Cytotoxic Potential of the Novel Horseshoe Crab Peptide Polyphemusin III

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identificantion of Antimicrobial Peptide
2.2. Expression and Purification of the Recombinant Peptides
2.3. Antimicrobial Activity
2.4. Cytotoxic Effects on Human Cells
2.5. Hemolytic Activity
2.6. Trypan Blue Assay for Dead Cells
2.7. Mechanism of Cell Death
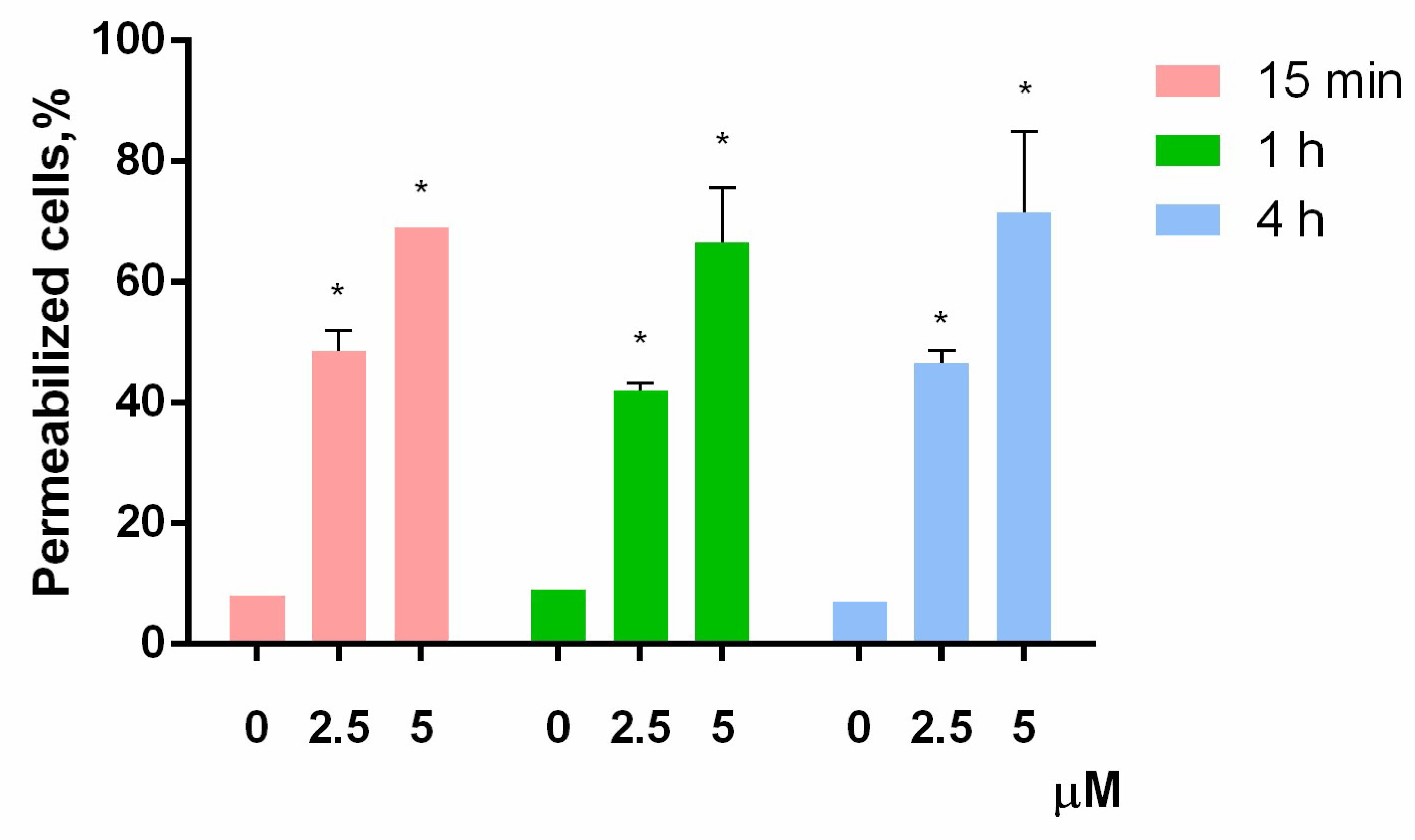
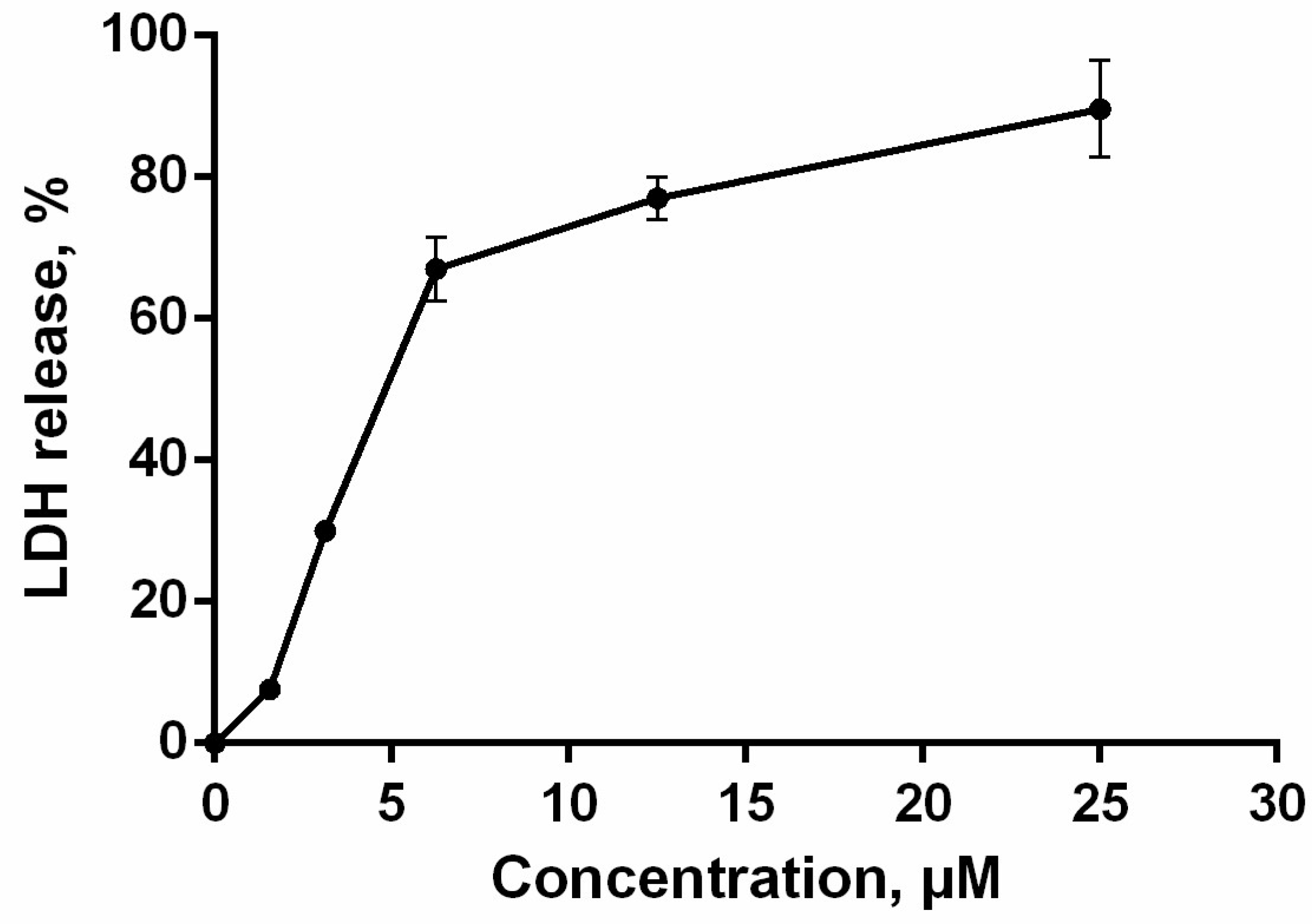
2.8. Cell Membrane Integrity
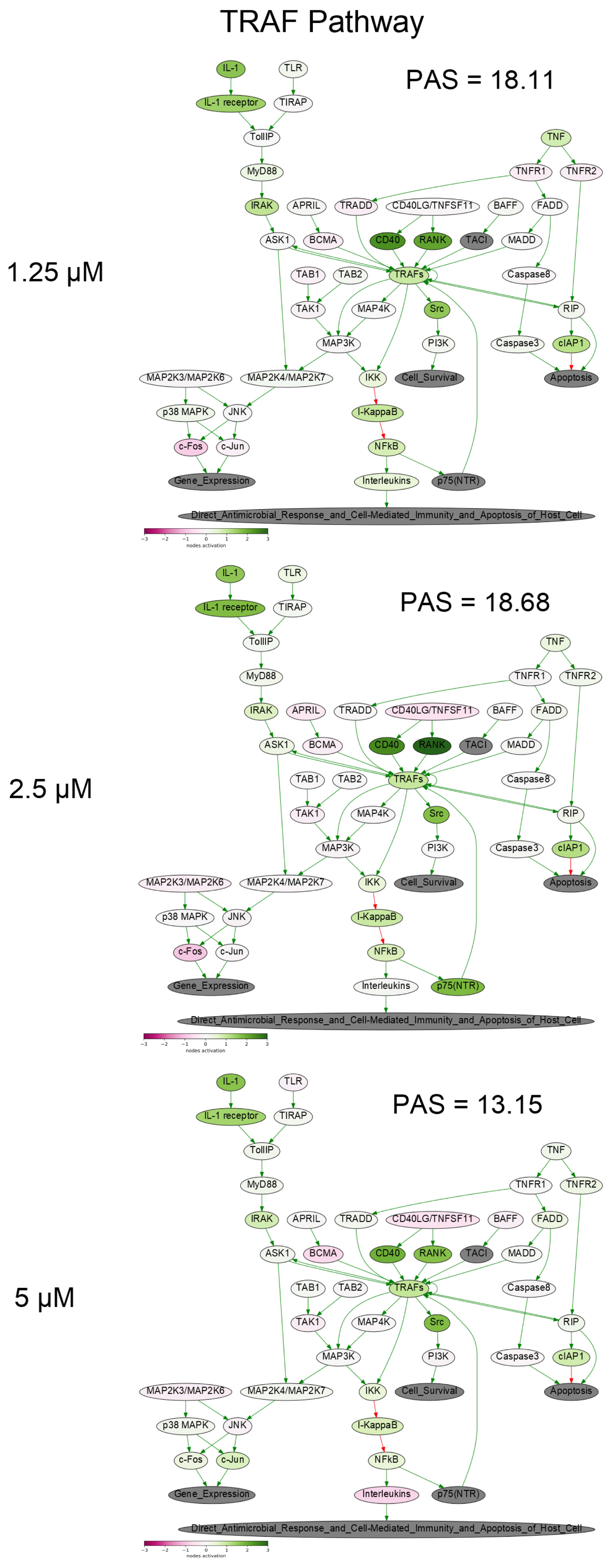
2.9. Gene Expression Profiling and Oncobox Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Peptides
4.3. Antimicrobial Assay
4.4. Cytotoxic Activity Assay
4.5. Hemolytic Activity Assay
4.6. Trypan Blue Exclusion Assay
4.7. Annexin V-FITC/Propidium Iodide Double Staining and Flow Cytometry
4.8. Lactate Dehydrogenase (LDH)-Release Assay
4.9. Statistical Analysis
4.10. Gene Expression Profiling and Functional Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brandenburg, L.-O.; Merres, J.; Albrecht, L.-J.; Varoga, D.; Pufe, T. Antimicrobial peptides: Multifunctional drugs for different applications. Polymers 2012, 4, 539–560. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Roudi, R.; Syn, N.L.; Roudbary, M. Antimicrobial peptides as biologic and immunotherapeutic agents against cancer: A comprehensive overview. Front. Immunol. 2017, 8, 1320. [Google Scholar] [CrossRef] [PubMed]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with dual antimicrobial and anticancer activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.G.; Dobroff, A.S.; Cavarsan, C.F.; Paschoalin, T.; Nimrichter, L.; Mortara, R.A.; Santos, E.L.; Fázio, M.A.; Miranda, A.; Daffre, S.; et al. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia 2008, 10, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gamero, E.J.; Martins, M.N.C.; Cappabianco, F.A.M.; Ide, J.S.; Miranda, A. Characterization of dual effects induced by antimicrobial peptides: Regulated cell death or membrane disruption. Biochim. Biophys. Acta 2012, 1820, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buri, M.V.; Torquato, H.F.V.; Barros, C.C.; Ide, J.S.; Miranda, A.; Paredes-Gamero, E.J. Comparison of cytotoxic activity in leukemic lineages reveals important features of β-hairpin antimicrobial peptides. J. Cell Biochem. 2017, 118, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Balandin, S.V.; Ivanov, V.T.; Ovchinnikova, T.V. A therapeutic potential of animal β-hairpin antimicrobial peptides. Curr. Med. Chem. 2017, 24, 1724–1746. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: Chemical structures and biological activity. J. Biochem. 1989, 106, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar] [PubMed]
- Muta, T.; Nakamura, T.; Furunaka, H.; Tokunaga, F.; Miyata, T.; Niwa, M.; Iwanaga, S. Primary structures and functions of anti-lipopolysaccharide factor and tachyplesin peptide found in horseshoe crab hemocytes. Adv. Exp. Med. Biol. 1990, 256, 273–285. [Google Scholar] [PubMed]
- Zhang, L.; Scott, M.G.; Yan, H.; Mayer, L.D.; Hancock, R.E. Interaction of polyphemusin I and structural analogs with bacterial membranes, lipopolysaccharide, and lipid monolayers. Biochemistry 2000, 39, 14504–14514. [Google Scholar] [CrossRef] [PubMed]
- Ohta, M.; Ito, H.; Masuda, K.; Tanaka, S.; Arakawa, Y.; Wacharotayankun, R.; Kato, N. Mechanisms of antibacterial action of tachyplesins and polyphemusins, a group of antimicrobial peptides isolated from horseshoe crab hemocytes. Antimicrob. Agents Chemother. 1992, 36, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β-hairpin peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Oishi, O.; Yamashita, S.; Nishimoto, E.; Lee, S.; Sugihara, G.; Ohno, M. Conformations and orientations of aromatic amino acid residues of tachyplesin I in phospholipid membranes. Biochemistry 1997, 36, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- Katsu, T.; Nakao, S.; Iwanaga, S. Mode of action of an antimicrobial peptide, tachyplesin I, on biomembranes. Biol. Pharm. Bull. 1993, 16, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Hu, J.; Ke, F. Experimental induction of bacterial resistance to the antimicrobial peptide tachyplesin I and investigation of the resistance mechanisms. Antimicrob. Agents Chemother. 2016, 60, 6067–6075. [Google Scholar] [CrossRef] [PubMed]
- Zapotoczna, M.; Forde, É.; Hogan, S.; Humphreys, H.; O’Gara, J.P.; Fitzgerald-Hughes, D.; Devocelle, M.; O’Neill, E. Eradication of staphylococcus aureus biofilm infections using synthetic antimicrobial peptides. J. Infect. Dis. 2017, 215, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, T.; Muta, T.; Toh, Y.; Tokunaga, F.; Iwanaga, S. Antimicrobial tachyplesin peptide precursor. cDNA cloning and cellular localization in the horseshoe crab (Tachypleus tridentatus). J. Biol. Chem. 1990, 265, 21350–21354. [Google Scholar] [PubMed]
- Janko, C.; Munoz, L.; Chaurio, R.; Maueröder, C.; Berens, C.; Lauber, K.; Herrmann, M. Navigation to the graveyard-induction of various pathways of necrosis and their classification by flow cytometry. Methods Mol. Biol. 2013, 1004, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, C.J. The history of Z-VAD-FMK, a tool for understanding the significance of caspase inhibition. Acta Histochem. 2001, 103, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Cao, L.; Chen, X.; Xiao, J.; Zou, Y.; Chen, Q. PTEN inhibits cell proliferation, promotes cell apoptosis, and induces cell cycle arrest via downregulating the PI3K/AKT/hTERT pathway in lung adenocarcinoma A549 Cells. Biomed. Res. Int. 2016, 2016, 2476842. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.-P.S.; Rozek, A.; Hancock, R.E.W. Structure-activity relationships for the beta-hairpin cationic antimicrobial peptide polyphemusin I. Biochim. Biophys. Acta 2004, 1698, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-B.; Wang, X.-F.; Wang, H.-Y.; Liu, Y.; Chen, Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Cancer Ther. 2011, 10, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Tachi, T.; Epand, R.F.; Epand, R.M.; Matsuzaki, K. Position-dependent hydrophobicity of the antimicrobial magainin peptide affects the mode of peptide-lipid interactions and selective toxicity. Biochemistry 2002, 41, 10723–10731. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Meyer, J.; Beyermann, M.; Maul, B.; Hoischen, C.; Bienert, M. General aspects of peptide selectivity towards lipid bilayers and cell membranes studied by variation of the structural parameters of amphipathic helical model peptides. Biochim. Biophys. Acta 2002, 1558, 171–186. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Beyermann, M.; Seelig, J. Binding of antibacterial magainin peptides to electrically neutral membranes: Thermodynamics and structure. Biochemistry 1999, 38, 10377–10387. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Dathe, M.; Beyermann, M.; Krause, E.; Maloy, W.L.; MacDonald, D.L.; Bienert, M. Peptide hydrophobicity controls the activity and selectivity of magainin 2 amide in interaction with membranes. Biochemistry 1997, 36, 6124–6132. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar] [CrossRef] [PubMed]
- Andreev, K.; Martynowycz, M.W.; Huang, M.L.; Kuzmenko, I.; Bu, W.; Kirshenbaum, K.; Gidalevitz, D. Hydrophobic interactions modulate antimicrobial peptoid selectivity towards anionic lipid membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, E.; Burrows, L.L.; Deber, C.M. Membrane interactions of designed cationic antimicrobial peptides: The two thresholds. Biopolymers 2008, 89, 360–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasupuleti, M.; Walse, B.; Svensson, B.; Malmsten, M.; Schmidtchen, A. Rational design of antimicrobial C3a analogues with enhanced effects against Staphylococci using an integrated structure and function-based approach. Biochemistry 2008, 47, 9057–9070. [Google Scholar] [CrossRef] [PubMed]
- Gottler, L.M.; de la Salud Bea, R.; Shelburne, C.E.; Ramamoorthy, A.; Marsh, E.N.G. Using fluorous amino acids to probe the effects of changing hydrophobicity on the physical and biological properties of the β-hairpin antimicrobial peptide protegrin-1. Biochemistry 2008, 47, 9243–9250. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jin, G.; Zhang, L.; Dai, J.; Dang, J.; Han, Y. Effects of tachyplesin I on human U251 glioma stem cells. Mol. Med. Rep. 2015, 11, 2953–2958. [Google Scholar] [CrossRef] [PubMed]
- Carriel-Gomes, M.C.; Kratz, J.M.; Barracco, M.A.; Bachére, E.; Barardi, C.R.M.; Simões, C.M.O. In vitro antiviral activity of antimicrobial peptides against herpes simplex virus 1, adenovirus, and rotavirus. Mem. Inst. Oswaldo Cruz 2007, 102, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wu, J.; Zhang, H.; Zhu, Q. Efflux of potassium ion is an important reason of HL-60 cells apoptosis induced by tachyplesin. Acta Pharmacol. Sin. 2006, 27, 1367–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Jiang, J.; Sottile, V.; McWhir, J.; Lebkowski, J.; Carpenter, M.K. Immortalized fibroblast-like cells derived from human embryonic stem cells support undifferentiated cell growth. Stem Cells 2004, 22, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Gendelman, H.E. Isolation and culture of human neurons, microglia, and astrocytes. In Current Laboratory Methods in Neuroscience Research; Xiong, H., Gendelman, H.E., Eds.; Springer: New York, NY, USA, 2014; ISBN 978-1-4614-8793-7. [Google Scholar]
- Panteleev, P.V.; Ovchinnikova, T.V. Improved strategy for recombinant production and purification of antimicrobial peptide tachyplesin I and its analogs with high cell selectivity. Biotechnol. Appl. Biochem. 2017, 64, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Bolosov, I.A.; Balandin, S.V.; Ovchinnikova, T.V. Design of antimicrobial peptide arenicin analogs with improved therapeutic indices. J. Pept. Sci. 2015, 21, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Bolosov, I.A.; Ovchinnikova, T.V. Bioengineering and functional characterization of arenicin shortened analogs with enhanced antibacterial activity and cell selectivity. J. Pept. Sci. 2016, 22, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, M.; Kholodenko, R.; Suntsova, M.; Malakhova, G.; Garazha, A.; Kholodenko, I.; Poddubskaya, E.; Lantsov, D.; Stilidi, I.; Arhiri, P.; et al. Oncobox bioinformatical platform for selecting potentially effective combinations of target cancer drugs using high-throughput gene expression data. Cancers 2018, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation—New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence 1 | Calculated [M+H]+ Monoisotopic Mass, Da 2 | Measured Monoisotopic m/z Value 3 | Net Charge | RP-HPLC Retention Time 4, min | Recombinant Peptide Final Yield, mg/L |
|---|---|---|---|---|---|---|
| PM I | RRWCFRVCYRGFCYRKCR | 2454.18 | 2453.97 | +7 | 35.5 | 2.4 |
| PM II | RRWCFRVCYKGFCYRKCR | 2426.18 | 2426.19 | +7 | 35.4 | 1.5 |
| PM III | RRGCFRVCYRGFCFQRCR | 2309.09 | 2309.01 | +6 | 36.2 | 5.9 |
| TP I | KWCFRVCYRGICYRRCR | 2264.10 | 2264.25 | +6 | 35.5 | 3.4 |
| TP II | RWCFRVCYRGICYRKCR | 2264.10 | 2264.14 | +6 | 35.9 | 3.0 |
| TP III | KWCFRVCYRGICYRKCR | 2236.09 | 2236.07 | +6 | 35.5 | 3.0 |
| Strain | Gram | MICs, μM | |||||
|---|---|---|---|---|---|---|---|
| PM I | PM II | PM III | TP I | TP II | TP III | ||
| E. coli ML-35p | - | 0.062 | 0.031 | 0.25 | 0.062 | 0.062 | 0.062 |
| K. pneumoniae (CI 287) | - | 0.5 | 0.5 | 2 | 0.5 | 1 | 0.5 |
| P. aeruginosa PAO1 | - | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| S. aureus ATCC 29213 | + | 4 | 4 | 16 | 8 | 8 | 16 |
| S. aureus 209P | + | 0.5 | 0.5 | 2 | 0.5 | 0.5 | 0.5 |
| B. subtilis B-886 | + | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 | 1 |
| M. luteus B-1314 | + | 0.5 | 1 | 0.5 | 1 | 1 | 2 |
| Cell Line | IC50, μM | |||||
|---|---|---|---|---|---|---|
| PM I | PM II | PM III | TP I | TP II | TP III | |
| HL-60 | 7.2 ± 0.5 | 7.2 ± 0.5 | 2.5 ± 0.1 | 4.8 ± 0.3 | 5.6 ± 0.5 | 5.0 ± 0.4 |
| HeLa | 12.5 ± 0.9 | 16.4 ± 1.3 | 6.0 ± 0.2 | 24.2 ± 1.7 | 24.4 ± 3.0 | 13.7 ± 1.3 |
| SK-BR-3 | 16.0 ± 0.6 | 17.3 ± 0.6 | 9.9 ± 0.1 | 30.1 ± 1.9 | 34.0 ± 1.3 | 27.5 ± 0.8 |
| A549 | 8.8 ± 1.3 | 10.8 ± 1.9 | 7.3 ± 0.5 | 26.5 ± 1.2 | 28.1 ± 1.7 | 15.3 ± 1.0 |
| HEK 293T | 9.4 ± 1.5 | 11.3 ± 1.6 | 7.3 ± 0.4 | 23.5 ± 1.2 | 24.6 ± 1.7 | 19.7 ± 1.9 |
| HEF | 8.3 ± 0.7 | 9.7 ± 0.2 | 7.0 ± 0.4 | 13.0 ± 1.5 | 17.7 ± 1.2 | 14.1 ± 1.2 |
| NHA | 14.5 ± 1.6 | 18.3 ± 1.4 | 7.5 ± 0.5 | 24.7 ± 1.8 | 29.4 ± 1.7 | 21.3 ± 1.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marggraf, M.B.; Panteleev, P.V.; Emelianova, A.A.; Sorokin, M.I.; Bolosov, I.A.; Buzdin, A.A.; Kuzmin, D.V.; Ovchinnikova, T.V. Cytotoxic Potential of the Novel Horseshoe Crab Peptide Polyphemusin III. Mar. Drugs 2018, 16, 466. https://doi.org/10.3390/md16120466
Marggraf MB, Panteleev PV, Emelianova AA, Sorokin MI, Bolosov IA, Buzdin AA, Kuzmin DV, Ovchinnikova TV. Cytotoxic Potential of the Novel Horseshoe Crab Peptide Polyphemusin III. Marine Drugs. 2018; 16(12):466. https://doi.org/10.3390/md16120466
Chicago/Turabian StyleMarggraf, Mariana B., Pavel V. Panteleev, Anna A. Emelianova, Maxim I. Sorokin, Ilia A. Bolosov, Anton A. Buzdin, Denis V. Kuzmin, and Tatiana V. Ovchinnikova. 2018. "Cytotoxic Potential of the Novel Horseshoe Crab Peptide Polyphemusin III" Marine Drugs 16, no. 12: 466. https://doi.org/10.3390/md16120466