Limonoids Containing a C1–O–C29 Moiety: Isolation, Structural Modification, and Antiviral Activity


1
Marine Drugs Research Center, College of Pharmacy, Jinan University, 601 Huangpu Avenue West, Guangzhou 510632, China
2
School of Pharmaceutical Sciences, Southern Medical University, 1838 Guangzhou Avenue North, Guangzhou 510515, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally.
Mar. Drugs 2018, 16(11), 434; https://doi.org/10.3390/md16110434
Submission received: 3 October 2018
/
Revised: 25 October 2018
/
Accepted: 31 October 2018
/
Published: 4 November 2018
Abstract
:Five new limonoids named thaigranatins A–E (1–5), containing a C1–O–C29 moiety, were isolated from seeds of the Thai Xylocarpus granatum, collected at the mangrove swamp of Trang Province, together with the known limonoid, granatumin L (6). The structures of these compounds were established by HR-ESIMS and extensive NMR spectroscopic data. The absolute configuration of 1 was unequivocally determined by single-crystal X-ray diffraction analysis, conducted with Cu Kα radiation; whereas that of 2 or 6 was established to be the same as that of 1 by the similarity of their electronic circular dichroism (ECD) spectra. In view of the marked antiviral activity of 6, its structure was modified via hydrolysis with alkaline KOH, esterification with diazomethane and various organic acids, and oximization with hydroxyamine. Finally, 18 derivatives, viz. 7–10, 8a–8i, 9a–9b, and 10a–10c, were obtained. In vitro antiviral activities of these derivatives against human immunodeficiency virus 1 (HIV-1) and influenza A virus (IAV) were evaluated. Most notably, 8i exhibited marked inhibitory activity against HIV-1 with an IC50 value of 15.98 ± 6.87 μM and a CC50 value greater than 100.0 μM; whereas 10b showed significant inhibitory activity against IAV with an IC50 value of 14.02 ± 3.54 μM and a CC50 value greater than 100.0 μM.
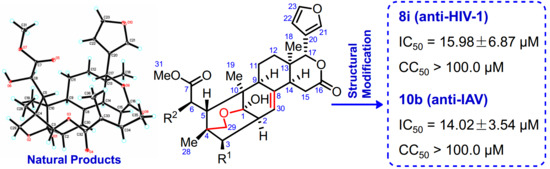
1. Introduction
Xylocarpus granatum, a true mangrove plant, belongs to the family Meliaceae, of which the main secondary metabolites are limonoids [1,2]. Krishnolide A, a khayanolide-type of limonoid isolated from the Indian mangrove, Xylocarpus moluccensis, showed moderate antiviral activity against human immunodeficiency virus 1 (HIV-1) with an IC50 value of 17.45 ± 1.65 μM and a CC50 value of 78.45 ± 1.69 μM, respectively [3]. Three other khayanolide-type of limonoids obtained from the Thai Xylocarpus moluccensis, viz. thaixylomolins I, K, and M, exhibited moderate antiviral activity against influenza A virus (IAV). The most potent one is thaixylomolin I with an IC50 value of 77.1 ± 8.7 μM [4].
Limonoids containing a C1–O–C29 moiety are a small group of natural products. To date, only 34 compounds of this group, including xyloccensin L, xylogranatin E, granaxylocarpin C, granatumins L–T and V–Y, sundarbanxylogranins C–E, krishnagranatins A–F, thaixylogranin D, godavarins D–G and K, moluccensin W, and erythrocarpines D–E, have been reported from mangrove plants of the genus Xylocarpus and the land plant Chisocheton erythrocarpus [5,6,7,8,9,10,11,12,13]. It is worth noting that 28 compounds of this group were obtained from X. granatum [5,6,7,8,9,10,11]. However, only one limonoid containing a C1–O–C29 moiety, i.e., thaixylogranin D, was identified from the Thai X. granatum [11]. To search for new antiviral natural compounds from mangrove plants, seeds of the Thai X. granatum, collected in the mangrove swamp of Trang Province, were investigated to afford five new limonoids containing a C1–O–C29 moiety, named thaigranatins A–E (1–5), along with the known one, granatumin L (6) (Figure 1), which exhibited antiviral activity against HIV-1. To find promising antiviral drug leads, structural modification was applied to granatumin L (6). Herein, we report the isolation and structural elucidation of thaigranatins A–E (1–5), the structural modification of granatumin L (6), and antiviral activities of the modified derivatives of 6 against HIV-1 and IAV.
2. Results and Discussion
2.1. Structure Identification of Natural Limonoids 1–5
Compound 1 was obtained as colorless crystals. Its molecular formula C32H40O10, indicating 13 degrees of unsaturation, was established by the positive HR-ESIMS ion peak at m/z 607.2509 (calcd. for [M + Na]+ 607.2514). According to the 1H and 13C NMR spectroscopic data (Table 1 and Table 2), 7 degrees of unsaturation were due to 3 carbonyl groups and 4 carbon-carbon double bonds; thus, the molecule was hexacyclic. The 13C NMR spectroscopic data and DEPT experiments indicated the presence of 6 methyl groups (a methoxy group, a methyl group linked to a secondary carbon and 4 methyl groups linked to tertiary carbons), 4 methylene groups, 12 methine groups (including 5 olefinic), and 10 quaternary carbons (including 3 carbonyl groups) in 1.
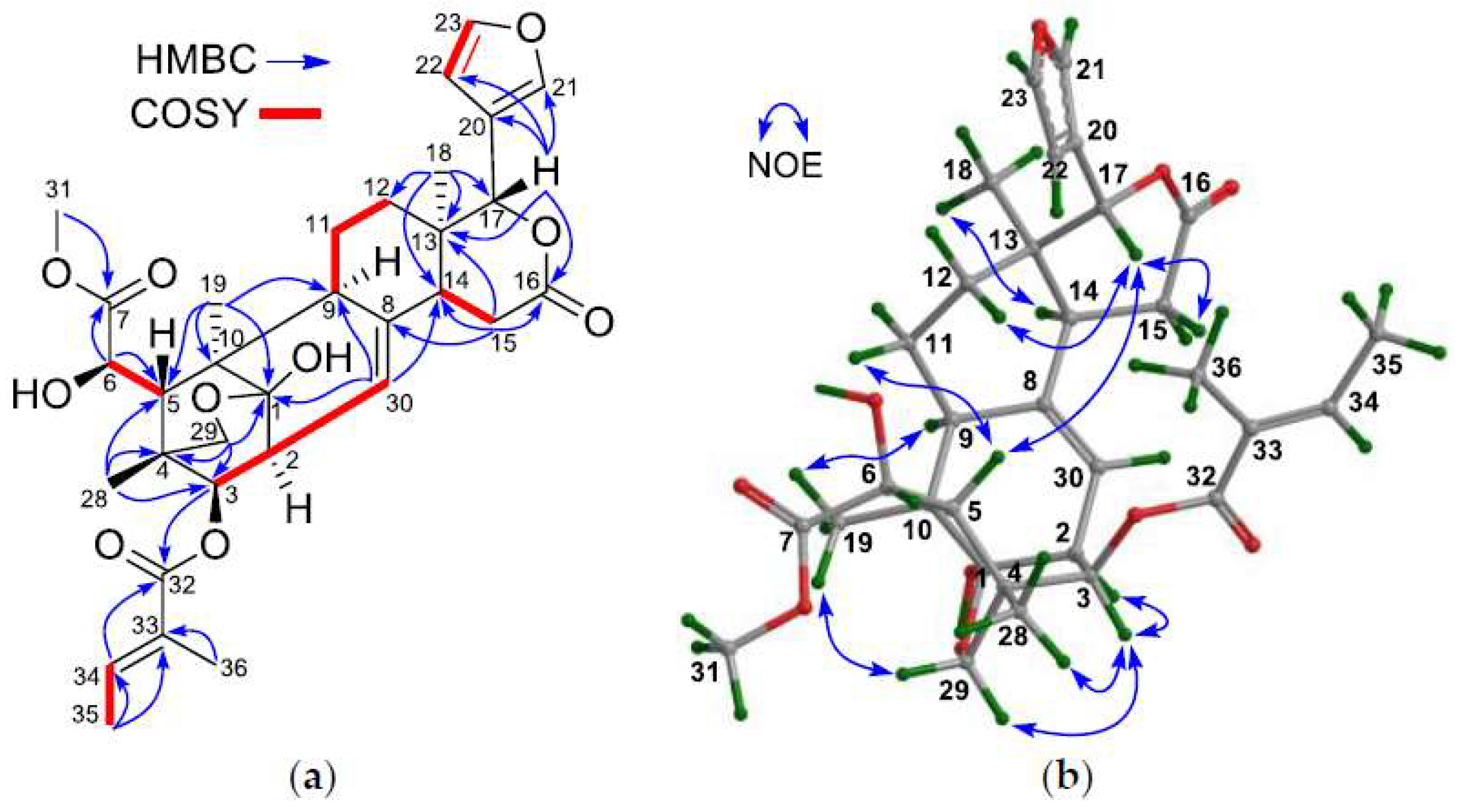
The NMR spectroscopic data of 1 (Table 1 and Table 2) and its 2D correlations (1H–1H COSY, HSQC, and HMBC) indicated the presence of a methoxycarbonyl moiety [δH 3.76 (s, H3-OMe); δC 53.2 (C-OMe), 176.1 (C-7)], a tigloyl group [δH 6.83 (q, J = 7.2 Hz), 1.67 (d, J = 7.2 Hz), 1.79 s; δC 167.2 qC, 127.8 qC, 138.7 CH, 14.6 CH3, 11.7 CH3], an oxygenated methylene moiety [δH 4.60 (d, J = 8.8 Hz, Hpro-S-29), 3.45 (d, J = 8.8 Hz, Hpro-R-29); δC 70.1 (C-29)], and a typical β-furyl ring [δH 7.50 (br s, H-21), 6.39 (br d, J = 1.2 Hz, H-22), 7.44 (t, J = 1.6 Hz, H-23); δC 121.5 (C-20, qC), 140.3 (C-21, CH), 109.3 (C-22, CH), 143.1 (C-23, CH)]. A δ-lactone ring (C-13, C-14, C-15, C-16, and C-17), characterized by NMR spectroscopic data [δH 2.31 (br s, H-14), 2.82 (m, H2-15); δC 36.7 (C-13, qC), 44.8 (C-14, CH), 29.6 (C-15, CH2), 169.3 (C-16, qC), and 76.6 (C-17, CH)], was corroborated by HMBC correlations between H2-15/C-13, H2-15/C-14, H2-15/C-16, H-17/C-13, H3-18/C-13, and H3-18/C-14 (Figure 2a). An olefinic methine group [δH 5.28 (d, J = 6.8 Hz); δC 120.2], exhibiting a 1H–1H COSY correlation to H-2 and HMBC correlations to C-9 and C-14, was attributed to CH-30.
The NMR spectroscopic data of 1 (Table 1 and Table 2) resembled those of granatumin L (6) [8], except for the presence of an additional 6-OH group in 1, being corroborated by the downshifted C-6 signal (δC 72.5 CH in 1; whereas δC 31.9 CH2 in granatumin L). 1H–1H COSY correlation between H-5/H-6 and HMBC correlations between H-6/C-5 and H-6/C-7 (Figure 2a) further confirmed the above result.
The relative configuration of 1, except for the chirality of C-6, was established to be the same as that of granatumin L (6) based on NOE interactions. Those from H-3 to Hpro-R-29 [δH 3.45 (d, J = 8.8 Hz)], but not from H-3 to H-5, established the α-oriented H-3 and the corresponding 3β-O-tigloyl group. NOE interactions between H3-18/H-14, H-9/H3-19, and H-3/H-2 indicated their mutual cis relationship and the α-oriented H-14, H-9, and H-2. Similarly, those between H-5/H-11β, H-5/H-17, and H-17/H-12β indicated the β-orientation of H-5 and H-17 (Figure 2b).
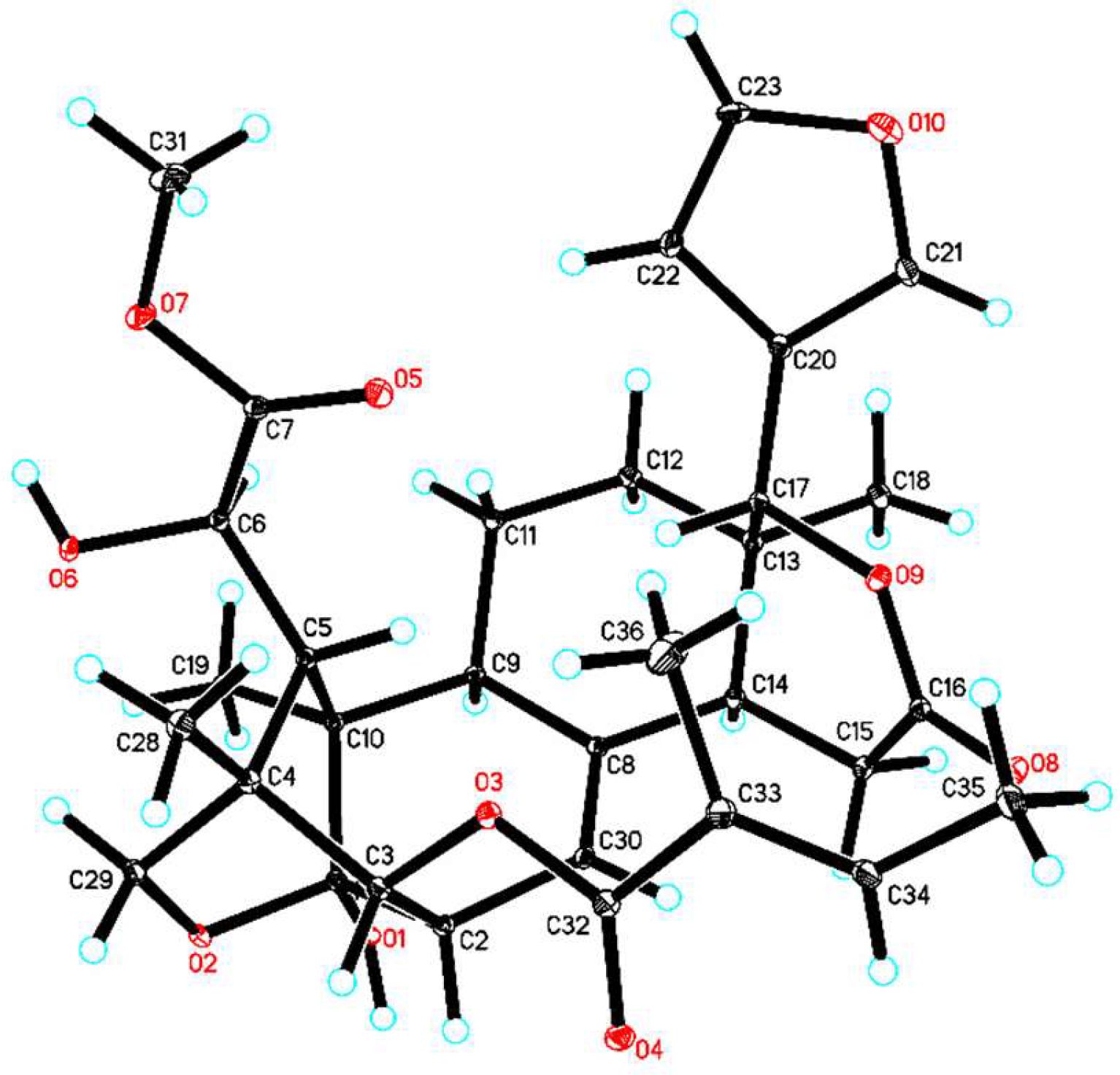
After considerable effort, suitable crystals of 1 were obtained in methanol/chloroform (4:1) at room temperature. Thus, the absolute configuration of 1 was unequivocally established as 1R,2S,3R,4S,5S,6R,9S,10R,13R,14S,17R (Figure 3, CCDC-1871152) by single-crystal X-ray diffraction analysis, conducted with CuKα radiation [Flack parameter of 0.02(3)]. Thus, the structure of 1, thaigranatin A, was assigned as depicted.
Compound 2, an amorphous powder, had the molecular formula C30H38O9 as established by the positive HR-ESIMS ion peak at m/z 543.2593 (calcd. for [M + H]+ 543.2589). The 1H and 13C NMR spectroscopic data of 2 (Table 1 and Table 2) were similar to those of 1, except for the absence of the 6-OH group and the replacement of the 3-O-tigloyl group in 1 by a 3-O-propionyl moiety [δH 2.37 (m, 2H), 1.09 (t, J = 7.2 Hz, 3H); δC 174.5 qC, 27.1 CH2, 8.7 CH3]. The HMBC correlation from H-3 [δH = 4.84 (d, J = 10.0 Hz)] to the carbonyl carbon (δC = 174.5 qC) of the propionyl group confirmed its location at C-3. The absence of the 6-OH group in 2 was corroborated by the upshifted CH2-6 [δH 2.35 (m H2-6); δC 31.9 qC] in 2. 1H–1H COSY correlations between H2-6/H-5 and HMBC correlations from H2-6 to C-5 and C-7 confirmed the above result.
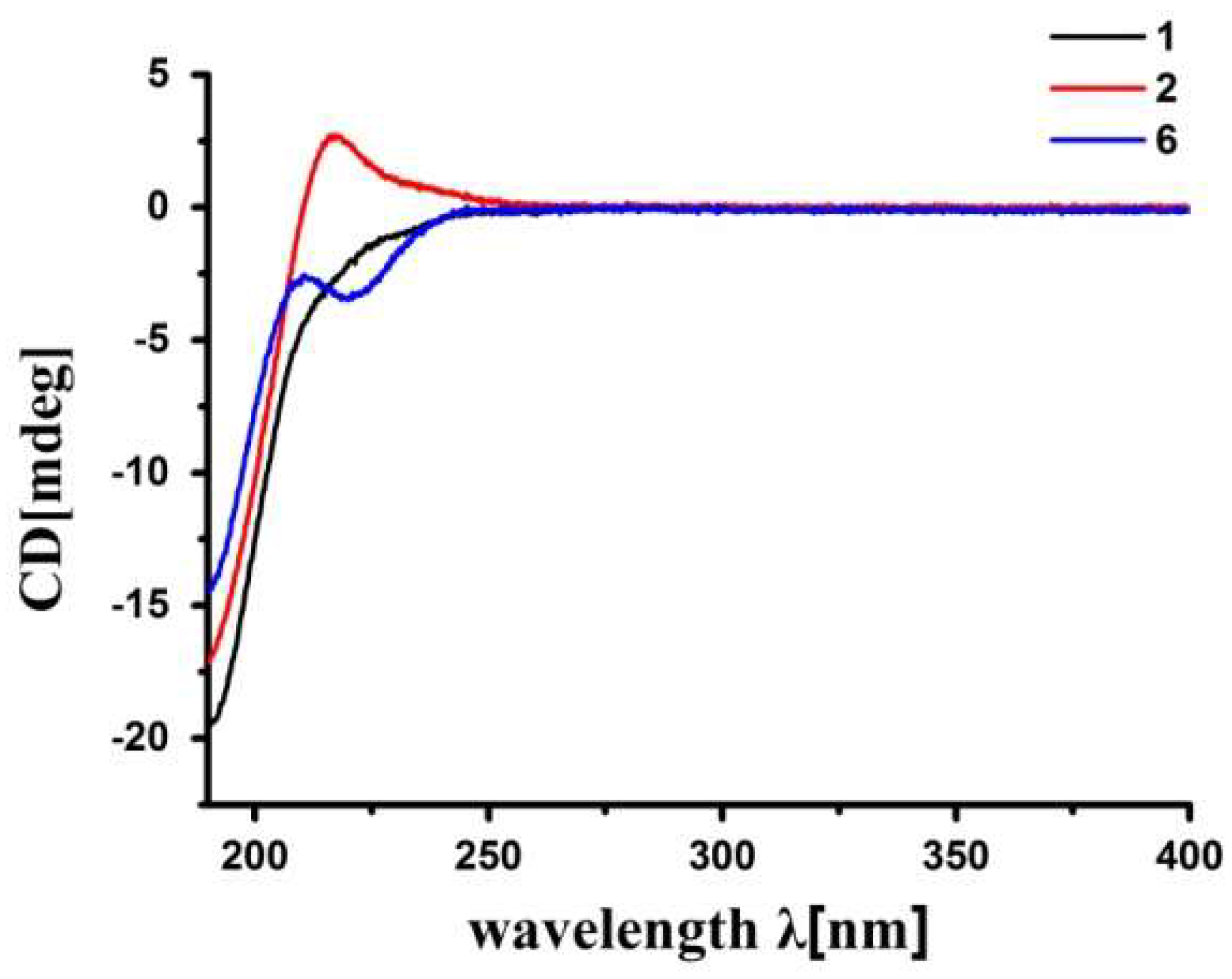
The relative configuration of 2 was determined to be the same as that of 1 by NOE interactions between H-17/H-11β, H-11β/H-5, and H-17/H-5, and those between H3-19/H-9, H3-19/Hpro-S-29, H-3/Hpro-R-29, H-3/H-2, and H3-18/H-14. The absolute configuration of 2, except for the deficiency of the chiral C-6, was established to be the same as that of 1, i.e., (1R,2S,3R,4S,5S,9S,10R,13R,14S,17R), by the accurate fit of their experimental electronic circular dichroism (ECD) spectra (Figure 4). Thus, the structure of 2, thaigranatin B, was assigned as depicted.
The molecular formula of 3 was determined to be C32H40O10 by HR-ESIMS ion peak at m/z 607.2512 (calcd. for [M + Na]+ 607.2514). The NMR spectroscopic dataof 3 (Table 1 and Table 2) were similar to those of godavarin D [12], the difference being the presence of an additional 30-OH group, which was corroborated by the downshifted C-30 (δC 66.3 CH in 3, whereas δC 26.3 CH2 in godavarin D) and 1H–1H COSY correlation between H-2/H-30. HMBC correlations from H-30 to C-1, C-2, C-3, C-8, C-9, and C-14 confirmed the above result (Figure 5a).
The relative configuration of 3 was established by NOE interactions (Figure 5b). Those between H-3/Hpro-R-29, H-3/H-2, H3-19/H-9, H3-18/H-11α assigned the α-oriented H-2, H-3, H3-18, and H3-19; whereas those between H-17/H-12β, H-12β/H-5, and H-17/H-5 established the β-oriented H-5 and H-17. The small coupling constant of J2,30, i.e., 2.4 Hz, revealed the Je,e coupling for H-2 and H-30, and thus the corresponding equatorial bond for H-30, i.e., the β-oriented H-30, which was further corroborated by NOE interactions between H-30/H-15α and H-30/H-15β of equal intensity, and no observation of NOE interaction between H-30/H-9. Thus, the structure of 3, thaigranatin C, was assigned as shown.
The molecular formula of 4 was established as C32H40O10 by the positive HR-ESIMS ion peak at m/z 585.2694 (calcd. for [M + H]+ 585.2694). The NMR spectroscopic data of 4 (Table 1 and Table 2) were similar to those of godavarin D [12], except for the presence of an additional 6-OH group, which was corroborated by the downshifted C-6 (δC 73.1 CH in 4; whereas δC 32.2 CH2 in godavarin D) [12]. 1H–1H COSY correlation between H-6/H-5 and HMBC correlations from H-6 to C-5 and C-7 confirmed the above result.
NOE interactions between H-17/H-12β and H-17/H-5 established the β-oriented H-5 and H-17. In turn, those between H3-18/H-11α, H3-19/H-9, H-3/Hpro-R-29, assigned the α-oriented H3-19, H-9, H3-18, and H-3. Thus, the structure of 4, thaigranatin D, was assigned as depicted.
Compound 5 gave the molecular formula C32H40O10 as obtained from the positive HR-ESIMS ion at m/z 607.2511 [M + Na]+ (calcd. for C32H40NaO10, 607.2514). The NMR spectroscopic data of 5 (Table 1 and Table 2) resembled those of 3, except for the replacement of the Δ8,14 double bond in 3 by a Δ8,9 double bond [δC 127.1 (C-8, qC), 139.2 (C-9, qC)]. HMBC correlations between H-15/C-8, H-15/C-9, H-30/C-8, H-30/C-9, and H3-19/C-9 confirmed the above deduction.
The relative configuration of 5 was established based on NOE interactions. Those between H-17/H-12β and H-5/H-11β assigned the β-oriented H-17 and H-5. NOE interactions between H3-18/H-14, H3-18/H-11α, and H-3/Hpro-R-29 assigned the α-orientation for H-14, H3-18, H3-19, and H-3. The broad singlet of H-30 revealed the Je,e coupling for H-2 and H-30, and thus assigned the corresponding equatorial bond for H-30, i.e., the β-oriented H-30. Thus, the structure of 5, thaigranatin E, was assigned as depicted.
2.2. Structural Modifications of Granatumin L (6)
In our previous bioassay, granatumin L (6) showed an inhibitory rate of 67.10 ± 3.04% against HIV-1 at the concentration of 20 μM. Thus, structural modification of the natural compound 6, particularly the bioequivalent substitution of the C-3 natural substituent, was investigated to find its potent and selective antiviral analogs. To achieve this purpose, 3 different protocols, viz. hydrolysis with alkaline KOH, esterification with diazomethane and various organic acids, and oximization with hydroxyamine, were employed (Scheme 1).
Compound 7 was obtained by the hydrolysis of 6 with an alkaline KOH. The ester functions at the C-3 and C-7 positions of 7 were hydrolyzed to give a hydroxy group at the C-3 position and a carboxyl group at the C-7 position, respectively, in the yield of 96.8%. Owing to the formation of the stable six-membered ring, the δ-lactone function of ring-D was not hydrolyzed. Then, 7 was treated with diazomethane to afford 8 as expected. Afterwards, the hydroxy group at the C-3 position of 8 was oxidized with IBX to give 9 with a carbonyl group at the C-3 position. Fortunately, the hemiacetal function of 8 was retained under the reaction condition.
Alternatively, 7 was first oxidized and then methylated to prepare 9. An intermediate compound 10 was obtained in the high yield of 98.8%. However, the final yield of 9 was 76.2%, which made this strategy inadvisable. Finally, the hydroxy group at the C-3 position of 8 was esterified with various organic acids to afford a series of compounds 8a–8i. The bioisosteric substitution of the ketone function at the C-3 position of 9 and 10 with different oximes led to the generation of derivatives 9a–9b and 10a–10c, respectively.
2.3. Antiviral Activity Bioassay
The inhibitory activities of natural limonoid 6 and all the modified derivatives 7–10, 8a–8i, 9a–9b, and 10a–10c against HIV-1 were tested [14]. Efavirenz was used as the positive control. The results were summarized in Table 3. The lack of substituent at the C-3 position of 8, 9, and 10 combined with the disappearance of their anti-HIV-1 activities indicated that the substituent at the C-3 position is essential for anti-HIV-1 activity of this limonoid skeleton. The oxidation of the hydroxy group at the C-3 position into a carbon-oxygen or carbon-nitrogen double bond did not improve anti-HIV-1 activity. For example, derivatives 9a–9b and 10a–10c containing a C=O or C=N double bond exhibited no activity.
Anti-HIV-1 bioassay (Table 3) revealed that the C-3 esterified products, particularly derivatives with a fatty acid ester at the C-3 position, markedly enhanced anti-HIV-1 activity. The derivative 8i exhibited the highest inhibition rate of 99.95 ± 0.01% at the concentration of 20.0 μM; whereas 8g showed an inhibition rate of 50.84 ± 6.96% at the same concentration. The IC50 values for 8i and 8g are 15.98 ± 6.87 and 21.98 ± 4.65 μM, respectively, among which the former is better than that of 6 (19.23 ± 0.12 μM). The results indicated that the introduction of alkyl groups at the C-3 position helped to maintain anti-HIV-1 activity. However, compounds with aromatic substitution at the C-3 position showed decreased activity.
Antiviral activities of all the modified derivatives of 6 against IAV were also evaluated [14]. Ribavirin was used as the positive control. Derivatives 8i and 8g exhibited no inhibitory activities against IAV at the concentration of 20.0 μM; whereas 8b, 8c, 8d, and 10b showed marked anti-IAV activities with IC50 values in the range of 14.0–22.8 μM (Table 4). The strongest inhibitory activity of 10b revealed the importance of a C-3–substituted O-methyl oxime group.
The above anti-HIV-1 and anti-IAV activities of modified derivatives shed light on the structural optimization of natural limonoid 6, particularly suitable substituted groups at the C-3 position. In-depth structural modification and structure-activity relationship studies of the limonoid skeleton of 6 will be proceeded by us in the near future.
3. Materials and Methods
3.1. General
Optical rotations were measured on a MCP200 modular circular polarimeter (Anton Paar OptoTec GmbH, Seelze, Germany). UV spectra were obtained on a GENESYS 10S UV–Vis spectrophotometer (Thermo Fisher Scientific, Shanghai, China). NMR spectra were recorded on a Bruker AV-400 spectrometer (Bruker Scientific Technology Co. Ltd., Karlsruhe, Germany) with TMS as the internal standard. HR-ESIMS were measured on a Bruker maXis ESI-QTOF mass spectrometer (Bruker Daltonics, Bremen, Germany). Semi-preparative HPLC was performed on C18 reversed-phase silica gel columns (YMC 250 × 10 mm i.d., or 250 × 4.6 mm i.d.) using a Waters 2535 pump equipped with a Waters 2489 UV detector (Waters Corporation, Milford, MA, USA). Silica gel (100–200 mesh) (Qingdao Marine Chemical Industrial Co. Ltd., Qingdao, China) and C18 reversed-phase silica gel (ODS-A-HG 12 nm, 50 μm, YMC Co. Ltd., Kyoto, Japan) were used for column chromatography. ECD spectra were measured on a Jasco J-810 spectropolarimeter (JASCO Corporation, Tokyo, Japan) in MeCN.
3.2. Plant Material
Seeds of Xylocarpus granatum were collected in August 2012 from the Thai mangrove swamps of the Trang Province. Identification of the mangrove was done by one of the authors (J.W.). A voucher sample (No. ThaiXG-02) is maintained in Marine Drugs Research Center, College of Pharmacy, Jinan University.
3.3. Extraction and Isolation
The air-dried and powdered seeds (9.0 kg) of Xylocarpus granatum were extracted 5 times with EtOH (95%, v/v) at room temperature to afford the resulting extract (1363.6 g). After removal of the solvent under vacuum, the residue was partitioned between water and EtOAc to yield the EtOAc portion (307.7 g). The EtOAc portion (150 g) was chromatographed on a silica gel column (70 × 15 cm i.d.), eluted with a gradient mixture of CHCl3/MeOH (100:0 to 5:1, v/v) to afford 116 fractions. Fractions 40 to 50 (19.4 g) were combined and further purified by C18 reversed-phase silica gel column chromatography (72 × 6.5 cm i.d.), eluted with a gradient mixture of acetone/H2O (40:60 to 100:0, v/v), to yield 58 subfractions, among which the subfraction 15 (1.27 g) was purified by preparative HPLC (MeCN/H2O, 40:60) to give compound 1 (25.0 mg). The subfraction 17 (1.50 g) was subjected to preparative HPLC (MeOH/H2O, 65:35) to yield compounds 2 (14.0 mg) and 4 (1.0 mg); whereas the subfraction 18 (1.36 g) was purified by preparative HPLC (MeOH/H2O, 69:31) to afford compounds 3 (1.3 mg) and 5 (34.3 mg). Fractions 19–20 (0.59 g) were combined and then recrystallized to afford crystals of 6 (500.0 mg) in the mixture of methanol/acetone (2:1) at room temperature.
Thaigranatin A (1): Colorless crystal, = −134 (c 0.1, acetone); UV (MeCN) λmax (logε) 203.6 (4.46) nm; ECD (0.34 mM, MeCN) λmax (Δε) 190.9 (−17.3) nm; HR-ESIMS m/z 607.2509 [M + Na]+ (calcd. for C32H40NaO10, 607.2514); 1H (400 MHz, CDCl3) and 13C NMR spectroscopic data (100 MHz, CDCl3) see Table 1 and Table 2.
Thaigranatin B (2): Amorphous solid, = −110 (c 0.1, acetone); UV (MeCN) λmax (logε) 200.0 (4.25) nm; ECD (0.37 mM, MeCN) λmax (Δε) 190.0 (−14.1), 217.6 (+2.2) nm; HR-ESIMS m/z 543.2593 [M + H]+ (calcd. for C30H39O9, 543.2589); 1H (400 MHz, CDCl3) and 13C NMR spectroscopic data (100 MHz, CDCl3) see Table 1 and Table 2.
Thaigranatin C (3): Amorphous solid, = −94 (c 0.1, acetone); UV (MeCN) λmax (logε) 203.8 (4.14), 287.2 (2.97) nm; ECD (0.34 mM, MeCN) λmax (Δε) 198.6 (−9.27), 219.6 (+3.7) nm; HR-ESIMS m/z 607.2512 [M + Na]+ (calcd. for C32H40NaO10, 607.2514); 1H (400 MHz, CDCl3) and 13C NMR spectroscopic data (100 MHz, CDCl3) see Table 1 and Table 2.
Thaigranatin D (4): Amorphous solid, = −130.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 201.6 (4.50) nm; ECD (0.34 mM, MeCN) λmax (Δε) 198.4 (−12.0), 220.2 (+8.2) nm; HR-ESIMS m/z 585.2694 [M + H]+ (calcd. for C32H41O10, 585.2694); 1H (400 MHz, CDCl3) and 13C NMR spectroscopic data (100 MHz, CDCl3) see Table 1 and Table 2.
Thaigranatin E (5): Amorphous solid, = −12.0 (c 0.1, acetone); UV (MeCN) λmax (logε) 197.8 (4.21); ECD (0.34 mM, MeCN) λmax (Δε) 195.2 (+8.7), 220.8 (−2.4), 251.8 (+0.6) nm; HR-ESIMS m/z 607.2511 [M + Na]+ (calcd. for C32H40NaO10, 607.2514); 1H (400 MHz, CDCl3) and 13C NMR spectroscopic data (100 MHz, CDCl3) see Table 1 and Table 2.
3.4. X-ray Crystal Data for Thaigranatin A (1)
Orthorhombic, C32H42O11 (C33H40O10·H2O), space group P2(1)2(1)2(1), a = 12.1554 (1) Å, b = 14.2125 (1) Å, c = 17.2234 (2) Å, α = 90°, β = 90°, γ = 90°, V = 2975.49 (5) Å3, Z = 4, Dcalcd = 1.323 Mg/m3, μ = 0.839 mm−1. Crystal size: 0.15 × 0.13 × 0.12 mm3. 36,359 measured reflections, 5945 [Rint = 0.0396] independent reflections, 400 parameters, 0 restraints, F(000) = 1248, R1 = 0.0398, wR2 = 0.1133 (all data), R1 = 0.0396, wR2 = 0.1131 [I > 2σ(I)], and goodness-of-fit (F2) = 1.100. The absolute structural parameter Flack x is −0.02(4), and Hooft y is −0.00(3).
CCDC-1871152 (1) contains the supplementary crystallographic data for this paper (excluding structure factors). These data are provided free of charge by The Cambridge Crystallographic Data Centre.
3.5. Procedure for Structural Modification of Granatumin L (6)
3.5.1. General Procedure for the Preparation of 7
Granatumin L (6) (500.0 mg) was stirred in methanol (25 mL) at room temperature for 20 min. Then 10% aqueous KOH solution was slowly added, and the mixture was stirred at room temperature for 48 h. Thin-layer chromatography (TLC) was used to monitor the products. After the reaction was completed, methanol was removed under reduced pressure. The remaining solution was neutralized with 10% HCl to pH 5–6, followed by C18 reversed-phase (RP18) HPLC to afford 7.
7: White powder, Yield 96.8%; 1H NMR (400 MHz, CD3OD): δ 7.74 (br s, 1H), 7.47 (t, J = 1.6 Hz, 1H), 6.50 (d, J = 1.4 Hz, 1H), 5.68 (s, 1H), 5.58 (br d, J = 7.0 Hz, 1H), 3.93 (d, J = 10.0 Hz, 1H), 3.83 (d, J = 10.0 Hz, 1H), 3.34 (m, 1H), 2.97 (dd, J = 18.8, 6.0 Hz, 1H), 2.93 (d, J = 18.8 Hz, 1H), 2.81 (m, 1H), 2.78 (m, 1H), 2.38 (m, 1H), 2.36 (m, 1H), 2.33 (m, 1H), 2.09 (dd, J = 12.0, 4.0 Hz, 1H), 1.82 (qd, J = 12.0, 4.0 Hz, 1H), 1.68 (m, 1H), 1.54 (m, 1H), 1.48 (m, 1H), 1.10 (s, 3H), 1.06 (s, 3H), 0.70 (s, 3H); 13C NMR (100 MHz, CD3OD): δ 177.7, 173.8, 144.2, 143.3, 138.2, 122.7, 122.3, 110.9, 98.5, 78.8, 74.5, 69.6, 49.4, 48.3, 46.1, 43.1, 38.6, 38.1, 35.9, 34.9, 33.1, 30.8, 22.2, 20.6, 15.6, 14.8; LR-ESIMS: [M + Na]+ calcd. for C26H32NaO8 495.20, found: 495.20.
3.5.2. General Procedure for the Preparation of 8
TMSCHN2 (5.0 mL) was added to a solution of 7 (50.0 mg) in methanol (2.0 mL) at room temperature [15]. The solution was stirred at room temperature for 10 h and the solvent was then removed under reduced pressure. The resulting residue was dissolved in acetone and purified by RP18 HPLC to give 8 (44.8 mg).
8: White powder, Yield 89.7%; 1H NMR (400 MHz, CDCl3): δ 7.69 (br s, 1H), 7.40 (t, J = 1.6 Hz, 1H), 6.45 (br s, 1H), 5.62 (br d, J = 6.8 Hz, 1H), 5.57 (s, 1H), 3.90 (d, J = 9.6 Hz, 1H), 3.88 (dd, J = 10.0, 6.8 Hz, 1H), 3.68 (s, 3H), 3.39 (dd, J = 9.6, 2.0 Hz, 1H), 2.96 (dd, J = 18.8, 2.0 Hz, 1H), 2.90 (dd, J = 18.8, 5.6 Hz, 1H), 2.82 (br t, J = 8.4 Hz, 1H), 2.70 (br d, J = 10.4 Hz, 1H), 2.45 (s, 1H), 2.39 (dd, J = 16.8, 10.4 Hz, 1H), 2.32 (d, J = 16.8 Hz, 1H), 2.31 (m, 1H), 2.15 (br d, J = 12.8 Hz, 1H), 1.84 (d, J = 6.8 Hz, 1H), 1.77 (m, 1H), 1.65 (m, 1H), 1.61 (m, 1H), 1.45 (m, 1H), 1.10 (s, 3H), 1.05 (s, 3H), 0.66 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.1, 170.4, 142.9, 141.8, 139.3, 120.7, 120.1, 109.7, 97.0, 77.1, 73.2, 68.5, 52.1, 48.0, 46.9, 45.2, 41.8, 37.6, 36.9, 34.5, 33.9, 32.0, 30.0, 21.9, 19.5, 14.8, 14.4; LR-ESIMS: [M + H]+ calcd. for C27H35O8 487.23, found: 487.17.
3.5.3. General Procedure for the Preparation of 8a–8i
Compound 8 (5.0 mg, 0.01 mmol) and various organic acid (1.5 eq.) were dissolved in dry dichloromethane (2.0 mL). Then the mixture was combined with DCC (1.5 eq.) and DMAP (1.0 eq.), and then stirred at room temperature for 3–5 h [16]. TLC was used to monitor the products. After the reaction was completed, the reaction solvent was evaporated. The resulting residue was dissolved in acetone or acetonitrile; whereas the insoluble matter was precipitated by centrifugation. The supernatant was purified by RP18 HPLC to afford the pure corresponding esterified products (8a–8i).
8a: white powder, yield 74.5%; 1H NMR (400 MHz, CDCl3): δ 8.06 (dd, J = 8.0 Hz, 1.2 Hz, 2H), 7.78 (br s, 1H), 7.48 (tt, J = 7.6 Hz, 1.2 Hz, 1H), 7.43 (t, J = 1.6 Hz, 1H), 7.29 (t, J = 8.0 Hz, 2H), 6.44 (br d, J = 1.2 Hz, 1H), 5.46 (s, 1H), 5.37 (br d, J = 7.2 Hz, 1H), 5.13 (d, J = 10.0 Hz, 1H), 4.00 (d, J = 10.0 Hz, 1H), 3.72 (s, 3H), 3.59 (dd, J = 10.0 Hz, 1.6 Hz, 1H), 3.11 (br d, J = 10.4 Hz, 1H), 3.05 (br t, J = 8.4 Hz, 1H), 2.72 (br d, J = 4.0 Hz, 2H), 2.46 (dd, J = 16.8 Hz, 10.8 Hz, 1H), 2.38 (dd, J = 16.8 Hz, 1.6 Hz, 1H), 2.24 (br s, 1H), 2.14 (dd, J = 12.4, 4.8 Hz, 1H), 1.65 (m, 1H), 1.81 (qd, J = 13.2, 4.0 Hz, 1H), 1.65 (m, 1H), 1.59 (m, 1H), 1.43 (td, J = 14.0, 4.0 Hz, 1H), 1.11 (s, 3H), 1.02 (s, 3H), 0.73 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.0, 168.9, 166.1, 143.0, 141.8, 139.0, 133.4, 129.7, 128.9, 128.6, 120.8, 119.8, 109.8, 97.0, 76.1, 75.4, 67.9, 52.2, 48.2, 45.5, 45.0, 41.6, 37.1, 36.6, 35.0, 34.5, 31.9, 29.6, 21.5, 19.7, 15.0, 14.5; HR-ESIMS: [M + H]+ calcd. for C34H39O9 591.2589, found: 591.2592.
8b: colorless crystal, yield 70.3%; 1H NMR (400 MHz, CDCl3): δ 7.89 (br d, J = 8.8 Hz, 2H), 7.72 (br s, 1H), 7.45 (t, J = 1.6 Hz, 1H), 7.40 (br d, J = 8.8 Hz, 2H), 6.43 (br d, J = 1.2 Hz, 1H), 5.42 (s, 1H), 5.36 (br d, J = 7.2 Hz, 1H), 5.11 (d, J = 10.0 Hz, 1H), 3.99 (d, J = 9.6 Hz, 1H), 3.72 (s, 3H), 3.58 (dd, J = 9.6 Hz, 1.6 Hz, 1H), 3.08 (br d, J = 10.4 Hz, 1H), 3.03 (br t, J = 8.4 Hz, 1H), 2.76 (dd, J = 18.8, 5.2 Hz, 1H), 2.71 (dd, J = 18.8, 2.0 Hz, 1H), 2.45 (dd, J = 16.8, 10.4 Hz, 1H), 2.37 (br d, J = 16.8 Hz, 1H), 2.36 (br s, 1H), 2.24 (br s, 1H), 2.15 (dd, J = 12.4, 4.8 Hz, 1H), 1.80 (qd, J = 13.2, 4.4 Hz, 1H), 1.67 (m, 1H), 1.63 (m, 1H), 1.44 (td, J = 14.0, 4.4 Hz, 1H), 1.10 (s, 3H), 1.01 (s, 3H), 0.70 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.0, 168.9, 165.4, 143.0, 141.5, 139.2, 132.0, 131.2, 128.6, 127.9, 120.9, 119.6, 109.7, 97.0, 76.4, 75.7, 67.8, 52.2, 48.1, 45.4, 45.0, 41.6, 37.0, 36.6, 34.9, 34.4, 31.8, 29.8, 21.5, 19.7, 15.0, 14.5; HR-ESIMS: [M + H]+ calcd. for C34H38BrO9 669.1694, found: 669.1699.
8c: colorless crystal, yield 96.4%; 1H NMR (400 MHz, CDCl3): δ 7.76 (br s, 1H), 7.42 (t, J = 1.6 Hz, 1H), 6.85 (m, 4H), 6.45 (br d, J = 1.2 Hz, 1H), 5.58 (s, 1H), 5.35 (br d, J = 6.8 Hz, 1H), 4.99 (d, J = 10.0 Hz, 1H), 4.67 (s, 2H), 3.92(d, J = 10.0 Hz, 1H), 3.66 (s, 3H), 3.49 (dd, J = 10.0, 1.6 Hz, 1H), 2.95 (br t, J = 8.4 Hz, 1H), 2.92 (dd, J = 18.8, 5.2 Hz, 1H), 2.86 (dd, J = 18.8, 1.6 Hz, 1H), 2.82 (br d, J = 10.4 Hz, 1H), 2.36 (m, 1H), 2.34 (m, 1H), 2.31 (m, 2H), 2.18 (m, 1H), 1.81 (qd, J = 13.2, 4.4 Hz, 1H), 1.66 (m, 1H), 1.61 (m, 1H), 1.46 (td, J = 14.4, 4.8 Hz, 1H), 1.10 (s, 3H), 1.07 (s, 3H), 0.51 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 173.7, 170.4, 169.1, 158.8, 156.4, 153.9, 142.9, 141.8, 138.8, 120.7, 119.5, 116.0, 115.9, 115.7, 115.5, 109.8, 96.7, 77.3, 76.2, 67.8, 65.4, 52.1, 47.5, 45.1, 44.8, 41.7, 36.8, 36.2, 35.2, 34.3, 31.8, 30.3, 22.0, 19.3, 14.7, 14.5; LR-ESIMS: [M + Na]+ calcd. for C35H39FNaO10 661.24, found: 661.31.
8d: light yellow powder, yield 85.2%; 1H NMR (400 MHz, CDCl3): δ 8.22 (br d, J = 9.2 Hz, 2H), 8.08 (br d, J = 9.2 Hz, 2H), 7.72 (br s, 1H), 7.45 (t, J = 1.6 Hz, 1H), 6.36 (br d, J = 1.2 Hz, 1H), 5.36 (s, 1H), 5.31 (br d, J = 6.8 Hz, 1H), 5.10 (d, J = 10.0 Hz, 1H), 4.01 (d, J = 10.0 Hz, 1H), 3.76 (s, 3H), 3.60 (dd, J = 10.0, 1.6 Hz, 1H), 3.10 (br d, J = 10.8 Hz, 1H), 3.06 (m, 1H), 2.73 (dd, J = 18.8, 5.6 Hz, 1H), 2.66 (dd, J = 18.8, 2.0 Hz, 1H), 2.47 (dd, J = 17.2, 10.8 Hz, 1H), 2.39 (br d, J = 17.2 Hz, 1H), 2.23 (br s, 1H), 2.16 (dd, J = 13.2, 4.8 Hz, 1H), 1.79 (qd, J = 13.2, 4.4 Hz, 1H), 1.69 (m, 1H), 1.65 (m, 1H), 1.43 (td, J = 10.0, 5.2 Hz, 1H), 1.12 (s, 3H), 0.98 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.2, 168.8, 164.2, 150.6, 143.3, 141.4, 139.8, 134.2, 130.9, 123.8, 120.7, 119.2, 109.6, 96.9, 76.7, 76.5, 67.7, 52.3, 48.1, 45.2, 45.0, 41.6, 36.9, 36.4, 34.9, 34.3, 31.7, 29.9, 21.5, 19.7, 15.0, 14.5; HR-ESIMS: [M + H]+ calcd. for C34H38NO11 636.2439, found: 636.2449.
8e: light yellow crystal, yield 74.0%; 1H NMR (400 MHz, CDCl3): δ 8.85 (t, J = 1.6 Hz, 1H), 8.38 (dt, J = 7.6, 1.2 Hz, 1H), 8.32 (dq, J = 8.4, 1.2 Hz, 1H), 7.68 (br s, 1H), 7.48 (t, J = 8.0 Hz, 1H), 7.41(t, J = 1.6 Hz, 1H), 6.33 (br d, J = 1.2 Hz, 1H), 5.32 (s, 1H), 5.29 (br d, J = 6.8 Hz, 1H), 5.09 (d, J = 10.4 Hz, 1H), 4.02 (d, J = 9.6 Hz, 1H), 3.80 (s, 3H), 3.60 (dd, J = 9.6 Hz, 2.0 Hz, 1H), 3.09 (br d, J = 10.4 Hz, 1H), 3.08 (overlapped, 1H), 2.70 (dd, J = 18.8, 5.2 Hz, 1H), 2.65 (dd, J = 18.8, 2.8 Hz, 1H), 2.48 (dd, J = 16.8, 10.4 Hz, 1H), 2.42 (br s, 1H), 2.40 (br d, J = 16.8 Hz, 1H), 2.22 (br s, 1H), 2.15 (dd, J = 12.4, 5.2 Hz, 1H), 1.79 (qd, J = 13.2 Hz, 4.4 Hz, 1H), 1.66 (m, 1H), 1.56 (m, 1H), 1.42 (td, J = 14.0, 4.4 Hz, 1H), 1.12 (s, 3H), 0.96 (s, 3H), 0.79 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.0, 168.9, 164.2, 148.3, 143.1, 141.7, 139.7, 135.5, 130.6, 129.9, 127.8, 124.7, 120.5, 119.2, 109.5, 96.9, 76.9, 76.4, 67.8, 52.4, 48.0, 45.2, 44.9, 41.6, 36.9, 36.3, 35.0, 34.4, 31.8, 29.7, 21.6, 19.6, 15.1, 14.5; HR-ESIMS: [M + H]+ calcd. for C34H38NO11 636.2439, found: 636.2441.
8f: white powder, yield 63.7%; 1H NMR (400 MHz, CDCl3): δ 7.98 (dd, J = 7.6, 1.6 Hz, 1H), 7.73 (br s, 1H), 7.40 (t, J = 1.6 Hz, 1H), 7.40 (overlapped, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.76 (t, J = 7.6 Hz, 1H), 6.42 (br d, J = 1.2 Hz, 1H), 5.39 (s, 1H), 5.38 (br d, J = 6.8 Hz, 1H), 5.10 (d, J = 10.0 Hz, 1H), 4.0 (d, J = 9.6 Hz, 1H), 3.91 (s, 3H), 3.72 (s, 3H), 3.57 (dd, J = 9.6, 1.6 Hz, 1H), 3.06 (overlapped, 1H), 3.04 (br d, J = 10.8 Hz, 1H), 2.76 (dd, J = 18.8, 2.4 Hz, 1H), 2.71 (dd, J = 18.8, 5.6 Hz, 1H), 2.45 (dd, J = 16.8, 10.4 Hz, 1H), 2.37 (dd, J = 16.8, 1.6 Hz, 1H), 2.22 (br d, J = 2.4 Hz, 1H), 2.13 (dd, J = 12.4, 5.2 Hz, 1H), 1.79 (qd, J = 13.2, 4.4 Hz, 2H), 1.63 (m, 1H), 1.55 (m, 1H), 1.41 (td, J = 14.0, 4.4 Hz, 1H), 1.10 (s, 3H), 1.00 (s, 3H), 0.75 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.0, 169.0, 164.9, 160.5, 142.8, 141.7, 138.6, 134.5, 132.2, 120.9, 120.3, 120.0, 117.4, 112.0, 109.9, 97.0, 76.1, 75.3, 68.1, 55.8, 52.1, 48.1, 45.5, 44.9, 41.7, 37.1, 36.5, 35.2, 34.5, 31.9, 29.6, 21.5, 19.6, 15.0, 14.6; HR-ESIMS: [M + H]+ calcd. for C35H41O10 621.2694, found: 621.2709.
8g: pink powder, yield 55.6%; 1H NMR (400 MHz, CDCl3): δ 7.76 (br s, 1H), 7.42 (t, J = 1.6 Hz, 1H), 6.47 (br d, J = 1.2 Hz, 1H), 5.57 (s, 1H), 5.34 (br d, J = 6.8 Hz, 1H), 4.96 (d, J = 10.4 Hz, 1H), 3.93 (d, J = 9.6 Hz, 1H), 3.68 (s, 3H), 3.50 (dd, J = 9.6, 1.6 Hz, 1H), 2.89 (m, 1H), 2.88 (overlapped, 1H), 2.88 (overlapped, 1H), 2.87 (br d, J = 10.4 Hz, 1H), 2.39 (dd, J = 16.8, 10.4 Hz, 1H), 2.37 (m, 1H), 2.32 (dd, J = 16.8, 2.4 Hz, 1H), 2.30 (br s, 1H), 2.14 (dd, J = 12.4, 4.8 Hz, 1H), 1.96 (m, 1H), 1.92 (m, 1H), 1.77 (qd, J = 13.2, 4.4 Hz, 1H), 1.67 (m, 1H), 1.63 (m, 1H), 1.54 (m, 1H), 1.50 (m, 1H), 1.46 (m, 1H), 1.37 (m, 1H), 1.33 (m, 1H), 1.32 (m, 1H), 1.22 (m, 1H), 1.17 (m, 1H), 1.11 (s, 3H), 1.07 (s, 3H), 0.61 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 175.5, 173.7, 169.9, 143.0, 141.8, 138.3, 120.7, 120.0, 109.7, 96.9, 76.7, 74.2, 67.9, 52.0, 48.0, 45.2, 45.2, 42.7, 41.6, 37.1, 36.6, 34.9, 34.5, 31.9, 30.0, 29.2, 28.4, 25.7, 25.4, 25.1, 21.7, 19.6, 14.8, 14.5; HR-ESIMS: [M + H]+ calcd. for C34H45O9 597.3058, found: 597.3060.
8h: white powder, yield 54.2%; 1H NMR (400 MHz, CDCl3): δ 8.36 (d, J = 4.8 Hz, 1H), 7.88 (br s, 1H), 7.79 (dd, J = 5.2, 1.2 Hz, 1H), 7.73 (br s, 1H), 7.42 (t, J = 1.6 Hz, 1H), 6.37 (br d, J = 1.2 Hz, 1H), 5.36 (s, 1H), 5.26 (br d, J = 7.2 Hz, 1H), 5.05 (d, J = 10.0 Hz, 1H), 4.00 (d, J = 9.6 Hz, 1H), 3.78 (s, 3H), 3.58 (dd, J = 9.6, 1.6 Hz, 1H), 3.06 (br d, J = 10.0 Hz, 1H), 3.05 (overlapped, 1H), 2.73 (dd, J = 18.8, 5.6 Hz, 1H), 2.66 (br d, J = 18.8 Hz, 1H), 2.47 (dd, J = 16.8, 10.4 Hz, 1H), 2.40 (dd, J = 16.8, 1.6 Hz, 1H), 2.23 (br d, J = 2.8 Hz, 1H), 2.16 (dd, J = 13.0, 5.2 Hz, 1H), 1.77 (qd, J = 13.2, 4.0 Hz, 1H), 1.67 (m, 1H), 1.62 (m, 1H), 1.43 (td, J = 14.0, 4.0 Hz, 1H), 1.11 (s, 3H), 0.99 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 174.1, 168.8, 163.5, 152.5, 150.7, 143.0, 141.7, 139.9, 138.9, 124.1, 121.7, 120.6, 119.0, 109.7, 96.9, 77.2, 76.5, 67.7, 52.3, 48.0, 45.0, 45.0, 41.6, 36.9, 36.3, 34.9, 34.3, 31.8, 29.9, 21.6, 19.6, 15.1, 14.4; HR-ESIMS: [M + H]+ calcd. for C33H37ClNO9 626.2151, found: 626.2156.
8i: white crystal, yield 84.7%; 1H NMR (400 MHz, CDCl3): δ 7.76 (br s, 1H), 7.41 (t, J = 1.6 Hz, 1H), 6.46 (br d, J = 1.2 Hz, 1H), 5.56 (s, 1H), 5.32 (br d, J = 6.8 Hz, 1H), 4.87 (d, J = 10.0 Hz, 1H), 3.94 (d, J = 9.6 Hz, 1H), 3.69 (s, 3H), 3.50 (dd, J = 9.6, 1.6 Hz, 1H), 2.93 (br t, J = 8.4 Hz, 1H), 2.89 (dd, J = 18.8, 3.2 Hz, 1H), 2.85 (overlapped, 1H), 2.85 (br d, J = 10.4 Hz, 1H), 2.40 (dd, J = 16.8, 10.8 Hz, 1H), 2.36 (overlapped, 1H), 2.33 (dd, J = 16.8, 3.2 Hz, 1H), 2.31 (overlapped, 1H), 2.29 (br s, 1H), 2.15 (dd, J = 12.4, 5.2 Hz, 1H), 1.79 (qd, J = 13.2, 4.4 Hz, 1H), 1.63 (m, 1H), 1.56 (m, 1H), 1.45 (td, J = 13.2, 4.4 Hz, 1H), 1.25 (m, 2H), 1.24 (m, 2H), 1.22 (m, 2H), 1.20 (m, 2H), 1.19 (m, 2H), 1.10 (s, 3H), 1.07 (s, 3H), 0.83(t, J = 7.2 Hz, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 173.8, 173.8, 169.9, 142.9, 141.8, 138.4, 120.8, 120.0, 109.8, 96.8, 76.9, 75.0, 68.0, 52.0, 47.8, 45.1, 45.0, 41.6, 36.9, 36.2, 35.1, 34.5, 33.9, 31.9, 31.6, 30.1, 29.1, 28.9, 24.7, 22.6, 21.8, 19.4, 14.8, 14.5, 14.1; HR-ESIMS: [M + H]+ calcd. for C35H49O9 613.3371, found: 613.3380.
3.5.4. General Procedure for the Preparation of 9
Compound 8 (50.0 mg) was dissolved in DMSO (2.0 mL) at room temperature. Then IBX (42.0 mg) was added [17]. The mixture was stirred at room temperature for 7.5 h. The solvent was then removed by lyophilization. The resulting residue was dissolved in acetone and purified by RP18 HPLC to give 9 (49.2 mg).
9: White powder, Yield 98.4%; 1H NMR (400 MHz, CDCl3): δ 7.64 (br s, 1H), 7.39 (t, J = 1.6 Hz, 1H), 6.42 (br d, J = 1.2 Hz, 1H), 5.77 (dt, J = 7.2, 2.0 Hz, 1H), 5.36 (s, 1H), 4.02 (d, J = 9.6 Hz, 1H), 3.69 (s, 3H), 3.55 (dd, J = 9.6, 1.6 Hz, 1H), 3.01 (br d, J = 7.2 Hz, 1H), 2.92 (s, 1H), 2.91 (d, J = 2.8 Hz, 1H), 2.74 (s, 1H), 2.58 (br d, J = 10.4 Hz, 1H), 2.51 (dd, J = 16.0, 10.4 Hz, 1H), 2.43 (br d, J = 16.0 Hz, 1H), 2.33 (overlapped, 1H), 2.30 (m, 1H), 1.72 (m, 1H), 1.67 (m, 1H), 1.63 (m, 1H), 1.45 (m, 1H), 1.20 (s, 3H), 1.11 (s, 3H), 0.79 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 207.2, 173.2, 169.6, 143.0, 141.8, 138.4, 120.5, 119.1, 109.6, 97.8, 76.6, 66.1, 57.0, 52.4, 48.6, 46.4, 44.9, 42.2, 36.9, 36.8, 34.3, 31.7, 29.6, 21.8, 19.1, 14.5, 11.4; LR-ESIMS: [M + H]+ calcd. for C27H33O8 485.21, found: 485.14.
3.5.5. General Procedure for the Preparation of 9a and 9b
Compound 9 (5.0 mg, 0.01 mmol) and oxyamine (4 eq.) in ethanol (2 mL) were added with pyridine (4 eq.), and then stirred at room temperature for 48 h [18]. TLC was used to monitor the products. After the reaction was completed, the reaction solvent was precipitated by centrifugation. The resulting supernatant was purified by RP18 HPLC to give the pure corresponding derivatives, 9a and 9b.
9a: White powder, Yield 51.0%; 1H NMR (400 MHz, CDCl3): δ 7.65 (br s, 1H), 7.39 (t, J = 1.6 Hz, 1H), 6.42 (br d, J = 1.2 Hz, 1H), 6.07 (br d, J = 6.4 Hz, 1H), 5.40 (s, 1H), 3.97 (d, J = 9.2 Hz, 1H), 3.70 (s, 3H), 3.52 (overlapped, 1H), 3.52 (dd, J = 9.2, 1.2 Hz, 1H), 2.97 (br d, J = 18.8 Hz, 1H), 2.90 (dd, J = 18.8, 6.0 Hz, 1H), 2.54 (br d, J = 10.8 Hz, 1H), 2.48 (dd, J = 16.0, 10.8 Hz, 1H), 2.38 (br d, J = 16.0 Hz, 1H), 2.31 (br d, J = 2.8 Hz, 1H), 2.18 (dd, J = 12.0, 5.6 Hz, 1H), 1.71 (m, 1H), 1.64 (m, 1H), 1.59 (m, 1H), 1.42 (m, 1H), 1.13 (s, 3H), 1.09 (s, 3H), 0.86 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 173.6, 170.4, 161.0, 143.0, 141.8, 136.6, 120.6, 119.9, 109.7, 97.2, 76.9, 68.4, 52.3, 47.9, 45.4, 44.8, 42.1, 39.5, 38.6, 36.8, 34.4, 32.0, 29.8, 21.8, 19.2, 14.7, 13.0; LR-ESIMS: [M + H]+ calcd. for C27H34NO8 500.22, found: 500.16.
9b: White powder, Yield 60.0%; 1H NMR (400 MHz, CDCl3): δ 7.67 (br s, 1H), 7.40 (t, J = 1.6 Hz, 1H), 6.43 (br d, J = 1.2 Hz, 1H), 5.97 (dt, J = 6.8, 2.0 Hz, 1H), 5.42 (s, 1H), 3.97 (d, J = 9.2 Hz, 1H), 3.86 (s, 3H), 3.69 (s, 3H), 3.51 (dd, J = 9.2 Hz, 1.6 Hz, 1H), 3.43 (br d, J = 6.8 Hz, 1H), 2.97 (dd, J = 18.8, 1.6 Hz, 1H), 2.89 (dd, J = 18.8, 6.4 Hz, 1H), 2.53 (br d, J = 10.8 Hz, 1H), 2.48 (dd, J = 15.6, 10.8 Hz, 1H), 2.41 (s, 1H), 2.37 (br d, J = 15.6 Hz, 1H), 2.31 (m, 1H), 2.19 (dd, J = 12.4, 5.2 Hz, 1H), 1.71 (qd, J = 13.2, 4.0 Hz, 1H), 1.66 (m, 1H), 1.58 (m, 1H), 1.43 (td, J = 14.0, 4.8 Hz, 1H), 1.12 (s, 3H), 1.09 (s, 3H), 0.86 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 173.5, 170.0, 159.5, 142.9, 141.8, 136.4, 120.6, 120.2, 109.6, 97.2, 76.8, 68.6, 61.8, 52.2, 47.9, 46.0, 44.9, 42.1, 39.6, 38.6, 36.8, 34.4, 32.1, 29.8, 21.8, 19.2, 14.6, 13.0; LR-ESIMS: [M + Na]+ calcd. for C28H35NNaO8 536.23, found: 536.30.
3.5.6. General Procedure for the Preparation of 10
Compound 7 (50.0 mg) was dissolved in DMSO (2.0 mL) at room temperature. Then IBX (42.0 mg) was added [17]. Then the mixture was stirred for 7.5 h. TLC was used to monitor the products. Then the solvent was removed by lyophilization. The resulting residue was dissolved in acetone and purified by RP18 HPLC to afford 10 (49.4 mg).
10: White powder, Yield 98.8%; 1H NMR (400 MHz, CD3COCD3): δ 7.70 (br s, 1H), 7.54 (br s, 1H), 6.51 (br s, 1H), 5.69 (br d, J = 5.2 Hz, 1H), 5.35 (s, 1H), 4.14 (d, J = 9.6 Hz, 1H), 3.45 (d, J = 9.6 Hz, 1H), 3.00 (dd, J = 18.4, 6.4 Hz, 1H), 2.95 (d, J = 6.4 Hz, 1H), 2.82 (br d, J = 18.4 Hz, 1H), 2.65 (m, 1H), 2.60 (m, 1H), 2.59 (overlapped, 1H), 2.42 (br d, J = 6.4 Hz, 1H), 2.35 (dd, J = 12.0, 4.8 Hz, 1H), 1.78 (m, 1H), 1.71 (m, 1H), 1.55 (m, 1H), 1.52 (m, 1H), 1.26 (s, 3H), 1.14 (s, 3H), 0.79 (s, 3H); 13C NMR (100 MHz, CD3COCD3): δ 209.4, 175.6, 169.5, 144.1, 142.8, 139.7, 122.1, 120.1, 110.8, 98.4, 77.1, 66.3, 57.9, 49.5, 47.2, 45.6, 43.0, 37.7, 37.4, 35.1, 31.7, 30.4, 22.2, 19.8, 14.9, 12.0; LR-ESIMS: [M + H]+ calcd. for C26H31O8 471.20, found: 471.14.
3.5.7. General Procedure for the Preparation of 10a–10c
Compound 10 (5.0 mg, 0.01 mmol) and oxyamine (4 eq.) in ethanol (2 mL) were added with pyridine (4 eq.), and then stirred at room temperature for 48 h [18]. TLC was used to monitor the products. After the reaction was completed, the solvent was precipitated by centrifugation. The supernatant was purified by RP18 HPLC to give the pure corresponding derivatives (10a–10c).
10a: white powder, yield 65.8%; 1H NMR (400 MHz, CD3COCD3): δ 7.73 (br s, 1H), 7.54 (t, J = 1.6 Hz, 1H), 6.52 (d, J = 1.2 Hz, 1H), 6.02 (dt, J = 6.8 Hz, 1H), 5.39 (s, 1H), 4.05 (d, J = 9.2 Hz, 1H), 3.52 (br d, J = 6.8 Hz, 1H), 3.37 (dd, J = 9.2, 1.6 Hz, 1H), 2.95 (dd, J = 18.4, 6.4 Hz, 1H), 2.83 (br d, J = 18.4 Hz, 1H), 2.58 (br t, J = 7.2 Hz, 1H), 2.56 (dd, J = 20.8, 8.2 Hz, 1H), 2.51 (dd, J = 20.8, 7.2 Hz, 1H), 2.37 (m, 1H), 2.22 (m, 1H), 1.74 (m, 1H), 1.71 (m, 1H), 1.51 (m, 1H), 1.48 (m, 1H), 1.19 (s, 3H), 1.13 (s, 3H), 0.89 (s, 3H); 13C NMR (100 MHz, CD3COCD3): δ 175.7, 169.5, 161.5, 144.0, 142.8, 137.6, 122.2, 120.9, 110.8, 97.8, 77.0, 69.0, 49.0, 46.4, 45.6, 42.8, 40.2, 39.4, 37.4, 35.3, 32.0, 30.5, 22.2, 19.9, 15.1, 13.7; LR-ESIMS: [M + H]+ calcd. for C26H32NO8 486.21, found: 486.16.
10b: transparent crystal, yield 74.3%; 1H NMR (400 MHz, CD3COCD3): δ 7.73 (br s, 1H), 7.54 (t, J = 1.6 Hz, 1H), 6.53 (d, J = 1.6 Hz, 1H), 5.89 (dt, J = 6.4, 2.0 Hz, 1H), 5.38 (br s, 1H), 4.06 (d, J = 9.2 Hz, 1H), 3.78 (s, 3H), 3.43 (br d, J = 6.4 Hz, 1H), 3.37 (d, J = 8.8 Hz, 1H), 2.95 (dd, J = 18.4, 6.4 Hz, 1H), 2.78 (d, J = 18.4 Hz, 1H), 2.57 (br t, J = 7.2 Hz, 1H), 2.55 (d, J = 16.4 Hz, 1H), 2.53 (dd, J = 16.4, 7.2 Hz, 1H), 2.37 (br d, J = 6.4 Hz, 1H), 2.22 (m, 1H), 1.75 (m, 1H), 1.71 (m, 1H), 1.52 (m, 1H), 1.49 (m, 1H), 1.18 (s, 3H), 1.13 (s, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, CD3COCD3): δ 175.7, 169.6, 162.4, 144.0, 142.8, 138.0, 122.2, 120.7, 110.8, 97.6, 77.1, 68.8, 61.7, 48.9, 46.7, 45.6, 42.8, 40.2, 39.4, 37.4, 35.2, 31.9, 30.6, 22.2, 19.8, 15.0, 13.6; LR-ESIMS: [M + Na]+ calcd. for C27H34NO8 500.22, found: 500.17.
10c: transparent oil, yield 23.6%; 1H NMR (400 MHz, CD3COCD3): δ 7.75 (br s, 1H), 7.55 (t, J = 1.6 Hz, 1H), 7.41 (m, 1H), 7.39 (overlapped, 1H), 7.37 (m, 1H), 7.28 (m, 1H), 6.55 (d, J = 1.6 Hz, 1H), 5.97 (dt, J = 4.8, 2.0 Hz, 1H), 5.43 (s, 1H), 5.05 (dd, J = 12.0, 4.0 Hz, 2H), 4.07 (d, J = 9.2 Hz, 1H), 3.47 (d, J = 6.8 Hz, 1H), 3.37 (d, J = 8.8 Hz, 1H), 2.98 (dd, J = 18.4, 6.4 Hz, 1H), 2.80 (d, J = 18.4 Hz, 1H), 2.62 (d, J = 8.6 Hz, 1H), 2.58 (d, J = 5.8 Hz, 1H), 2.53 (d, J = 13.8 Hz, 1H), 2.39 (d, J = 6.4 Hz, 1H), 2.23 (t, J = 9.6 Hz, 1H), 1.76 (m, 1H), 1.73 (m, 1H), 1.53 (m, 1H), 1.50 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, CD3COCD3): δ 175.7, 169.6, 162.8, 144.1, 142.8, 139.0, 138.3, 129.2, 129.0, 128.4, 122.2, 120.5, 110.8, 97.7, 77.1, 76.4, 68.9, 49.0, 46.9, 45.6, 42.8, 40.2, 39.6, 37.5, 35.3, 32.0, 30.6, 22.2, 19.8, 15.0, 13.6; LR-ESIMS: [M + H]+ calcd. for C33H38NO8 576.25, found: 576.18.
3.6. Antiviral Bioassay
Antiviral activities of 6, 7–10, 8a–8i, 9a–9b, and 10a–10c against HIV-1 and IAV were performed according to references [14,19]. 293T cells (2 × 105) were co-transfected with 0.6 μg of pNL-Luv-E−-Vpu− and 0.4 μg of pHIT/G. After 48 h, the VSV-G pseudo-typed viral supernatant was harvested by filtration through a 0.45 μm filter, and the concentration of viral capsid protein was determined by p24 antigen capture ELISA (Biomerieux). SupT1 cells were exposed to VSV-G pseudo-typed virus (MOI = 1) at 37 °C for 48 h in the absence or presence of compounds at different concentrations. The inhibition rate was determined by using a firefly luciferase assay system (Promega, Beijing). Efavirenz was used as the positive control for HIV-1; whereas ribavirion was used as the positive control for IAV.
4. Conclusions
In conclusion, five new limonoids named thaigranatins A–E (1–5), containing a C1–O–C29 moiety, were isolated from seeds of the Thai Xylocarpus granatum, collected at the mangrove swamp of Trang Province, together with the known one, granatumin L (6). The structures of these limonoids, including absolute configurations of 1, 2, and 6 were established by HR-ESIMS, extensive NMR investigations, single-crystal X-ray diffraction analysis (Cu Kα radiation), and comparison of experimental ECD spectra. The structural modification of 6 via hydrolysis, esterification, and oximization, afforded 18 derivatives, viz. 7–10, 8a–8i, 9a–9b, and 10a–10c. The derivative 8i exhibited marked inhibitory activity against HIV-1 with an IC50 value of 15.98 ± 6.87 μM and a CC50 value greater than 100.0 μM; whereas 10b showed significant inhibitory activity against IAV with an IC50 value of 14.02 ± 3.54 μM and a CC50 value greater than 100.0 μM. These results demonstrate that the mangrove X. granatum continues to be an abundant resource to produce new antiviral limonoids. In-depth structural modification and structure-activity relationship studies of the limonoid skeleton of 6 will be proceeded by us for the discovery of antiviral drug leads.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/16/11/434/s1. Copies of HR-ESIMS, 1D and 2D NMR spectra of compounds 1–5; and copies of LR/HR-ESIMS and 1D NMR spectra of compounds 7, 8, 9, 10, 8a–8i, 9a, 9b, and 10a–10c.
Author Contributions
L.S. and J.W. conceived and designed the experiments; J.-L.R., X.-P.Z., and W.-S.L. performed the experiments and analyzed the data; J.-L.R. and X.-P.Z. wrote the draft; L.S. and Jun Wu revised the paper. All authors have read and approved the manuscript.
Funding
This work was financially supported by grants from National Natural Science Foundation of China (NSFC) (U1501221, 31770377, and 81661148049) and the Fundamental Research Funds for the Central Universities, P.R. China (21617474).
Acknowledgments
We thank Patchara Pedpradab (Rajamangala University of Technology Srivijaya, Trang Province, Thailand) for providing the plant materials in this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wu, J.; Xiao, Q.; Xu, J.; Li, M.Y.; Pan, J.Y.; Yang, M.H. Natural Products from True Mangrove Flora: Source, Chemistry and Bioactivities. Nat. Prod. Rep. 2008, 25, 955–981. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Li, X.W.; Guo, Y.W. Recent Progress on the Mangrove Plants: Chemistry and Bioactivity. Curr. Org. Chem. 2016, 20, 1923–1942. [Google Scholar] [CrossRef]
- Zhang, Q.; Satyanandamurty, T.; Shen, L.; Wu, J. Krishnolides A–D: New 2-Ketokhayanolides from the Krishna Mangrove, Xylocarpus moluccensis. Mar. Drugs 2017, 15, 333. [Google Scholar] [CrossRef] [PubMed]
- Li, W.S.; Jiang, Z.Z.; Shen, L.; Pedpradab, P.; Bruhn, T.; Wu, J.; Bringmann, G. Antiviral Limonoids Including Khayanolides from the Trang Mangrove Plant Xylocarpus moluccensis. J. Nat. Prod. 2015, 78, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, S.; Xiao, Q.; Li, Q.X.; Huang, H.S.; Long, L.J.; Huang, L.M. Xlyogranatin L, a novel limonoid from Xylocarpus granatum. Tetrahedron. Lett. 2004, 45, 591–593. [Google Scholar] [CrossRef]
- Wu, J.; Ding, H.X.; Li, M.Y.; Zhang, S. Xylogranatin E, a New Phragmalin with a Rare Oxygen Bridge Between C1 and C29, from the Fruit of a Chinese Mangrove Xylocarpus granatum. Z. Naturforsch. 2007, 62, 569–572. [Google Scholar] [CrossRef]
- Yin, S.; Wang, X.N.; Fan, C.Q.; Lin, L.P.; Ding, J.; Yue, J.M. Limonoids from the Seeds of the Marine Mangrove Xylocarpus granatum. J. Nat. Prod. 2007, 70, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Xiao, Q.; Satyanandamurty, T.; Wu, J. Limonoids with an oxygen bridge between C(1) and C(29) from the seeds of a Krishna mangrove, Xylocarpus granatum. Chem. Biodivers. 2014, 11, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.G.; Wu, J.; Padmakumar, K.P.; Shen, L. Sundarbanxylogranins A-E, five new limonoids from the Sundarban Mangrove, Xylocarpus granatum. Fitoterapia 2017, 122, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.X.; Liao, Q.; Shen, L.; Wu, J. Krishnagranatins A-I: New limonoids from the mangrove, Xylocarpus granatum, and NF-κB inhibitory activity. Fitoterapia 2018, 131, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.H.; Pedpradab, P.; Wu, J. Thaixylogranins A–H: Eight new limonoids from the Thai mangrove, Xylocarpus granatum. Phytochem. Lett. 2017, 19, 126–131. [Google Scholar] [CrossRef]
- Li, J.; Li, M.Y.; Feng, G.; Xiao, Q.A.; Sinkkonen, J.; Satyanandamurty, T.; Wu, J. Limonoids from the seeds of Godavari mangrove, Xylocarpus moluccensis. Phytochemistry 2010, 71, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Khalijah, A.; Chong, S.L.; Khalit, M.; Morita, H.; Hirasawa, Y.; Takeya, K.; Thoisone, O.; Hadi, A.H.A. Erythrocarpines A-E, new cytotoxic limonoids from Chisocheton erythrocarpus. Bioorg. Med. Chem. 2007, 15, 5997–6002. [Google Scholar]
- Zhao, J.; Feng, J.; Tan, Z.; Liu, J.; Zhao, J.; Chen, R.; Xie, K.; Zhang, D.; Li, Y.; Yu, L.; et al. Stachybotrysins A−G, Phenylspirodrimane Derivatives from the Fungus Stachybotrys chartarum. J. Nat. Prod. 2017, 80, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Ushitora, H. Trapping of carbamic acid species with (trimethylsilyl)diazomethane. Tetrahedron 2006, 62, 226–235. [Google Scholar] [CrossRef]
- D’yakonov, V.A.; Dzhemileva, L.U.; Tuktarova, R.A.; Ishmukhametova, S.R.; Yunusbaeva, M.M.; Ramazanov, I.R.; Dzhemilev, U.M. Novel Hybrid Molecules on the Basis of Steroids and (5Z,9Z)-Tetradeca-5,9-dienoic Acid: Synthesis, Anti-Cancer Studies and Human Topoisomerase I Inhibitory Activity. Anti-Cancer Agents Med. Chem. 2017, 17, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Ishihara, K. Hypervalent iodine-mediated oxidation of alcohols. Chem. Commun. 2009, 16, 2086–2099. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Maddox, M.M.; Adhikari, S.; Bruhn, D.F.; Kumar, M.; Lee, R.E.; Hurdle, J.G.; Lee, R.E.; Sun, D.Q. Syntheses and evaluation of macrocyclic engelhardione analogs as antitubercular and antibacterial agents. J. Antibiot. 2013, 66, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, Z.; Mi, Z.; Li, X.; Jia, P.; Zhou, J.; Yin, X.; You, X.; Yu, L.; Guo, F.; et al. High-throughput assay to identify inhibitors of Vpu-mediated down-regulation of cell surface BST-2. Antivir. Res. 2011, 91, 321–329. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of compounds 1–6.

Figure 2.
(a) Selected 1H–1H COSY and HMBC correlations for 1; (b) Diagnostic NOE interactions for 1 (measured in CDCl3).
Figure 2.
(a) Selected 1H–1H COSY and HMBC correlations for 1; (b) Diagnostic NOE interactions for 1 (measured in CDCl3).

Figure 3.
ORTEP illustration of the X-ray structure of 1. Ellipsoids are given at the 10% probability level.
Figure 3.
ORTEP illustration of the X-ray structure of 1. Ellipsoids are given at the 10% probability level.

Figure 4.
Comparison of the experimental ECD spectra of 1, 2, and the known compound, granatumin L (6), containing a Δ8,30 double bond (recorded at the concentration of 200 μg mL−1 in MeCN).
Figure 4.
Comparison of the experimental ECD spectra of 1, 2, and the known compound, granatumin L (6), containing a Δ8,30 double bond (recorded at the concentration of 200 μg mL−1 in MeCN).

Figure 5.
(a) Selected 1H–1H COSY and HMBC correlations for 3; (b) Diagnostic NOE interactions for 3 (measured in CDCl3).
Figure 5.
(a) Selected 1H–1H COSY and HMBC correlations for 3; (b) Diagnostic NOE interactions for 3 (measured in CDCl3).

Scheme 1.
Summary of the structural modification approaches undertaken in this work. Reagent and conditions: (i) 10% KOH, CH3OH, r.t., 30.0 h; (ii) TMSCHN2, CH3OH, r.t., 2.0 h; (iii) organic acids, DMAP, DCC, CH2Cl2, r.t., 3–12 h; (iv) IBX (2-iodoxybenzoic acid), DMSO, r.t., 7.5 h; (v) TMSCHN2, CH3OH, r.t., 4.0 h; (vi) IBX, DMSO, r.t., 7.5 h; (vii) (viii) RONH2, Pyridine, CH3CH2OH, r.t., 24–36 h.
Scheme 1.
Summary of the structural modification approaches undertaken in this work. Reagent and conditions: (i) 10% KOH, CH3OH, r.t., 30.0 h; (ii) TMSCHN2, CH3OH, r.t., 2.0 h; (iii) organic acids, DMAP, DCC, CH2Cl2, r.t., 3–12 h; (iv) IBX (2-iodoxybenzoic acid), DMSO, r.t., 7.5 h; (v) TMSCHN2, CH3OH, r.t., 4.0 h; (vi) IBX, DMSO, r.t., 7.5 h; (vii) (viii) RONH2, Pyridine, CH3CH2OH, r.t., 24–36 h.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
1H (400 MHz) NMR spectroscopic data of compounds 1–5 in CDCl3 (δ in ppm, J in Hz).
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 2 | 3.00 t (8.4) | 2.95 t (8.4) | 2.77 dd (10.8, 2.4) | 2.57 m | 2.78 dd (10.4, 1.6) |
| 3 | 4.71 d (10.0) | 4.84 d (10.0) | 5.14 d (10.8) | 4.82 d (10.4) | 5.27 d (10.8) |
| 5 | 2.93 s | 2.84 d (10.0) | 2.98 d (10.8) | 2.92 br s | 2.64 a |
| 6a | 4.56 s | 2.35 m | 2.42 dd (16.8, 2.0) | 4.59 s | 2.35 m |
| 6b | 2.35 m | 2.42 dd (16.8, 10.8) | 2.64 a | ||
| 9 | 2.24 m | 2.15 dd (12.2, 5.2) | 2.65 br s | 2.46 m | |
| 11α | 1.76 m | 1.62 a | 1.73 m | 1.73 m | 1.97 m |
| 11β | 1.76 m | 1.77 m | 1.79 m | 1.73 m | 2.23 m |
| 12α | 1.52 m | 1.45 m | 1.11 m | 1.20 m | 1.51 m |
| 12β | 1.71 m | 1.62 a | 1.62 m | 1.62 m | 1.51 m |
| 14 | 2.31 br s | 2.30 br s | 2.52 mdd (11.6, 5.2) | ||
| 15α | 2.80 d (21.6) | 2.84 d (21.6) | 3.41 dd (21.2, 3.2) | 3.25 dt (21.6, 2.8) | 2.69 dd (16.0, 3.2) |
| 15β | 2.84 d (21.6) | 2.91 d (21.6) | 3.59 dd (21.2, 1.6) | 3.44 d (21.6) | 2.18 dd (16.0, 3.2) |
| 17 | 5.42 s | 5.55 s | 5.41 s | 5.33 s | 4.90 s |
| 18 | 0.98 s | 1.09 s | 1.08 s | 0.98 s | 0.78 s |
| 19 | 1.40 s | 1.07 s | 1.13 s | 1.38 s | 1.11 s |
| 21 | 7.50 br s | 7.74 br s | 7.53 br s | 7.44 br s | 7.44 br s |
| 22 | 6.38 br s | 6.45 br s | 6.46 br s | 6.38 br s | 6.41 br s |
| 23 | 7.44 br s | 7.41 br s | 7.42 br s | 7.42 br s | 7.42 br s |
| 28 | 0.77 s | 0.63 s | 0.56 s | 0.71 s | 0.70 s |
| Hpro-S-29 | 4.60 d (8.8) | 3.93 d (9.6) | 3.92 d (9.6) | 4.59 d (8.8) | 3.80 d (10.0) |
| Hpro-R-29 | 3.45 d (8.8) | 3.48 dd (9.6, 1.6) | 3.51 dd (9.6, 1.6) | 3.42 (8.8, 1.6) | 3.69 (10.0) |
| 30β | 5.28 d (6.8) | 5.31 d (6.8) | 4.51 d (2.4) | 2.24 m | 3.94 br s |
| 30α | 2.24 m | ||||
| 31 | 3.76 s | 3.69 s | 3.70 s | 3.84 s | 3.72 s |
| 3-Acyl | 3-Acyl | 3-Acyl | 3-Acyl | 3-Acyl | |
| 33 | 2.37 m | ||||
| 2.37 m | |||||
| 34 | 6.83 q (7.2) | 1.09 t (7.2) | 6.99 q (7.2) | 6.92 q (7.2) | 6.89 q (7.2) |
| 35 | 1.67 d (7.2) | 1.80 dd (7.2, 1.2) | 1.80 dd (7.2, 0.8) | 1.86 d (7.2) | |
| 36 | 1.79 s | 1.87 s | 1.86 s | 1.88 s |
a Overlapped signals assigned by 1H–1H COSY, HSQC, and HMBC spectra without designating multiplicity.
Table 2.
13C (100 MHz) NMR spectroscopic data of compounds 1–5 in CDCl3 (δ in ppm).
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 96.9 qC | 96.9 qC | 97.4 qC | 97.1 qC | 97.4 qC |
| 2 | 45.3 CH | 45.1 CH | 48.2 CH | 43.6 CH | 46.2 CH |
| 3 | 76.2 CH | 75.2 CH | 75.2 CH | 78.5 CH | 74.8 CH |
| 4 | 36.6 qC | 36.2 qC | 37.1 qC | 36.6 qC | 36.8 qC |
| 5 | 39.6 CH | 35.0 CH | 34.5 CH | 39.3 CH | 40.6 CH |
| 6 | 72.5 CH | 31.9 CH2 | 32.1 CH2 | 73.1 CH | 31.9 CH2 |
| 7 | 176.1 qC | 173.9 qC | 174.2 qC | 175.8 qC | 174.3 qC |
| 8 | 138.3 qC | 138.4 qC | 131.3 qC | 130.6 qC | 127.1 qC |
| 9 | 48.9 CH | 47.8 CH | 38.7 CH | 43.9 CH | 139.2 qC |
| 10 | 42.1 qC | 41.6 qC | 45.1 qC | 44.7 qC | 43.6 qC |
| 11 | 20.4 CH2 | 19.4 CH2 | 17.7 CH2 | 18.2 CH2 | 21.1 CH2 |
| 12 | 34.6 CH2 | 34.5 CH2 | 29.4 CH2 | 30.3 CH2 | 29.4 CH2 |
| 13 | 36.7 qC | 36.9 qC | 38.2 qC | 37.8 qC | 35.3 qC |
| 14 | 44.8 CH | 45.0 CH | 134.3 qC | 129.0 qC | 36.8 CH |
| 15 | 29.6 CH2 | 30.1 CH2 | 32.3 CH2 | 33.1 CH2 | 32.2 CH2 |
| 16 | 169.3 qC | 170.1 qC | 169.5 qC | 169.8 qC | 172.7 qC |
| 17 | 76.6 CH | 77.0 CH | 80.7 CH | 81.6 CH | 81.3 CH |
| 18 | 21.2 CH3 | 21.9 CH3 | 17.4 CH3 | 18.0 CH3 | 20.2 CH3 |
| 19 | 14.9 CH3 | 14.4 CH3 | 14.2 CH3 | 15.1 CH3 | 14.3 CH3 |
| 20 | 121.5 qC | 120.8 qC | 120.7 qC | 121.1 qC | 121.2 qC |
| 21 | 140.3 CH | 141.8 CH | 141.6 CH | 140.9 CH | 140.4 CH |
| 22 | 109.3 CH | 109.7 CH | 109.9 CH | 109.8 CH | 109.7 CH |
| 23 | 143.1 CH | 143.0 CH | 142.9 CH | 143.1 CH | 143.2 CH |
| 28 | 15.8 CH3 | 14.8 CH3 | 15.6 CH3 | 16.4 CH3 | 18.2 CH3 |
| 29 | 70.1 CH2 | 68.0CH2 | 67.6 CH2 | 70.0 CH2 | 68.6 CH2 |
| 30 | 120.2 CH | 119.9 CH | 66.3 CH | 26.4 CH2 | 66.1 CH |
| 31 | 53.2 CH3 | 52.1 CH3 | 52.1 CH3 | 53.1 CH3 | 52.0 CH3 |
| 3-Acyl | 3-Acyl | 3-Acyl | 3-Acyl | 3-Acyl | |
| 32 | 167.2 qC | 174.5 qC | 167.6 qC | 167.5 qC | 167.6 qC |
| 33 | 127.8 qC | 27.2 CH2 | 128.8 qC | 128.9 qC | 128.4 qC |
| 34 | 138.7 CH | 8.8 CH3 | 139.6 CH | 138.7 CH | 139.2 CH |
| 35 | 14.6 CH3 | 14.6 CH3 | 14.6 CH3 | 14.8 CH3 | |
| 36 | 11.7 CH3 | 12.3 CH3 | 12.2 CH3 | 12.4 CH3 |
Table 3.
Inhibitory activities of natural limonoid 6 and its modified derivatives against HIV-1.
| cpd No. | Inhibition Rate (20.0 μM) (%) | IC50 Value (μM) | CC50 Value (μM) | cpd No. | Inhibition Rate (20.0 μM) (%) | IC50 Value (μM) | CC50 Value (μM) |
|---|---|---|---|---|---|---|---|
| 6 | 67.10 ± 3.04 | 19.23 ± 0.12 | > 100 | 8f | NA | NT | NT |
| 7 | NA | NT | NT | 8g | 50.84 ± 6.96 | 21.98 ± 4.65 | 86.76 ± 4.76 |
| 8 | NA | NT | NT | 8h | NA | NT | NT |
| 9 | NA | NT | NT | 8i | 99.95 ± 0.01 | 15.98 ± 6.87 | > 100 |
| 10 | NA | NT | NT | 9a | NA | NT | NT |
| 8a | NA | NT | NT | 9b | NA | NT | NT |
| 8b | 39.87 ± 2.83 | NT | NT | 10a | NA | NT | NT |
| 8c | 45.47 ± 6.84 | NT | NT | 10b | NA | NT | NT |
| 8d | 35.63 ± 8.85 | NT | NT | 10c | NA | NT | NT |
| 8e | 10.58 ± 3.55 | NT | NT | EFV | 88.78 ± 4.54 (4 nM) | NT | NT |
cpd: compound; NA: No activity; NT = Not tested.
Table 4.
Inhibitory activities of natural limonoid 6 and its modified derivatives against IAV.
| cpd No. | Inhibition Rate (20.0 μM) (%) | IC50 Value (μM) | CC50 Value (μM) | cpd No. | Inhibition Rate (20.0 μM) (%) | IC50 Value (μM) | CC50 Value (μM) |
|---|---|---|---|---|---|---|---|
| 7 | 38.25 ± 9.67 | NT | NT | 8e | 43.42 ± 1.79 | NT | NT |
| 8 | NA | NT | NT | 8f | 44.94 ± 9.73 | NT | NT |
| 9 | NA | NT | NT | 8h | 48.15 ± 1.64 | NT | NT |
| 10 | NA | NT | NT | 9a | 44.72 ± 1.43 | NT | NT |
| 8a | 23.46 ± 2.42 | NT | NT | 10a | NA | NT | NT |
| 8b | 54.65 ± 1.43 | 17.93 ± 4.76 | 85.90 ± 4.65 | 10b | 63.40 ± 7.43 | 14.02 ± 3.54 | > 100 |
| 8c | 68.61 ± 0.99 | 15.87 ± 6.54 | 77.98 ± 4.65 | 10c | NA | NT | NT |
| 8d | 51.41 ± 1.54 | 22.78 ± 1.86 | 87.65 ± 4.76 | ribavirin | 87.54 ± 2.86 (60 μM) | NT | NT |
cpd: compound; NA: No activity; NT = Not tested.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ren, J.-L.; Zou, X.-P.; Li, W.-S.; Shen, L.; Wu, J. Limonoids Containing a C1–O–C29 Moiety: Isolation, Structural Modification, and Antiviral Activity. Mar. Drugs 2018, 16, 434. https://doi.org/10.3390/md16110434
AMA Style
Ren J-L, Zou X-P, Li W-S, Shen L, Wu J. Limonoids Containing a C1–O–C29 Moiety: Isolation, Structural Modification, and Antiviral Activity. Marine Drugs. 2018; 16(11):434. https://doi.org/10.3390/md16110434
Chicago/Turabian StyleRen, Jing-Ling, Xiao-Peng Zou, Wan-Shan Li, Li Shen, and Jun Wu. 2018. "Limonoids Containing a C1–O–C29 Moiety: Isolation, Structural Modification, and Antiviral Activity" Marine Drugs 16, no. 11: 434. https://doi.org/10.3390/md16110434
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.