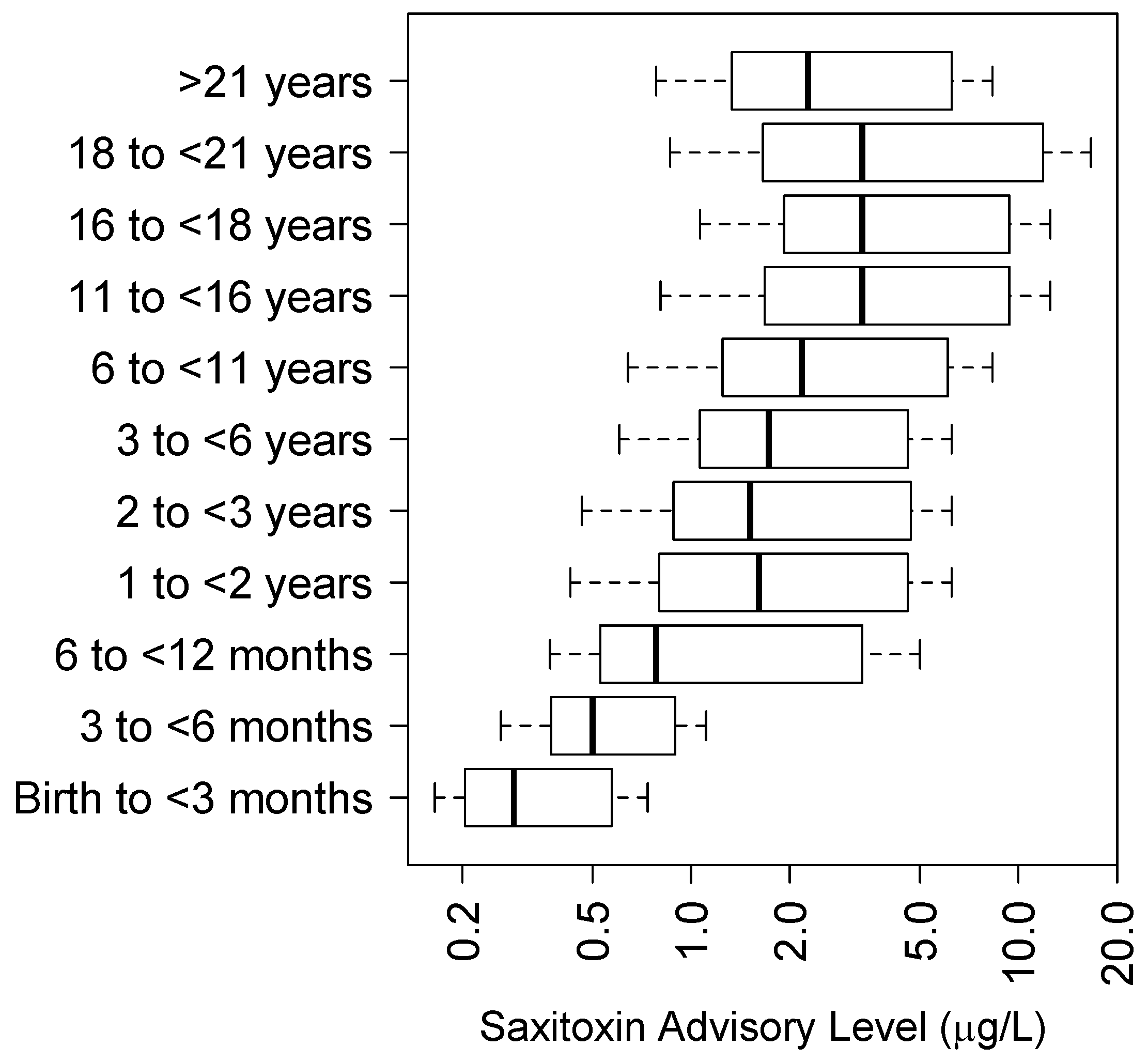{kind=link}
| Site | Lake Erie Surface Water | Mean a | Max a | N b |
|---|---|---|---|---|
| 1 | Lake Erie @ Gibralter Island Docks | 3144.69 | 3144.69 | 1 |
| 2 | Maumee Bay State Park Lake Erie Beach | 21.38 | 570.00 | 52 |
| 3 | Camp Perry Beach-Lake Erie | 2.10 | 2.10 | 1 |
| 4 | Lake Erie (Open Lake) East of Fairport Harbor | 1.70 | 1.70 | 1 |
| 5 | Lake Erie between Toledo/Oregon WTP Intakes | 0.62 | 2.20 | 6 |
| 6 | Lake Erie Ambient Site-Off Maumee Bay | 0.58 | 3.20 | 30 |
| 7 | Lake Erie Ambient Station-West Sister Island | 0.31 | 2.80 | 34 |
| 8 | Lake Erie Ambient Site-Port Clinton | 0.28 | 2.00 | 33 |
| 9 | Lake Erie North of Port Clinton | 0.26 | 2.00 | 16 |
| 10 | Lake Erie Off Detroit Near Canadian Border | 0.19 | 2.00 | 22 |
| 11 | Lake Erie @ Meinke Marina West | 0.13 | 0.96 | 12 |
| 12 | Lake Erie Ambient Station, Off Cedar Point | 0.11 | 0.79 | 13 |
| 13 | Lake Erie Ambient Station-Conneaut | 0.10 | 0.49 | 5 |
| 14 | Lake Erie Ambient Station-Fairport North | 0.08 | 0.31 | 4 |
| 15 | Lake Erie Ambient Site-Off Sandusky Bay | 0.03 | 0.49 | 17 |
| 16 | Lake Erie Ambient Station-Huron | 0.02 | 0.58 | 30 |
| 17 | Lake Erie Ambient Station-Rocky River | ND | ND | 7 |
| 18 | Lake Erie Ambient Station-Lorain West | ND | ND | 7 |
| 19 | Lake Erie Ambient Station-Wildwood | ND | ND | 4 |
| 20 | Lake Erie Fairport Transect Station 3 | ND | ND | 1 |
| 21 | Lake Erie @ Channel Grove Marina | ND | ND | 16 |
| 22 | Lake Erie@Wild Wings Marina | ND | ND | 10 |
| 23 | Lake Erie@Lakefront Marina | ND | ND | 3 |
| 24 | Lake Erie@Brands Marina | ND | ND | 4 |
| 25 | Lake Erie Ambient Station-Geneva North | ND | ND | 4 |
| Intake Site a | Mean a | Max a | N b |
|---|---|---|---|
| Put-In-Bay Water Treatment Plant Lake Erie Intake | 5.83 | 340.00 | 63 |
| Oregon Water Treatment Plant Lake Erie Intake | 3.93 | 37.20 | 99 |
| Camp Patmos Water Treatment Plant Lake Erie Intake | 2.27 | 28.00 | 53 |
| Carroll Water & Sewer Water Treatment Plant Lake Erie Intake | 2.08 | 18.20 | 77 |
| Toledo Water Treatment Plant Lake Erie Intake | 1.24 | 50.00 | 1377 |
| Painesville Water Treatment Plant Lake Erie Intake 2 | 1.10 | 3.90 | 9 |
| Ottawa County Water Treatment Plant Lake Erie Intake | 0.92 | 12.14 | 99 |
| Lake Erie Utilities Water Treatment Plant Lake Erie Intake | 0.69 | 4.33 | 54 |
| Aqua Ohio-Mentor Water Treatment Plant Lake Erie Intake | 0.67 | 2.20 | 8 |
| Lake Co West Water Treatment Plant Lake Erie Intake | 0.65 | 1.61 | 14 |
| Kelleys Island Water Treatment Plant Lake Erie Intake | 0.57 | 5.88 | 66 |
| Fairport Harbor Water Treatment Plant Lake Erie Intake | 0.39 | 1.20 | 7 |
| Huron Water Treatment Plant Lake Erie Intake | 0.34 | 4.62 | 31 |
| Marblehead Water Treatment Plant Lake Erie Intake 1 | 0.33 | 3.80 | 61 |
| Sandusky Water Treatment Plant Lake Erie Intake | 0.30 | 2.50 | 73 |
| Lake Co East Water Treatment Plant Lake Erie Intake | 0.23 | 0.73 | 14 |
| Avon Lake Water Treatment Plant Lake Erie 54 inch Intake | 0.13 | 0.67 | 28 |
| Lorain Water Treatment Plant Lake Erie Intake | 0.12 | 0.61 | 24 |
| Vermilion Water Treatment Plant Lake Erie Intake | ND | ND | 13 |
| Elyria Water Treatment Plant Lake Erie Intake | ND | ND | 3 |
| Painesville WTP Lake Erie Intake 1 | ND | ND | 1 |
| Plant | Mean | Max | N |
|---|---|---|---|
| Celina WTP | 0.039 | 11 | 295 |
| Carroll Water & Sewer WTP | 0.113 | 3.56 | 77 |
| Cadiz WTP | 0.050 | 3.4 | 68 |
| Toledo WTP | 0.021 | 3.19 | 690 |
| Kelleys Island WTP | 0.025 | 1.68 | 67 |
| Put-In-Bay WTP | 0.016 | 0.6 | 60 |
| Camp Patmos WTP | 0.009 | 0.5 | 54 |
| Campbell Soup Supply Co WTP | 0.038 | 0.35 | 18 |
| Oregon WTP | 0.002 | 0.23 | 96 |
| Toxin | Microcystins | Cylindrospermopsins | Saxitoxins | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Source | c WHO | d EPA (U.S. and Canada) | This Report | This Report | EPA (U.S. and Canada) | This Report | This Report | ||
| Critical study | Fawell et al. 1999 | Heinze 1999 | Li et al. 2015 | Chen et al. 2011 | Humpage and Falconer 2002, 2003 | Humpage and Falconer 2002, 2003 | e CONTAM | ||
| a LOAEL/NOAEL (µg/kg/day) | 40 | 50 | 5 | 1 | 30 | 30 | 0.5 | ||
| End point | Liver Toxicity | Liver Toxicity | Central Nervous System Toxicity | Male Reproductive Toxicity | Kidney Toxicity | Kidney Toxicity | Peripheral Nervous System Toxicity | ||
| b Age of Exposed | Adult | <6 years | >6 years | <6 years | <6 years | <6 years | >6 years | <6 years | <6 years |
| DWI/BW/day (L/kg/d) | 0.03 | 0.15 | 0.03 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 |
| Uncertainty Factor | 1000 | 1000 | 1000 | 1000 | 1000 | 300 | 300 | 1000 | 3 |
| Guideline Value (µg/kg) | 0.96 | 0.3 | 1.6 | 0.03 | 0.01 | 0.7 | 3 | 0.2 | 0.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, T.R.; Beversdorf, L.J.; Weirich, C.A.; Bartlett, S.L. Cyanobacterial Toxins of the Laurentian Great Lakes, Their Toxicological Effects, and Numerical Limits in Drinking Water. Mar. Drugs 2017, 15, 160. https://doi.org/10.3390/md15060160
Miller TR, Beversdorf LJ, Weirich CA, Bartlett SL. Cyanobacterial Toxins of the Laurentian Great Lakes, Their Toxicological Effects, and Numerical Limits in Drinking Water. Marine Drugs. 2017; 15(6):160. https://doi.org/10.3390/md15060160
Chicago/Turabian StyleMiller, Todd R., Lucas J. Beversdorf, Chelsea A. Weirich, and Sarah L. Bartlett. 2017. "Cyanobacterial Toxins of the Laurentian Great Lakes, Their Toxicological Effects, and Numerical Limits in Drinking Water" Marine Drugs 15, no. 6: 160. https://doi.org/10.3390/md15060160
APA StyleMiller, T. R., Beversdorf, L. J., Weirich, C. A., & Bartlett, S. L. (2017). Cyanobacterial Toxins of the Laurentian Great Lakes, Their Toxicological Effects, and Numerical Limits in Drinking Water. Marine Drugs, 15(6), 160. https://doi.org/10.3390/md15060160