1. Introduction
Linear terpenoids, represented by linear furano- and pyrrolo-terpenoids, are a unique family of natural products with widespread bioactivities [
1,
2]. Among all the isolated linear terpenoids, C
25 linear sesterterpenoids are the major components with more than 200 compounds been isolated, such as nitropyrrolins, heronapyrroles, fukanedones, and manoalide-type sesterterpenoids [
3,
4,
5,
6,
7]. Inspiringly, the manoalide-type C
25 sesterterpenoids, which are frequently obtained from marine sponges, have been studied as topical antipsoriatic lead drug candidates for their potent anti-inflammatory activity [
8,
9]. Apart from the common C
15 sesquiterpenoids, C
20 diterpenoids, and C
25 sesterterpenoids, a few linear terpenoids take irregular C
17 or C
21 carbon skeletons [
1,
2,
3,
4,
5,
6,
7]. To the best of our knowledge, there are only four C
17 terpenoids been isolated from red algae
Laurencia viridis [
10], marine spong
Fasciospongia cavernosa [
11], and Chloranthaceae plant
Chloranthus sessilifolius [
12]. Only one of them takes the linear structure. C
21 linear terpenoids are also a rare class of natural products which are mainly isolated from marine sponges of the genera
Cacospongia, Carteriospongia, Dysidea, Fasciospongia, Hippospongia, Leiosella, Spirastrella, and
Spongia [
1,
2].
Marine sponges of the genus
Cacospongia (order Dictyoceratida, family Thorectidae) draw much attention for its production of diverse terpenoids [
13], such as C
21-difuran terpenoid cacospongienone A from
C. scalaris [
14], manoalide-type sesterterpenoid cacospongionolide from
C. mollior [
15], and tetracyclic scalarane-type sesterterpenoid scalarin from
C. scalaris [
16]. However, only four out of the total 17 identified
Cacospongia species have been studied for their chemical constituents. Besides, the unidentified species of
Cacospongia (
Cacospongia sp.) with potential morphological challenge are found to be significant resource for novel terpenoids. For example, two new terpenoids, namely cacofuran and (+)-isojaspic acid with novel bridged tricyclic carbon skeletons were isolated from two unidentified
Cacospongia species collected from Okinawa and Papua New Guinea, respectively [
17,
18]. Furthermore, the
Cacospongia derived terpenoids usually exhibit excellent pharmacological potentials, such as antibacterial activity, anti-inflammatory, and cytotoxicity [
13,
14,
15,
16,
17,
18].
In the course of our continuing search for new bioactive metabolites from Xisha Island sponges [
19,
20], an unidentified
Cacospongia species was collected and investigated for its chemical components, yielding three pairs of rare C
17 γ-lactone norditerpenoid enantiomers (
1a,
1b,
2a,
2b and the inseparable
3a/
3b), an infrequent C
21 pyridine terpenoid (
7), and six notable C
25 manoalide-type sesterterpenoids (
4–
6,
8–
10) including two pairs of enantiomers (
4a,
4b,
5a and
5b) (
Figure 1). Among them, (±)-8,13-secoepicavernosine (
3a/
3b), (+)-hippolide E (
4a), (+)-(6
E)-neomanoalide (
5a), (3
R,4
R)-14,18-secoluffariolide C (
6), and cacospongine A (
7) were identified as new compounds, while the enantiomers
1a/
1b and
2a/
2b were separated by chiral HPLC. This is the first time to obtain manoalide-type enantiomers from nature. The structures with absolute configurations of the new compounds were unambiguously elucidated by a combinatorial methods, including HR-ESI-MS, 1D and 2D NMR spectra analysis, optical rotation comparison, experimental and calculated ECD comparison, and Mo
2(OAc)
4 induced circular dichroism (ICD) method. Furthermore, cytotoxicity of these compounds was tested against selected tumor cell lines. Herein, we report the isolation, chiral resolution, structure elucidation, and bioactivity of the isolated linear terpenoids.
2. Results and Discussion
Compound
1 was obtained as colorless oil, and its molecular formula was determined as C
17H
28O
3 by HR-ESI-MS (
m/
z 303.1936, [M+Na]
+, calcd 303.1931,
Supplementary Figure S8). A comparison of the
1H and
13C NMR data (
Table 1) with literature suggested
1 to be the reported cavernosine definitely [
11]. However, compound
1 showed neither optical activity (
) nor Cotton effect on its CD spectra, just as cavernosine did in the original report (
) [
11], which suggested
1 to be a racemic mixture. Then, chiral HPLC resolution of
1 afforded a pair of stereoisomers
1a (
) and
1b (
) with a ratio of approximately 1:1 (
Supplementary Figure S3). The relative configuration of the C-4–C-5 fragment in
1a and
1b were both assigned as
erythro type (4
S*,5
R*) by comparing the
1H and
13C NMR data (
Table 1,
Supplementary Figures S9 and S10) with those of the synthesized
erythro (±)-cavernosine and
threo (±)-epicavernosine [
21]. The absolute configurations of
1a and
1b were established by ECD methods. According to literature, the
γ-chiral center of a saturated
γ lactone would induce a Cotton effect at 213 nm, which is mainly attributed to the
πxπx* electronic transition of the carbonyl group in the
γ-lactone ring [
22]. Accordingly,
1a and
1b were found to take a negative and positive Cotton effects at 213 nm, respectively. Thus,
1a and
1b were proposed to have the 4
S and 4
R absolute configurations [
22]. The result was further confirmed by a comparison of the experimental ECD spectra with the calculated curves of the four possible candidate stereoisomers of
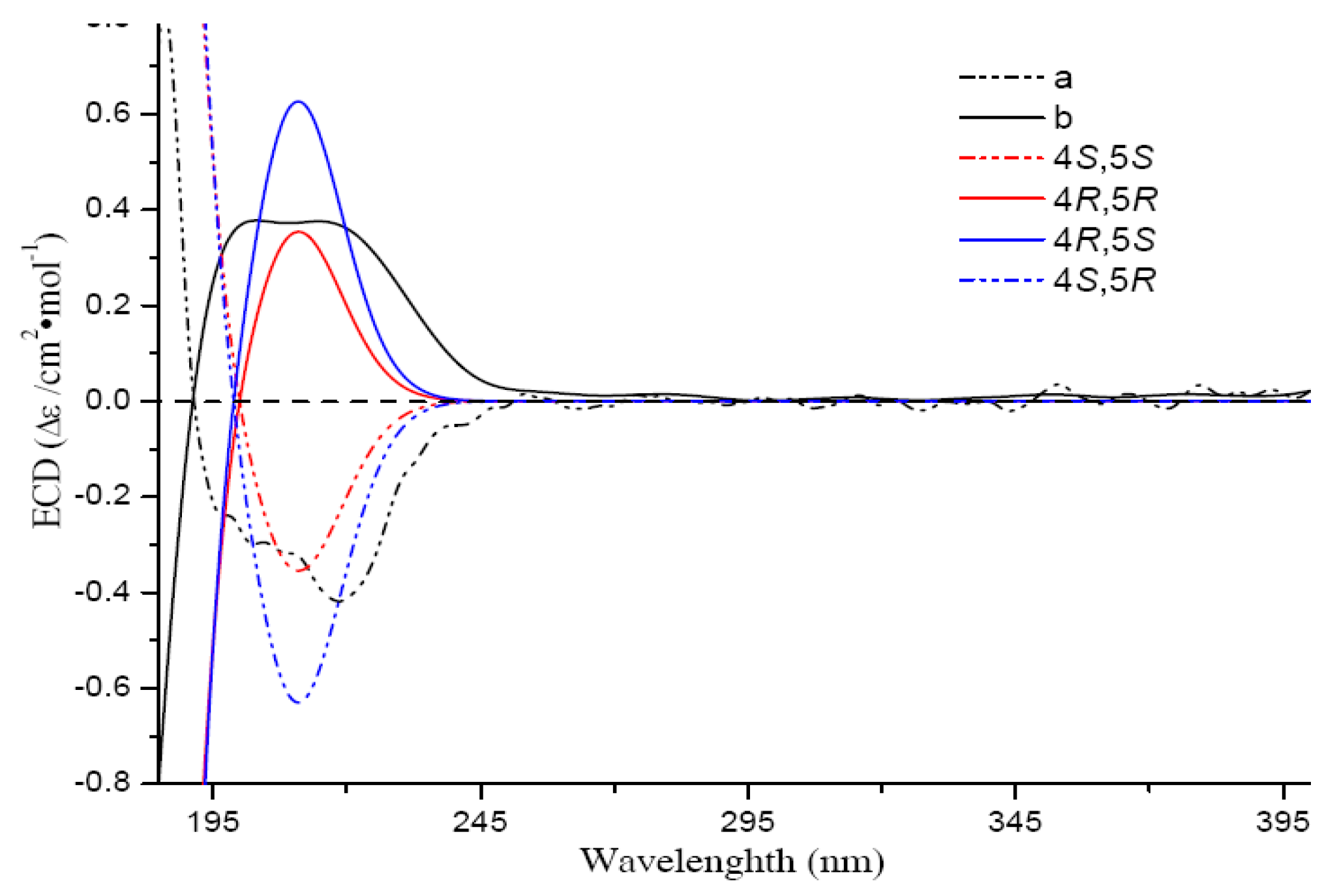
1. The calculated ECD data suggested that the valuence of the Cotton effect at 213 nm was solely related to the absolute configuration of C-4 (negative-4
S, positive-4
R) (
Figure 2), while the Cotton effect intensity could be affected by the auxochromic hydroxy group at C-5. Finally, the absolute configurations of
1a [(+)-cavernosine] and
1b [(−)-cavernosine] were assigned as 4
S,5
R and 4
R,5
S, respectively. This is the first time to separate enantiomers of cavernosine and to determine their absolute configurations.
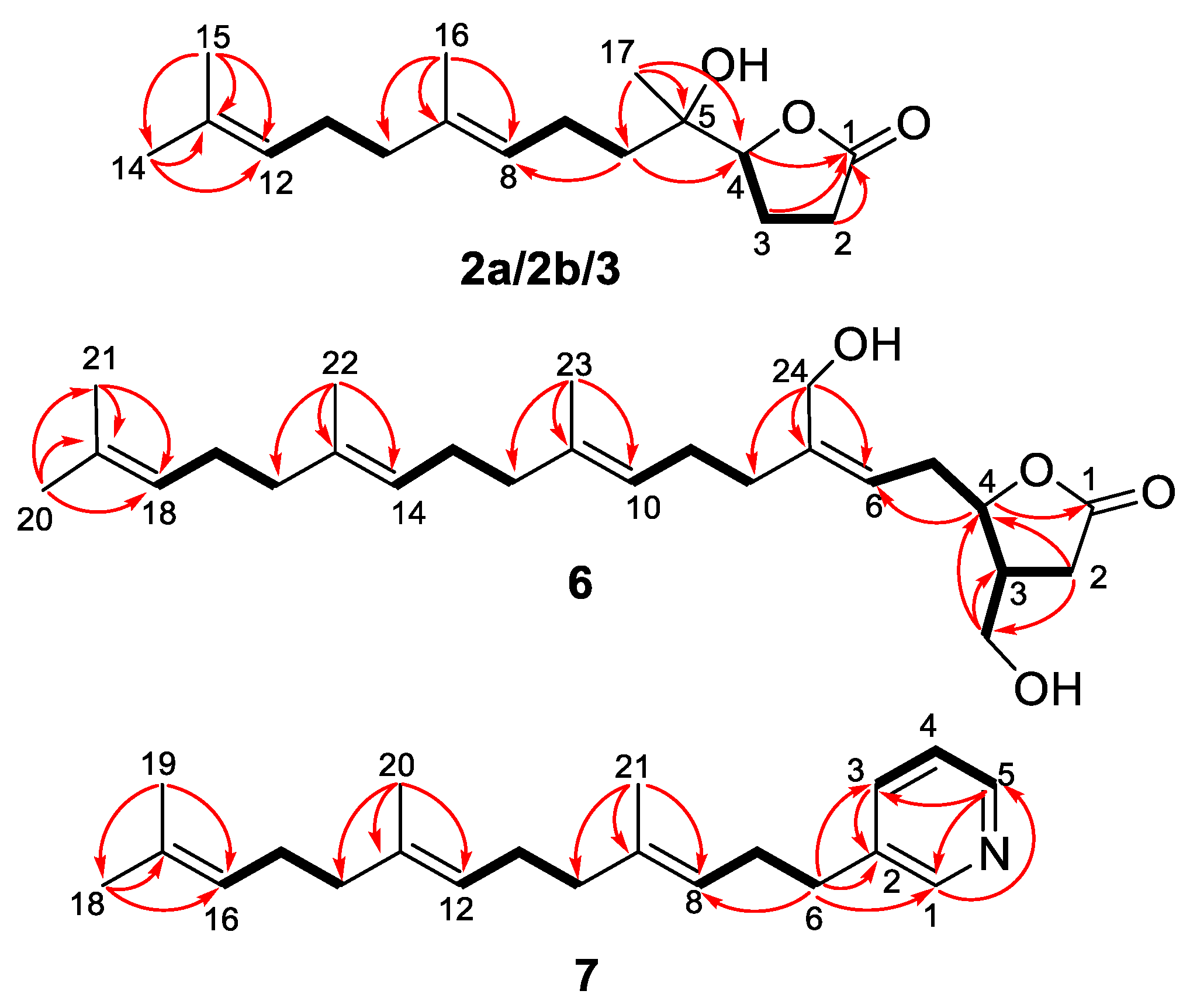
Compounds
2 and
3, having the same molecular formula of C
17H
28O
3 with
1, were isolated as colorless oil. A comparison of the 1D NMR data of
2 with those of
1 (
Table 1,
Supplementary Figures S13–S15, S21 and S22) suggested that they both take the same
γ-lactone ring and C-4-C-7 fragments. However, the rest fragment of
2 is obviously different with that of
1 according to their 1D data, especially the two methine signals (
δH 5.08, t,
J = 7.1 Hz and
δH 5.04, t,
J = 6.9 Hz) observed in
2. HMBC correlations from H
3-14/H
3-15 to two alkenyl carbon signals at
δC 124.0 (C-12) and
δC 131.2 (C-13) strongly suggested the formation of a double bound of C-12 = C-13 in
2 (
Supplementary Figure S18). Thus compound
2 was determined as a 8,13-seco product of
1. Compound
2 has been synthesized as a side product [
23]. The mostly identical 1D and 2D NMR data of
3 and
2 indicated that they both take the same planar structure. The slight difference of Me-17 in
2 and
3 (
δH 1.14,
δC 20.9 in
3 vs.
δH 1.26,
δC 23.0 in
2) suggested they may be a pair of epimers. Geometrical configurations of double bonds ∆
8 in
2 and
3 were both deduced as
E geometry, which was deduced from NOE correlations of H
3-16/H
2-7 and H
2-10/H-8, and from the chemical shifts of C-16 (<20 ppm, a methyl resonance appearing at a chemical shift less than 20 ppm is indicative of an ‘
E’ configuration, whereas a value larger than 20 ppm indicates a ‘
Z’ configuration) [
24]. Comparisons of chemical shifts of H
3-17 in
2 (
δH 1.02; C
6D
6) and
3 (
δH 0.74; C
6D
6) with that of the synthesized
erythro 8,13-secocavernosine (
δH 1.08; C
6D
6) revealed
2 possessing a
erythro-type relative configuration for the C-4–C-5 fragment [
23]. While chemical shifts of Me-17 in
3 (
δH 1.14 vs.
δH 1.26 in
2; CDCl
3) was in agreement with that of the synthesized
threo-type analog [(
S)-5-((
S)-2-hydroxy-6-methylhept-5-en-2-yl)-dihydrofuran-2(3H)-one] (
δH 1.16; CDCl
3) [
25]. Thus, the relative configurations of
2 and
3 were assigned as
erythro (4
S*,5
R*) and
threo (4
S*,5
S*), respectively. Optical rotation value and ECD experiment disclosed that
2 and
3 were both optical inactive. Further chiral resolution of
2 afforded a pair of stereoisomers
2a (
) and
2b (
) (
Supplementary Figure S4). The absolute configurations of
2a and
2b were established as 4
S,5
R and 4
R,5
S, respectively, by experimental and calculated ECD spectra comparison (
Figure 3) Thus,
2a and
2b were finally established as (+)-8,13-secocavernosine and (−)-8,13-secocavernosine, respectively.
However, the optical inactive compound
3 could not be further separated by chiral HPLC (
Supplementary Figure S5). To address the absolute configuration of
3, several chemical tailoring reactions were tried. During the NaOH hydrolysis of
3, the hydrolytic product
3-h was unstable and spontaneously formed the original
γ-lactone
3, even using CH
2N
2 as a protective agent for the carboxyl of
3-h (
Supplementary Scheme S1). Finally, compound
3 was successfully reduced to acyclic 1,4,5-triol derivative using LiAlH
4 (
Scheme 1) [
11]. The reduced product was further separated to be a pair of isomers
3a-r (
) and
3b-r (
), with a ratio of 1:1 on chiral HPLC (
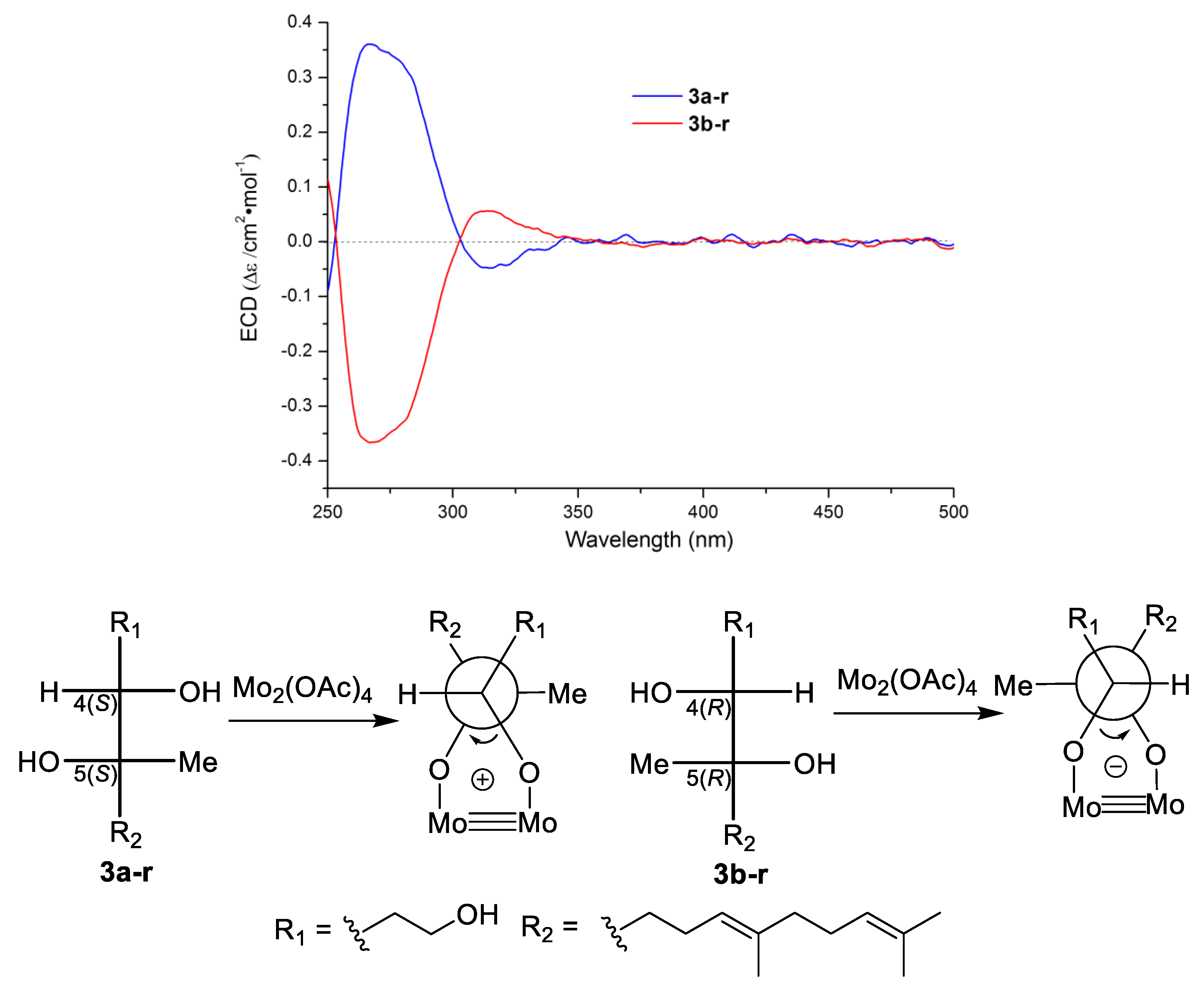
Scheme 1). On the basis of Snatzke’s theory [
26,
27], Mo
2(OAc)
4 induced circular dichroism (ICD) spectra of acyclic adjacent diol substrate (relative configuration determined) could be used to established the absolute configurations of the diol fragment according to the key diagnostic Cotton effect at 310 nm (band IV). Thus, Mo
2(OAc)
4 ICD experiments was carried out, and a positive and a negative Cotton effect at 310 nm on the ICD spectra of
3a-r and
3b-r were observed, respectively. The absolute configurations of
3a-r and
3b-r were determined to be 4
S,5
S and 4
R,5
R (
Figure 4), respectively [
26,
27]. This result confirmed
3 to be a pair of inseparable new enantiomers bearing the 4
S,5
S and 4
R,5
R absolute configurations.
Compounds
4a/
4b and
5a/
5b, having the same molecular formula of C
25H
40O
4 as deduced by HR-ESI-MS data (
Supplementary Figures S27 and S31), were obtained as two pairs of enantiomers both with an approximate ratio of 1:3 (
Supplementary Figure S7). Each pair of isomers possess the same NMR data (
Table 2), while their optical rotations (
4a + 4.1 vs.
4b − 7.4;
5a + 3.6 vs.
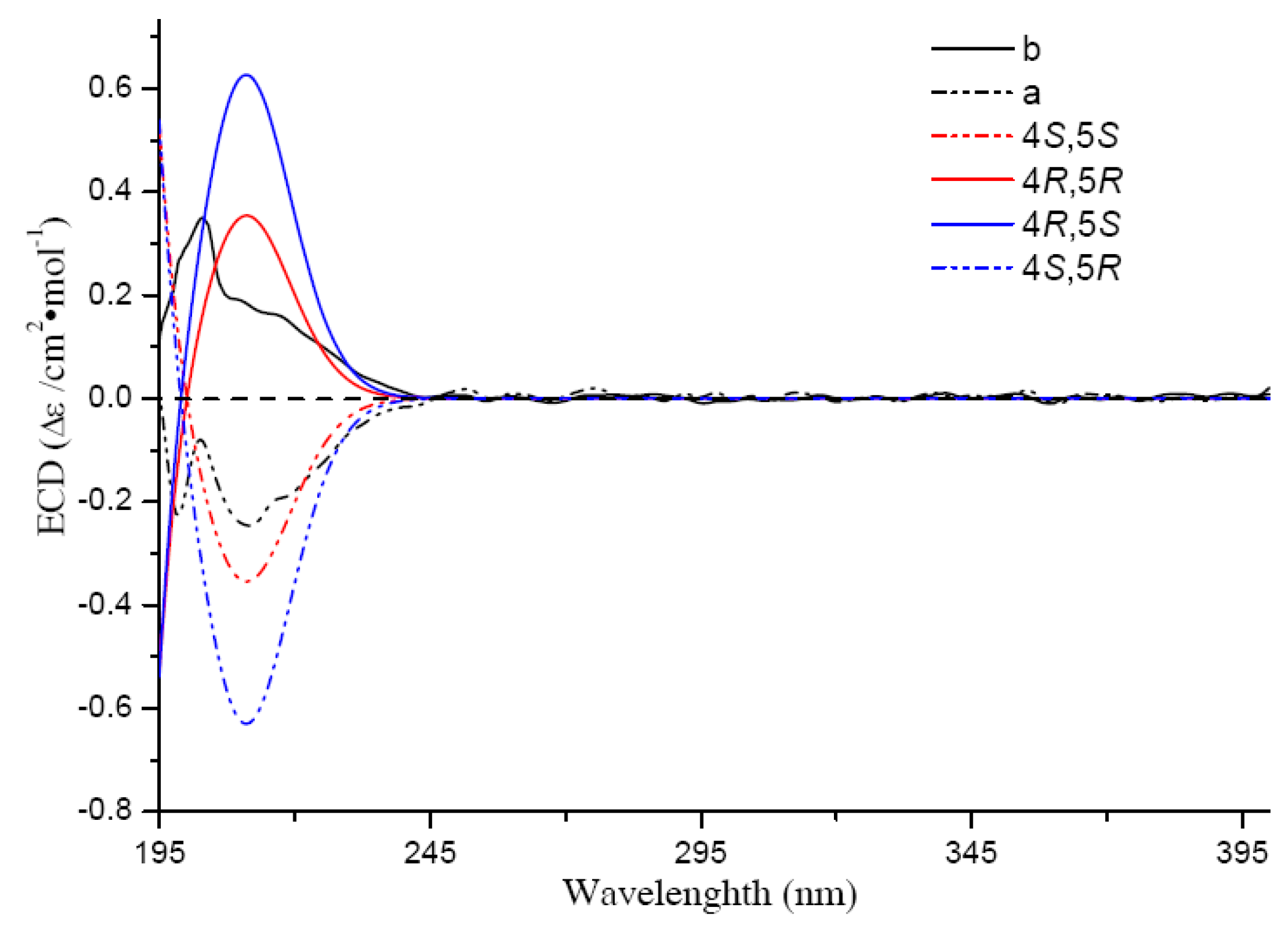
5b − 7.7) and ECD curves (
Figure 5) are almost opposite. Comparisons of 1D NMR data of
4a/
4b and
5a/
5b (
Table 2,
Supplementary Figures S28–S30 and S32–S34) with those of literatures revealed their planar structure to be hippolide E and (
E)-neomanoalide, respectively [
28,
29]. The absolute configurations of
4b [(−)-hippolide E] and
5b [(−)-(
E)-neomanoalide] were assigned to be the same as hippolide E and (6
E)-neomanoalide-24-ol [
28,
29] by CD spectra comparisons. Their enantiomers
4a [(+)-hippolide E] and
5a [(+)-(6
E)-neomanoalide-24-ol] were both deduced to be 4
S configuration according to their opposite CD absorptions (
Figure 5) and opposite optical rotations comparing with
4b and
5b. Thus,
4a and
5a were identified as two new enantiomers of hippolide E and (
E)-neomanoalide-24-ol, respectively. This is the first time to obtain the natural-occurring enantiomers of the well-known manoalide-type sesterterpenoids.
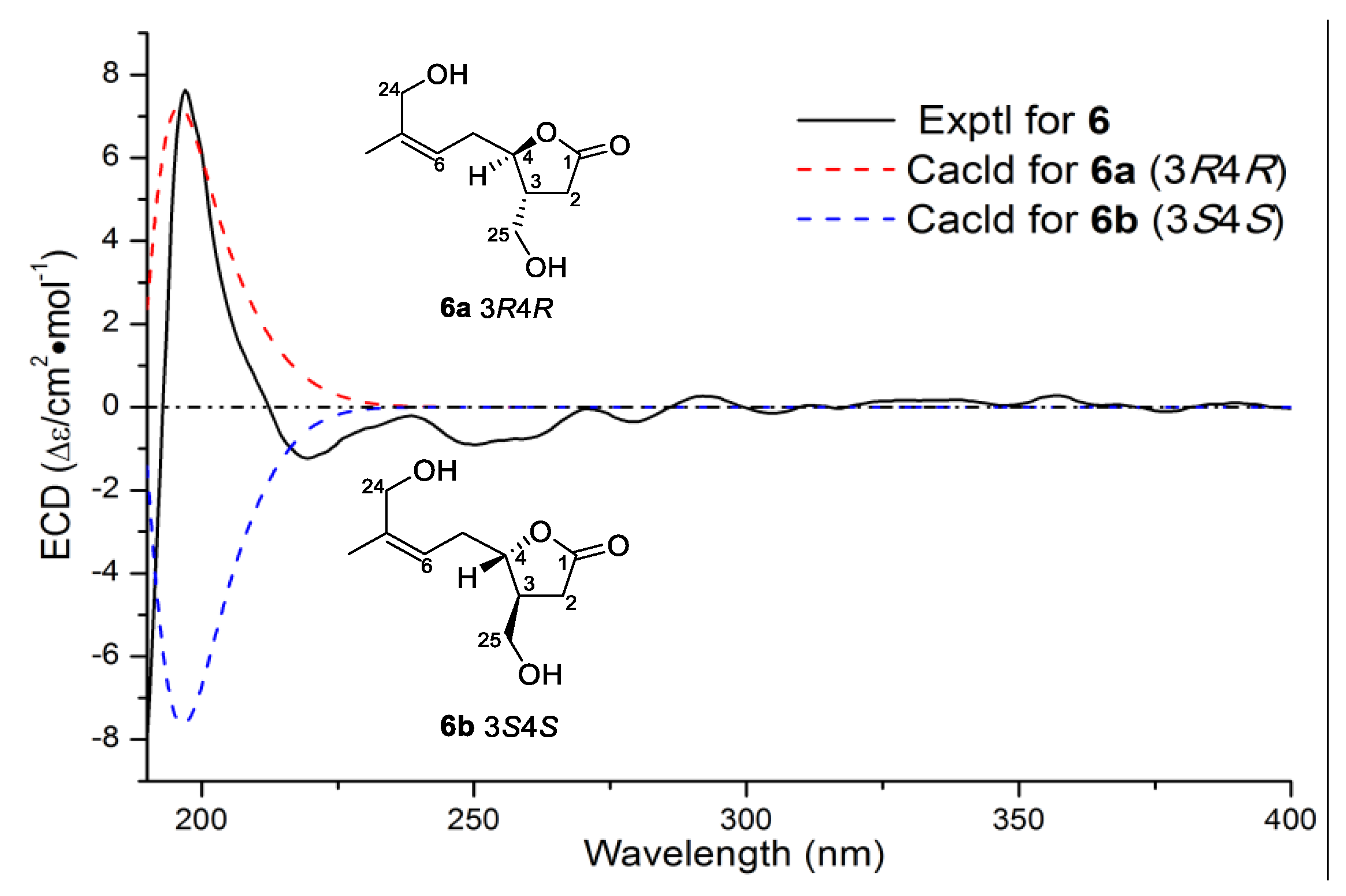
Compound
6, with the molecular formula of C
25H
40O
4, was also considered as a manoalide-type sesterterpenoid according to its similar 1D NMR data with those of
4a/
4b and
5a/
5b (
Table 2,
Supplementary Figures S36–S38). Further 1D NMR data comparison of
6 with those of co-isolated luffariolide C (
8) revealed their structural similarity, except for a slight difference of Me-20 (
δH 1.68 in
6 vs.
δH 0.99 in luffariolide C) and Me-21 (1.60 vs. 0.99) [
30]. HMBC correlations from H
3-20/H
3-21 to the olefinic carbons of C-18 (
δC 124.3) and C-19 (
δC 131.3) (
Figure 6,
Supplementary Figure S41) suggested that the cyclohexene ring in luffariolide C was cleaved and formed an extra double bound of C-18=C-19 in
6. Further 2D NMR analysis, such as
1H-
1H COSY correlations of H
2-16/H
2-17/H-18 and HMBC correlations from H
3-22 to C-14/C-15/C-16, (
Figure 6) confirmed the planar structure of
6 to be 14,18-seco luffariolide C. Double bounds of ∆
10 and ∆
14 were both deduced to be
E geometry according to NOE correlations of H
3-23/H
2-9 and H
3-22/H
2-13 (
Figure 7). While double bound of ∆
6 was assigned as
Z geometry by NOE correlation of H
2-24/H
2-5. Additionally, NOE correlations of H-3/H
2-5 and H-4/H
2-25 (
Figure 7), as well as the coupling constant between H-3 and H-4 (
J = 0 Hz) in
6, suggested it takes the same 3,4-
trans relative configuration with luffariolide C. Moreover, the similar optical rotations of
6 (
) and luffariolide C (
) revealed the same 3
R,4
R absolute configurations. The result was further supported by experimental and theoretical ECD comparisons of the main chiral lactone structure in
6 (
Figure 8). Thus,
6 was determined as (3
R,4
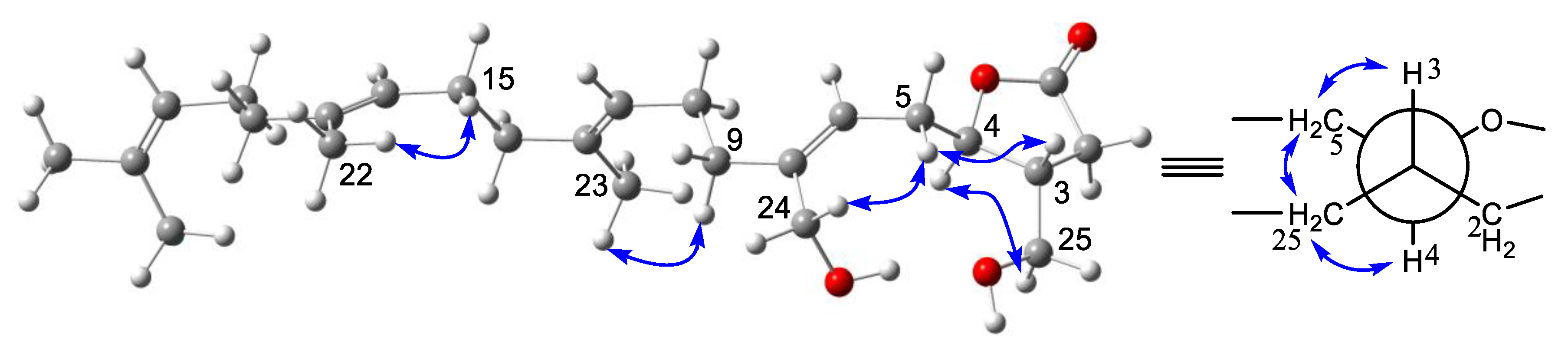
R)-14,18-secoluffariolide C.
The molecular formula of compound
7 was deduced as C
21H
31N by HR-ESI-MS at
m/
z 298.2532 ([M + H]
+ calcd 298.2529,
Supplementary Figure S43).
1H NMR spectrum of
7 showed four aromatic proton signals (
δH 8.40, s; 8.38, d,
J = 4.1 Hz; 7.60, d,
J = 7.6 Hz; 7.27, dd,
J = 6.7, 5.6 Hz). The remaining proton signals in upfield were similar with those of
6, especially the four olefinic methyls of H
3-18 (
δH 1.63 in
7 vs. 1.68 in
6), H
3-19 (1.54 vs. 1.60), H
3-20 (1.54 vs. 1.60) and H
3-21 (1.47 vs. 1.60).
1H-
1H COSY correlations of H
2-6/H
2-7/H-8, H
2-10/H
2-11/H-12 and H
2-14/H
2-15/H-16, together with HMBC correlations from H
3-18/ H
3-19 to C-16/C-17, from H
3-20 to C-12/C-13/C-14, and from H
3-21 to C-8/C-9/C-10, indicated a C
16 prenyl chain (C-6-C-18). Additionally, a monosubstituted pyridine ring was constructed based on
1H-
1H COSY correlations of H-3/H-4/H-5 and HMBC correlations from H-5 to C-1/C-3, from H-3 to C-2 and from H-1 to C-5 (
Figure 6,
Supplementary Figures S48 and S49). The C
16 linear chain was connected with the pyridine ring at C-2 by HMBC correlations from H
2-6 (
δH 2.60, t,
J = 7.6 Hz) to C-1 (
δC 149.6), C-2 (
δC 137.0) and C-3 (
δC 135.8) (
Figure 6). The
E geometry of double bounds ∆
8 and ∆
12 were both deduced from the chemical shifts of C-21 and C-20 (
δC both at 15.7, < 20 ppm) [
24]. Thus compound
7 was determined as a C
21 pyridine terpenoid and was named as cacospongine A.
The structures of the known analogs of luffariolide C (
8) [
30], (
Z)-neomanoalide (
9) [
29] and hippolide J (
10) [
31] were determined by comparison of their 1D/2D NMR data, ESI-MS data and optical rotations with those of reported values.
The cytotoxicity of the isolates against human tumor cell lines of K562, HCT116, Hep3B, A-549, and Jurkat was evaluated, using MTT method [
32] with adriamycin as positive control. However, all the tested compounds were inactive (IC
50 > 10 μM).
3. Experimental Section
3.1. General Methods
Optical rotations were measured on a JASCO P-1020 digital polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra were measured on a Beckman DU640 spectrophotometer. ECD spectra were obtained on a Jasco J-815 CD spectrometer. IR spectra were recorded on a Nicolet Nexus 470 (FT-IR) spectrophotometer (Thermo Electron Co., Madison, WI, USA), KBr pellets. NMR spectra were recorded on a Bruker DRX-500MHz instrument (Bruker BioSpin GmbH Co., Rheinstetten, Germany), 500 MHz for 1H NMR and 125 MHz for 13C NMR in CDCl3; chemical shifts δ in ppm referred to the solvent peaks at δH 7.26 and δC 77.0 for CDCl3, δH 2.50 and C 39.5 for DMSO-d6 and δH 7.16 for C6D6, and coupling constant J in Hz. HR-ESI-MS were obtained from a Micromass Q-Tof Ultima GLOBAL GAA076 LC-mass spectrometer (Waters Corporation, Milford, MA, USA). HPLC separation was performed on an Agilent 1100 series instrument with DAD detector (Agilent technologies, Santa Clara, CA, USA), equipped with a semi-preparative reversed-phased column (YMC-packed C18, 5 μm, 250 × 10 mm, 1.5 mL/min) or an analytic chiral column DAICEL IC-3 (DAICEL chiral technologies, Shanghai, China). Precoated silica gel plates (GF254, Qingdao Marine Chemical Inc., Qingdao, China) were used for TLC analyses. Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China) was used for column chromatography (CC).
3.2. Animal Material
The sponge
Cacospongia sp. is in irregular shape, with black-brownish color, and covered with a tight encrusting crust. There are irregularity distributed holes on the surface, wihch is thick and solid, and is in the dimensions of around 4 × 5 cm
6 × 8 cm (see
Supplementary I. Photos of sponge specimen). The specimen was collected from the coral-reef regions of Yong Xing Island (16°50′ N, 112°20′ E) in the South China Sea, at a depth ranging from 18 to 25 m, in November 2010. The specimen, with a voucher of no. XS-2009-34, was frozen immediately at −20 °C until it was examined. The voucher was deposited at the School of Medicine and Pharmacy, Ocean University of China, China. The sponge was identified by Dr. N.J.d.V. (Naturalis Biodiversity Centre, Leiden, The Netherlands).
3.3. Extraction and Isolation
The frozen sponge (2.6 kg, wet weight) was minced and extracted with MeOH for three times (each time for one day) at room temperature (5 L × 3). The combined solution was evaporated in vacuum and desalinated for three times to yield a residue (56.0 g). The crude extract was then subjected to a reduced pressure silica gel column eluting with a step-by-step gradient elution of acetone-petroleum ether (from 0:1 to 1:0, v/v), to give six fractions. Fraction 2 was then further separated by silica gel column eluting with petroleum ether/acetone (2:1, 1:1, 0:1, v/v) to give subfractions F2-1–F2-5. F2-2 was further purified by semi-preparative HPLC with a mobile phase of MeOH/H2O (70:30, v/v) to give three pairs of isomer mixtures 1 (6.0 mg; tR 53.0 min), 2 (30.1 mg; tR 59.0 min) and 3 (4.1 mg; tR 56.0 min). These isomers were finally separated by chiral HPLC with a mobile phase of n-hexane/ isopropanol (93:7, v/v), and yielded 1a (2.3 mg; tR 15.0 min), 1b (2.4mg; tR 28.0 min), 2a (12.1 mg; tR 15.0 min), 2b (12.0 mg; tR 24.5 min), 3a/3b (enantiomeric mixture, 4.1 mg; tR 17.5 min). HPLC purification (85:15, MeOH/H2O, v/v) of F2-3 afforded compound 7 (6.1 mg; tR 37.0 min). Fraction 3 was chromatographed on another silica gel column to give two subfractions F3-1 and F3-2, which were found to contain terpenoid components by TLC analyses. Then F3-1 was further separated by HPLC (85:15, MeOH/H2O, v/v) to give 6 (2.4 mg; tR 42.0 min), 8 (1.5 mg; tR 37.0 min), 9 (4.6 mg; tR 56.0 min) and 10 (5.0 mg; tR 61.0 min). The two pairs of isomers of 4a (1.6 mg; tR 47.0 min)/4b (4.2 mg; tR 42.0 min) and 5a (1.5 mg; tR 49.0 min) /and 5b (4.3 mg; tR 44.0 min) were ultimately separated by chiral HPLC with a mobile phase of n-hexane/ethanol (94:6, v/v) from F3-2.
Cavernosine (
1)
: colorless oil; (+)
-cavernosine (
1a),
(
c 0.1, MeOH);
(−)-cavernosine (
1b),
(
c 0.1, MeOH); IR (KBr)
vmax 3479, 2926, 1779, 1450, 1374, 1191 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 1; HRESIMS
m/
z 303.1936 [M + Na]
+ (calcd for C
17H
28O
3Na, 303.1931).
8,13-secocavernosine (
2): colorless oil;
(+)-8,13-secocavernosine (
2a)
: (
c 0.1, MeOH);
(−)-8,13-secocavernosine (
2b),
(
c 0.1, MeOH); IR (KBr)
vmax 3469, 2928, 1774, 1453, 1377, 1191 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 1;
1H NMR in C
6D
6 (500 MHz)
δ 5.22 (t,
J = 6.9 Hz, 1H), 5.16 (t,
J = 6.9 Hz, 1H), 3.64 (t,
J = 7.5 Hz, 1H), 1.68 (s, 3H), 1.57 (s, 3H), 1.57 (s, 3H), 1.02 (s, 3H); HRESIMS
m/
z 303.1938 [M + Na]
+ (calcd for C
17H
28O
3Na, 303.1931).
(±)-8,13-secoepicavernosine (
3a/3b, unseparated)
: colorless oil (MeOH);
(
c 0.1, MeOH); IR (KBr)
vmax 3473, 2919, 1774, 1649, 1456, 1379, 1190, 1004 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 1;
1H NMR in C
6D
6 (500 MHz)
δ 5.25 (t,
J = 6.9 Hz, 1H)), 5.20 (t,
J = 7.0 Hz, 1H), 3.62 (t,
J = 7.5 Hz, 1H), 1.70 (s, 3H), 1.61 (s, 3H), 1.59 (s, 3H), 0.74 (s, 3H); HRESIMS
m/
z 303.1936 [M + Na]
+ (calcd for C
17H
28O
3Na, 303.1931).
(+)-hippolide E (
4a)
: colorless oil (MeOH);
(
c 0.1, MeOH); IR (KBr)
vmax 3410, 2925, 2360, 2338, 1746, 1700, 1650, 1540, 1455, 1381 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 2; HRESIMS
m/
z 425.2671 [M + Na]
+ (calcd for C
25H
38O
4Na, 425.2662).
(+)-(6E)-neomanoalide (
5a)
: colorless oil (MeOH);
(
c 0.1, MeOH); IR (KBr)
vmax 3415, 2926, 2860, 2359, 2338, 1744, 1647, 1455, 1380, 1143, 1063 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 2; HRESIMS
m/
z 425.2674 [M + Na]
+ (calcd for C
25H
38O
4Na, 425.2662).
(3R,4R)-14,18-secoluffariolide C (
6)
: colorless oil (MeOH);
(
c 0.1, MeOH); IR (KBr)
vmax 3384, 2922, 2856, 2359, 2337, 1750, 1451, 1375, 1265, 1196, 1018 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 2; HRESIMS
m/
z 405.2995 [M + H]
+ (calcd for C
25H
41O
4, 405.2999).
Cacospongine A (
7)
: colorless oil (MeOH); IR (KBr)
vmax 2924, 2854, 2361, 2339, 1718, 1453, 1379, 1190 cm
−1;
1H NMR (DMSO-
d6, 500 MHz) and
13C NMR (DMSO-
d6, 125 MHz), see
Table 2; HRESIMS
m/
z 298.2532 [M + H]
+ (calcd for C
21H
32N, 298.2529).
3.4. LiAlH4 Reduction of 3
The solution of
3 (3 mg) in dry THF (2 mL) containing LiAlH
4 (3 mg) were stirred for 6 h at room temperature, under the protection of argon atmosphere. The reduction reaction was quenched by addition of 2 mL of 10% KOH aqueous solution, and the mixture was then extracted by EtOAc to yielded crude products (2.7 mg) [
11]. The isomer mixtures were further separated by chiral HPLC (n-hexane/ isopropanol, 93:7,
v/
v) to give
3a-r (1.3 mg;
tR 11.7 min) and
3b-r (1.2 mg;
tR 12.7 min).
Compounds3a-r and 3b-r: colorless oil from MeOH; for 3a-r and +11.1 for 3b-r (c 0.1, MeOH); 1H NMR (CDCl3, 500 MHz) δ 5.14 (1H, t, J = 6.7 Hz, H-8), 5.08 (1H, t, J = 6.7 Hz, H-12), 3.70 (2H, m, H2-1), 3.46 (1H, d, J = 10.9 Hz, H-4), 2.11 (1H, m, H-7a), 2.06 (1H, m, H-7b), 2.06 (2H, m, H2-11), 1.98 (2H, m, H2-10), 1.75 (2H, m, H2-2), 1.68 (3H, s, H3-14), 1.62 (3H, s, H3-16), 1.60 (3H, s, H3-15), 1.56 (1H, m, H-6a), 1.50 (each 1H, m, H-6b, H-3a), 1.40 (1H, m, H2-3b), 1.12 (3H, s, H3-17); 13C NMR (CDCl3, 125 MHz) δ 135.8 (C, C-9), 131.5 (C, C-13), 124.2 (CH, C-12), 124.1 (CH, C-8), 77.2 (CH, C-4), 75.1 (C, C-5), 62.9 (CH2, C-1), 39.7 (CH2, C-10), 38.8 (CH2, C-6), 30.1 (CH2, C-2), 28.6 (CH2, C-3), 26.6 (CH2, C-11), 25.7 (CH3, C-14), 20.8 (CH3, C-17), 22.0 (CH2, C-7), 17.7 (CH3, C-15), 16.0 (CH3, C-16).
3.5. Determination of the Absolute Configuration of the Diol Moiety in 3a-r and 3b-r by Snatzke’s Method
ICD spectra of Mo-complexes of
3a-r and
3b-r were obtained according to reported procedures [
26,
27]. The reduced products (each 0.5 mg) and Mo
2(OAc)
4 (1.0 mg) were dissolved in 1.5 mL of dry DMSO to give a solution, with the ligand to metal molar ratio being around 1:1.2. The electronic transitions of the metal complexes in DMSO were monitored by a Jasco J-815 CD spectrometer in the UV–vis region of 250–500 nm, and stationary ICD spectra were obtained after 50 min at 15 °C. Because there were no inherent absorptions for the reduced products, the observed ICD spectra could be directly used to analyze the absolute configurations of diol fragments in the ligands with the characteristic cotton effects around 310 nm, according to Snatzke’s theory [
26,
27].
3.6. Calculating Section
The quantum chemical calculations were performed using the density functional theory (DFT) by Gaussian 09 [
33]. The initial key chiral structures in compounds
1a,
1b,
2a,
2b and
6 were built with Spartan 10 software, and all trial structures were first minimized based on molecular mechanics calculations. Conformational search was performed by Spartan 10 software using MMFF force filed, and conformers occurring within a 10 kcal/mol energy window from the global minimum were chosen for geometry optimization in the gas phase with the DFT method at the B3LYP/DGDZVP level. The B3LYP/DGDZVP harmonic vibrational frequencies were further calculated to confirm their stability. The spin-allowed excitation energies and rotatory (
Rn) and oscillator strengths (
fn) of the lowest excited states of stable conformers were calculated for ECD spectra using TD-DFT method with the basis set RB3LYP/DGDZVP. Solvent effects of methanol solution were evaluated at the same DFT level by using the SCRF/PCM method in agreement with the experiment condition. Electronic transitions were expanded as Gaussian curves with a FQHM (full width at half maximum) for each peak of 0.32 eV. The ECD spectra were combined after Boltzmann weighting according to their population contribution.
3.7. Cytotoxicity
In vitro cytotoxicity was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric assay against K562, HCT116, Hep3B, A-549 and Jurkat cell lines by reported procedures [
32]. All the cell lines were purchased from Shanghai Institute of Cell Biology (Shanghai, China). Adriamycin (doxorubicin, ADM) was used as a positive control.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}