α-Glucosidase Inhibition and Molecular Docking Studies of Natural Brominated Metabolites from Marine Macro Brown Alga Dictyopteris hoytii

 , , ,
, , ,
Abstract
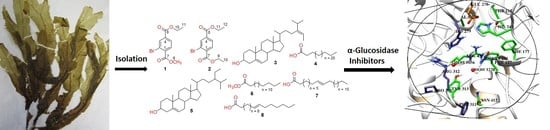
:
1. Introduction
2. Results and Discussion
2.1. Structure Elucidation of Compounds 1–8
2.2. α-Glucosidase Inhibition
2.3. Molecular Docking of α-Glucosidase Inhibitors
2.4. Pharmakinetic Prediction of α-Glucosidase Inhibitors
3. Material and Methods
3.1. General
3.2. Sample Collection and Identification
3.3. Extraction, Isolation and Purification
3.4. In Vitro α-Glucosidase Inhibition
3.5. Molecular Docking and Pharmacokinetic Prediction
3.6. Pharmacophore Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO (World Health Organization). Global Health Estimates: Deaths by Cause, Age, Sex and Country,2000–2012; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Rehman, N.U.; Khan, A.; Al-Harrasi, A.; Hussain, H.; Wadood, A.; Riaz, M.; Al-Abri, Z. New α-glucosidase inhibitors from the resins of Boswellia species with structure–glucosidase activity and molecular docking studies. Bioorg. Chem. 2018, 79, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Nam, K.A.; Kurihara, H.; Kim, S.M. Potent alpha-glucosidase inhibitors purified from the red alga Grateloupia elliptica. Phytochemistry 2008, 69, 2820–2825. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.E.; Montgomery, P.A. Acarbose: An α-glucosidase inhibitor. American Journal of Health System Pharmacy. Am. J. Health. Syst. Pharm. 1996, 53, 2277–2290. [Google Scholar] [CrossRef] [PubMed]
- Chougale, A.D.; Ghadyale, V.A.; Panaskar, S.N.; Arvindekar, A.U. Alpha glucosidase inhibition by stem extract of Tinospora cordifolia. J. Enyzm. Inhib. Med. Chem. 2009, 24, 998–1001. [Google Scholar] [CrossRef]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef]
- Shi, D.; Xu, F.; He, J.; Li, J.; Fan, X.; Han, L. Inhibition of bromophenols against PTP1B and anti-hyperglycemic effect of Rhodomela confervoides extract in diabetic rats. Chin. Sci. Bull. 2008, 53, 2476–2479. [Google Scholar] [CrossRef]
- Wang, W.; Okada, Y.; Shi, H.; Wang, Y.; Okuyama, T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2005, 68, 620–622. [Google Scholar] [CrossRef]
- Fisch, K.M.; Bohm, V.; Wright, A.D.; Konig, G.M. Antioxidative meroterpenoids from the brown alga Cystoseira crinita. J. Nat. Prod. 2003, 66, 968–975. [Google Scholar] [CrossRef]
- Jesus, A.; Correia-da-Silva, M.; Afonso, C.; Pinto, M.; Cidade, H. Isolation and potential biological applications of haloaryl secondary metabolites from macroalgae. Mar. Drugs 2019, 17, 73. [Google Scholar] [CrossRef]
- Liu, M.; Hansen, P.E.; Lin, X. Bromophenols in marine algae and their bioactivities. Mar. Drugs 2011, 9, 1273–1292. [Google Scholar] [CrossRef]
- Kim, K.Y.; Nguyen, T.H.; Kurihara, H.; Kim, S.M. Alpha-glucosidase inhibitory activity of bromophenol purified from the red alga Polyopes lancifolia. J. Food Sci. 2010, 75, H145–H150. [Google Scholar]
- Fan, X.; Xu, N.J.; Shi, J.G. Bromophenols from the red alga Rhodomela confervoides. J. Nat. Prod. 2003, 66, 455–458. [Google Scholar] [CrossRef]
- Li, K.; Li, X.M.; Ji, N.Y.; Wang, B.G. Natural bromophenols from the marine red alga Polysiphonia urceolata (Rhodomelaceae): structural elucidation and DPPH radical-scavenging activity. Bioorg. Med. Chem. 2007, 15, 6627–6631. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, X.M.; Ji, N.Y.; Wang, B.G. Bromophenols from the marine red alga Polysiphonia urceolata with DPPH radical scavenging activity. J. Nat. Prod. 2008, 71, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.J.; Li, X.M.; Wang, B.G. Highly brominated mono- and bis-phenols from the marine red alga Symphyocladia latiuscula with radical-scavenging activity. J. Nat. Prod. 2007, 70, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Zatelli, G.A.; Philippus, A.C.; Falkenberg, M. An overview of odoriferous marine seaweeds of the Dictyopteris genus: insights into their chemical diversity, biological potential and ecological roles. Braz. J. Pharmacog. 2018, 28, 243–260. [Google Scholar] [CrossRef]
- AlgaebBase. Available online: http://www.algaebase.org (accessed on 7 October 2019).
- Feng, M.T.; Wang, T.; Liu, A.H.; Li, J.; Yao, L.G.; Wang, B.; Guo, Y.W.; Mao, S.C. PTP1B inhibitory and cytotoxic C-24 epimers of Δ28-24-hydroxy stigmastane-type steroids from the brown alga Dictyopteris undulata Holmes. Phytochemistry 2018, 146, 25–35. [Google Scholar] [CrossRef]
- Segawa, M.; Yamano, K.; Shirahama, H. A germacrane-type sesquiterpene from the brown alga Dictyopteris divaricata. Phytochemistry 1990, 29, 973–974. [Google Scholar] [CrossRef]
- El Hattab, M.; Culioli, G.; Piovetti, L.; Chitour, S.E.; Valls, R. Comparison of various extraction methods for identification and determination of volatile metabolites from the brown alga Dictyopteris membranacea. J. Chromatogr. A. 2007, 1143, 1–7. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, L.W.; Feng, M.T.; Liu, A.H.; Li, J.; Zhao, T.S.; Lai, X.P.; Wang, B.; Guo, Y.W.; Mao, S.C. Dictyoptesterols A–C, ∆ 22-24-oxo cholestane-type sterols with potent PTP1B inhibitory activity from the brown alga Dictyopteris undulata Holmes. Fitoterapia 2018, 130, 241–246. [Google Scholar] [CrossRef]
- Ji, N.Y.; Song, Y.P.; Miao, F.P.; Liang, X.R. Three cadinane derivatives from the marine brown alga Dictyopteris divaricata. Magn. Reson. Chem. 2016, 54, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, X.; Li, S.; Hao, L.; Du, J.; Gao, D.; Kang, Q.; Lu, J. Extraction, characterization and biological activity of sulfated polysaccharides from seaweed Dictyopteris divaricata. Int. J. Biol. Macromol. 2018, 117, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Dimou, M.; Ioannou, E.; Daskalaki, M.G.; Tziveleka, L.A.; Kampranis, S.C.; Roussis, V. Disulfides with anti-inflammatory activity from the brown alga Dictyopteris membranacea. J. Nat. Prod. 2016, 79, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Fan, X.; Xu, X.; Zhao, J.; Yang, Y.; Shi, J. Cadinane sesquiterpenes from the brown alga Dictyopteris divaricata. J. Nat. Prod. 2004, 67, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.Y.; Wen, W.; Li, X.M.; Xue, Q.Z.; Xiao, H.L.; Wang, B.G. Brominated selinane sesquiterpenes from the marine brown alga Dictyopteris divaricata. Mar. Drugs 2009, 7, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Ali, L.; Khan, A.; Al-Broumi, M.; Al-Harrasi, R.; Al-Kharusi, L.; Hussain, J.; Al-Harrasi, A. New enzyme-inhibitory triterpenoid from marine macro brown alga Padina boergesenii Allender & Kraft. Mar. Drugs 2017, 15, E19. [Google Scholar] [PubMed]
- Ali, L.; Khan, A.; Al-Kharusi, L.; Hussain, J.; Al-Harrasi, A. New α-glucosidase inhibitory triterpenic acid from marine macro green alga Codium dwarkense Boergs. Mar. Drugs 2015, 13, 4344–4356. [Google Scholar] [CrossRef]
- Ali, L.; Al-Kharusi, L.; Al-Harrasi, A. Two new sulfonoglycolipids from green alga Codium dwarkense. Nat. Prod. Commun. 2017, 12, 583–585. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, L.; Fan, W.; Li, B.; Liu, G.; Liu, X.; Zhao, X. Structural diversity, luminescence and photocatalytic properties of six coordination polymers based on designed bifunctional 2-(imidazol-1-yl) terephthalic acid. CrystEngComm 2016, 18, 6914–6925. [Google Scholar] [CrossRef]
- Huang, X.; Yang, L.; Emanuelsson, R.; Bergquist, J.; Strømme, M.; Sjödin, M.; Gogoll, A. A versatile route to polythiophenes with functional pendant groups using alkyne chemistry. Beilstein J. Org. Chem. 2016, 12, 2682–2688. [Google Scholar] [CrossRef]
- Fast, C.D.; Woods, J.; Lentchner, J.; Makal, T.A. Stabilizing defects in metal–organic frameworks: pendant Lewis basic sites as capping agents in UiO-66-type MOFs toward highly stable and defective porous materials. Dalton Trans. 2019, 48, 14696–14704. [Google Scholar] [CrossRef] [PubMed]
- Aboutabl, E.A.; Zeid, A.H.A.; Sleem, A.A.; El Rafi, H.M. Secondary metabolites and certain bioactivities of Pterocladia capillacea (S. Gmelin) Bornet and Dictyopteris membranacea (Stackhouse) Batters. Med. Aromat. Plant Sci. Biotechnol. 2010, 4, 41–48. [Google Scholar]
- Nyemb, J.N.; Magnibou, L.M.; Talla, E.; Tchinda, A.T.; Tchuenguem, R.T.; Henoumont, C.; Laurent, S.; Mbafor, J.T. Lipids constituents from gardenia aqualla stapf & hutch. Open Chem. 2018, 16, 371–376. [Google Scholar]
- Hwang, S.H.; Jang, J.M.; Lim, S.S. Isolation of fucosterol from Pelvetia siliquosa by high-speed countercurrent chromatography. Fish. Aquat. Sci. 2012, 15, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.K.; Park, S.K.; Park, S.H.; Jhang, S.K. Lipids Constituents of the Korean marine sponges. J. Korean Chem. Soc. 1991, 35, 85–89. [Google Scholar]
- Ajoku, G.A.; Okwute, S.K.; Okogun, J.I. Isolation of hexadecanoic acid methyl ester and 1, 1, 2-ethanetricarboxylic acid-1-hydroxy-1, 1-dimethyl ester from the calyx of green Hibiscus sabdariffa (Linn). Nat. Prod. Chem. Res. 2015, 3, 169–174. [Google Scholar]
- Ododo, M.M.; Choudhury, M.K.; Dekebo, A.H. Structure elucidation of β-sitosterol with antibacterial activity from the root bark of Malva parviflora. SpringerPlus 2016, 5, 1210. [Google Scholar] [CrossRef] [Green Version]
- Ming, Y.; Russell, L.M. Predicted hygroscopic growth of sea salt aerosol. J. Geophys. Res. Atmos. 2001, 106, 28259–28274. [Google Scholar] [CrossRef]
- De Rosaa, S.; Seizova, K.; Kamenarska, Z.; Petrova, A.; Iodicea, C.; Stefanov, K.; Popov, S. Sterol and lipid composition of three adriatic sea sponges. Z. Naturforsch. C. J. Biosci. 2006, 61, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Sumitra, S.; Reddu, S.K.; Sharma, S.K.; Mohammad, A. New unsaturated fatty acid from roots of Bougainvillea spectabilis Willd. Asian J. Chem. 2009, 21, 4744–4748. [Google Scholar]
- Ahmad, M.Z.; Ali, M.; Mir, S.R. New phytoconstituents from the roots of Ocimum sanctum L. J. Pharm. Res. 2012, 5, 548–550. [Google Scholar]
- Chang, H.S.; Chen, Y.S.; Cheng, M.J.; Wu, H.C.; Chan, H.Y.; Hsieh, S.Y.; Yang, S.S.; Chen, I.S.; Chen, J.J. Chemical constituents of the fungus Mycoleptodiscus sp. 09F0149. Chem. Nat. Compd. 2018, 54, 396–398. [Google Scholar] [CrossRef]
- Al-Saif, S.S.; Abdel-Raouf, N.; El-Wazanani, H.A.; Aref, I.A. Antibacterial substances from marine algae isolated from Jeddah coast of Red sea, Saudi Arabia. Saudi J. Biol. Sci. 2014, 21, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase—a guide for the treatment of Pompe disease. Nat. Comm. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipo, J.R.; Feng, J.H.; Yamamoto, T.; Ohsaki, Y.; Nanba, E.; Tsujino, S.; Sakuragawa, N.; Martiniuk, F.; Ninomiya, H.; Oka, A.; et al. New GAA mutations in Japanese patients with GSDII (Pompe disease). Pediatr. Neurol. 2003, 29, 284–287. [Google Scholar] [CrossRef]
- Hermans, M.M.; Leenen, D.V.; Kroos, M.A.; Beesley, C.E.; Van der Ploeg, A.T.; Sakuraba, H.; Wevers, R.; Kleijer, W.; Michelakakis, H.; Kirk, E.P.; et al. Twenty-two novel mutations in the lysosomal α-glucosidase gene (GAA) underscore the genotype–phenotype correlation in glycogen storage disease type II. Hum. Mutat. 2004, 23, 47–56. [Google Scholar] [CrossRef]
- MOE (Molecular Operating Environment). Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite 910, Montreal, QC, Canada. 2014. [Google Scholar]
- Rizvi, T.S.; Hussain, I.; Ali, L.; Mabood, F.; Khan, A.L.; Shujah, S.; Rehman, N.U.; Al-Harrasi, A.; Hussain, J.; Khan, A.; et al. New gorgonane sesquiterpenoid from Teucrium mascatense Boiss, as α-glucosidase inhibitor. S. Afr. J. Bot. 2019, 124, 218–222. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) |
|---|---|
| 1 | 522.0 ± 0.51 |
| 2 | 234.2 ± 4.18 |
| 3 | 289.4 ± 4.91 |
| 4 | NA |
| 5 | ND |
| 6 | 659.78 ± 2.15 |
| 7 | 30.5 ± 0.41 |
| 8 | 480.1 ± 2.11 |
| Acarbose | 942 ± 0.74 |
| S. cerevisiae α-glucosidase | ||||||
| Compounds | IC50 (μM) | Docking score | Protein–ligand interactions | |||
| Ligand | Receptor | Interactions | Distances (Å) | |||
| 1 | 522.0 | −7.87 | O12 | NE2-HIS111 | HBA | 2.43 |
| 2 | 234.2 | −8.47 | O21 O12 | NH1-ARG439 O-WAT1174 | HBA HBA | 1.95 1.97 |
| 3 | 289.4 | −8.23 | O69 O69 O69 | OD1-ASP214 OD2-ASP214 NE2-HIS111 | HBD HBD HBA | 2.15 1.83 2.30 |
| 6 | 659.78 | −5.33 | O1 O7 | O-PHE157 N-TYR313 | HBD HBA | 1.88 2.33 |
| 7 | 30.5 | −10.62 | O8 O8 | OD1-ASP214 NE2-HIS111 | HBD HBA | 2.62 2.14 |
| 8 | 480.1 | −8.18 | O22 O24 | NH2-ARG212 O-WAT1026 | HBA HBA | 2.09 1.96 |
| Acarbose | 942 | −1.24 | O17 O19 O21 O40 O42 O87 O19 O21 O21 O40 O44 | OE1-GLU276 OD1-ASP214 OD1-ASP349 O-HOH1228 OD1-ASP408 OE2-GLU304 NH2-ARG212 NE2-HIS348 O-HOH1026 O-HOH1056 O-HOH1174 | HBD HBD HBD HBD HBD HBD HBA HBA HBA HBA HBA | 2.43 2.79 3.27 2.53 2.99 2.96 3.47 3.02 2.71 2.42 2.56 |
| H. Sapiens α-glucosidase | ||||||
| Compounds | IC50 (μM) | Docking score | Protein–ligand interactions | |||
| Ligand | Receptor | Interactions | Distances (Å) | |||
| 1 | ND | −6.41 | O12 O21 O23 | NH1-ARG600 NE2-HIS674 NE2-HIS674 | HBA HBA HBA | 1.95 2.44 1.91 |
| 2 | ND | −6.44 | O12 O21 | NH1-ARG600 NE2-HIS674 | HBA HBA | 2.03 1.95 |
| 3 | ND | −7.07 | O69 O69 O69 | OD1-ASP616 OD2-ASP616 NH1-ARG600 | HBD HBD HBA | 1.94 2.22 2.84 |
| 6 | ND | −6.26 | O1 | OD2-ASP518 | HBD | 1.85 |
| 7 | ND | −9.08 | O8 O10 | OD1-ASP404 O-Wat1106 | HBD HBA | 2.00 3.01 |
| 8 | ND | −7.23 | O22 O24 | OD2-ASP518 NE2-HIS674 | HBD HBA | 1.94 3.14 |
| Acarbose | ND | −9.65 | O15 O17 O19 O21 N37 O40 O42 O42 O15 O19 O42 O64 O87 N37 N37 | OD1-ASP616 O-HOH1164 OD2-ASP404 OD1-ASP404 OD2-ASP616 OD1-ASP282 OD2-ASP282 SD-MET519 NH1-ARG600 NE2-HIS674 NH1-ARG600 N-ALA284 O-HOH1276 OD1-ASP616 OD2-ASP616 | HBD HBD HBD HBD HBD HBD HBD HBD HBA HBA HBA HBA HBA IONIC IONIC | 2.60 2.84 2.90 2.86 2.75 3.12 2.78 3.21 3.05 3.38 3.08 3.06 3.14 3.36 2.75 |
| Compounds | Pharmacokinetic Properties |
|---|---|
| 1 | HIA = High, Caco-2 = Good, BBB = Yes, HOB = Yes, P-glycoprotein inh/subs = No, Carcinogenicity = No, Ames mutagenesis = No, Hepatotoxicity = No, Acute Oral Toxicity = III (2.28 kg/mol), Biodegradation = Yes, Log Kp = −6.14 cm/s, CYP3A4 subs = No, CYP2C9 subs = No, CYP2D6 subs = No, CYP3A4 inh = No, CYP2C9 inh = No, CYP2C19 inh = Yes, CYP2D6 inh = Yes, CYP1A2 inh = Yes, CYP inhibitory promiscuity = No |
| 2 | HIA = High, Caco-2 = Good, BBB = Yes, HOB = Yes, P-glycoprotein inh/subs = No, Carcinogenicity = No, Ames mutagenesis = No, Hepatotoxicity = No, Acute Oral Toxicity = III (1.695 kg/mol), Biodegradation = No, Log Kp = −5.97 cm/s, CYP3A4 subs = No, CYP2C9 subs = No, CYP2D6 subs = No, CYP3A4 inh = No, CYP2C9 inh = No, CYP2C19 inh = No, CYP2D6 inh = No, CYP1A2 inh = Yes, CYP inhibitory promiscuity = No |
| 3 | HIA = Low, Caco-2 = Good, BBB = No, HOB = No, P-glycoprotein inh/subs = No, Carcinogenicity = No, Ames mutagenesis = No, Hepatotoxicity = No, Acute Oral Toxicity = I (3.471 kg/mol), Biodegradation = No, Log Kp = −2.53 cm/s, CYP3A4 subs = Yes, CYP2C9 subs = No, CYP2D6 subs = No, CYP3A4 inh = No, CYP2C9 inh = No, CYP2C19 inh = No, CYP2D6 inh = No, CYP1A2 inh = No, CYP inhibitory promiscuity = No |
| 6 | HIA = Low, Caco-2 = No, BBB = No, HOB = No, P-glycoprotein inh/subs = No, Carcinogenicity = No, Ames mutagenesis = No, Hepatotoxicity = No, Acute Oral Toxicity = IV (1.499kg/mol), Biodegradation = Yes, Log Kp = −0.39cm/s, CYP3A4 subs = Yes, CYP2C9 subs = No, CYP2D6 subs = No, CYP3A4 inh = No, CYP2C9 inh = No, CYP2C19 inh = No, CYP2D6 inh = No, CYP1A2 inh = No, CYP inhibitory promiscuity = No |
| 7 | HIA = Low, Caco-2 = No, BBB = No, HOB = No, P-glycoprotein inh/subs = No, Carcinogenicity = No, Ames mutagenesis = No, Hepatotoxicity = No, Acute Oral Toxicity = IV (1.078kg/mol), Biodegradation = Yes, Log Kp = −0.11cm/s, CYP3A4 subs = No, CYP2C9 subs = Yes, CYP2D6 subs = No, CYP3A4 inh = No, CYP2C9 inh = No, CYP2C19 inh = No, CYP2D6 inh = No, CYP1A2 inh = Yes, CYP inhibitory promiscuity = No |
| 8 | HIA = High, Caco-2 = Good, BBB = Yes, HOB = No, P-glycoprotein inh/subs = No, Carcinogenicity = No, Ames mutagenesis = No, Hepatotoxicity = No, Acute Oral Toxicity = IV (1.699kg/mol), Biodegradation = Yes, Log Kp = −2.90cm/s, CYP3A4 subs = No, CYP2C9 subs = Yes, CYP2D6 subs = No, CYP3A4 inh = No, CYP2C9 inh = No, CYP2C19 inh = No, CYP2D6 inh = No, CYP1A2 inh = Yes, CYP inhibitory promiscuity = No |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ur Rehman, N.; Rafiq, K.; Khan, A.; Ahsan Halim, S.; Ali, L.; Al-Saady, N.; Hilal Al-Balushi, A.; Al-Busaidi, H.K.; Al-Harrasi, A. α-Glucosidase Inhibition and Molecular Docking Studies of Natural Brominated Metabolites from Marine Macro Brown Alga Dictyopteris hoytii. Mar. Drugs 2019, 17, 666. https://doi.org/10.3390/md17120666
Ur Rehman N, Rafiq K, Khan A, Ahsan Halim S, Ali L, Al-Saady N, Hilal Al-Balushi A, Al-Busaidi HK, Al-Harrasi A. α-Glucosidase Inhibition and Molecular Docking Studies of Natural Brominated Metabolites from Marine Macro Brown Alga Dictyopteris hoytii. Marine Drugs. 2019; 17(12):666. https://doi.org/10.3390/md17120666
Chicago/Turabian StyleUr Rehman, Najeeb, Kashif Rafiq, Ajmal Khan, Sobia Ahsan Halim, Liaqat Ali, Nadiya Al-Saady, Abdullah Hilal Al-Balushi, Haitham Khamis Al-Busaidi, and Ahmed Al-Harrasi. 2019. "α-Glucosidase Inhibition and Molecular Docking Studies of Natural Brominated Metabolites from Marine Macro Brown Alga Dictyopteris hoytii" Marine Drugs 17, no. 12: 666. https://doi.org/10.3390/md17120666