Isolation, Structure Elucidation and Biological Evaluation of Lagunamide D: A New Cytotoxic Macrocyclic Depsipeptide from Marine Cyanobacteria


Abstract
:
1. Introduction
2. Results and Discussion
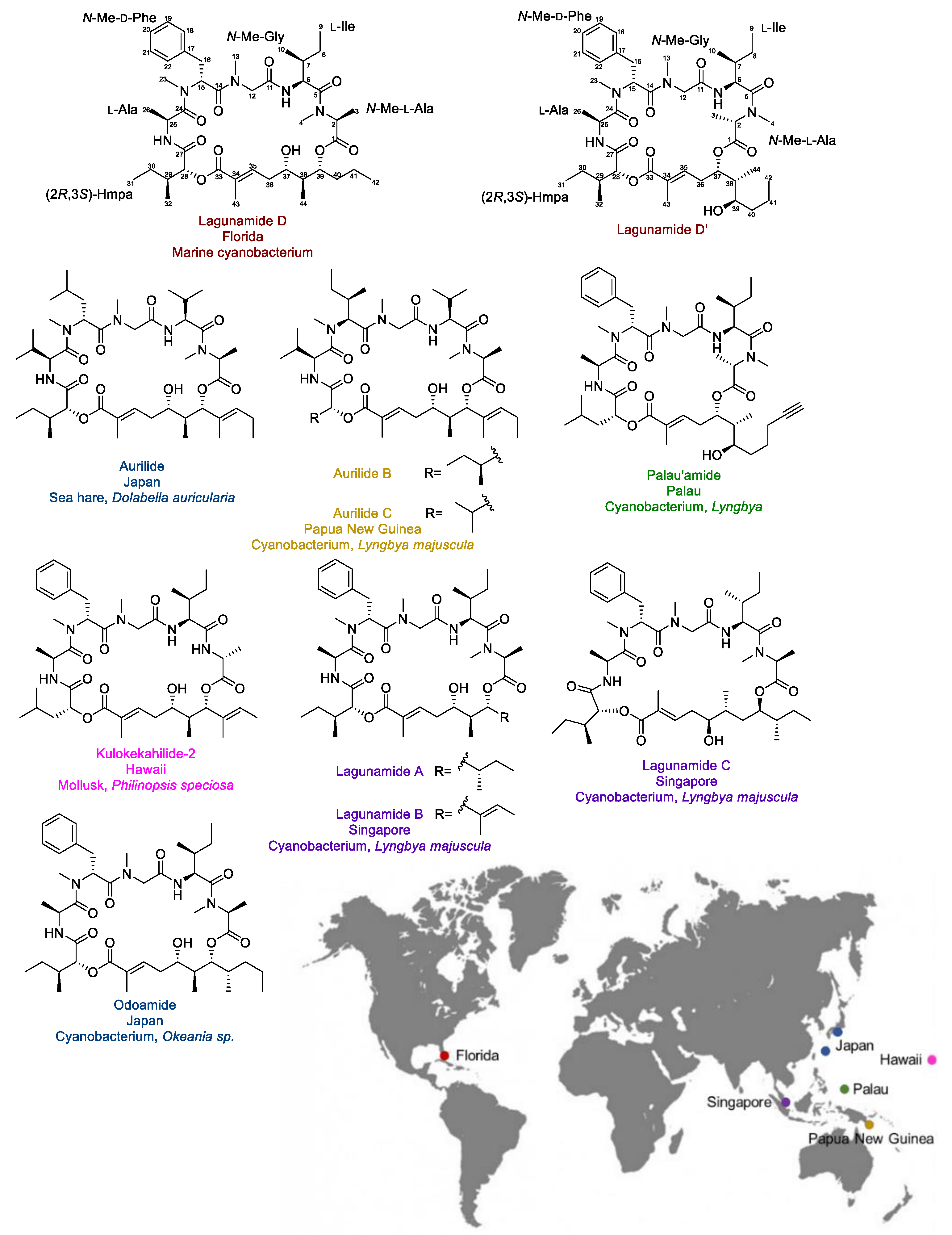
2.1. Isolation and Structure Elucidation
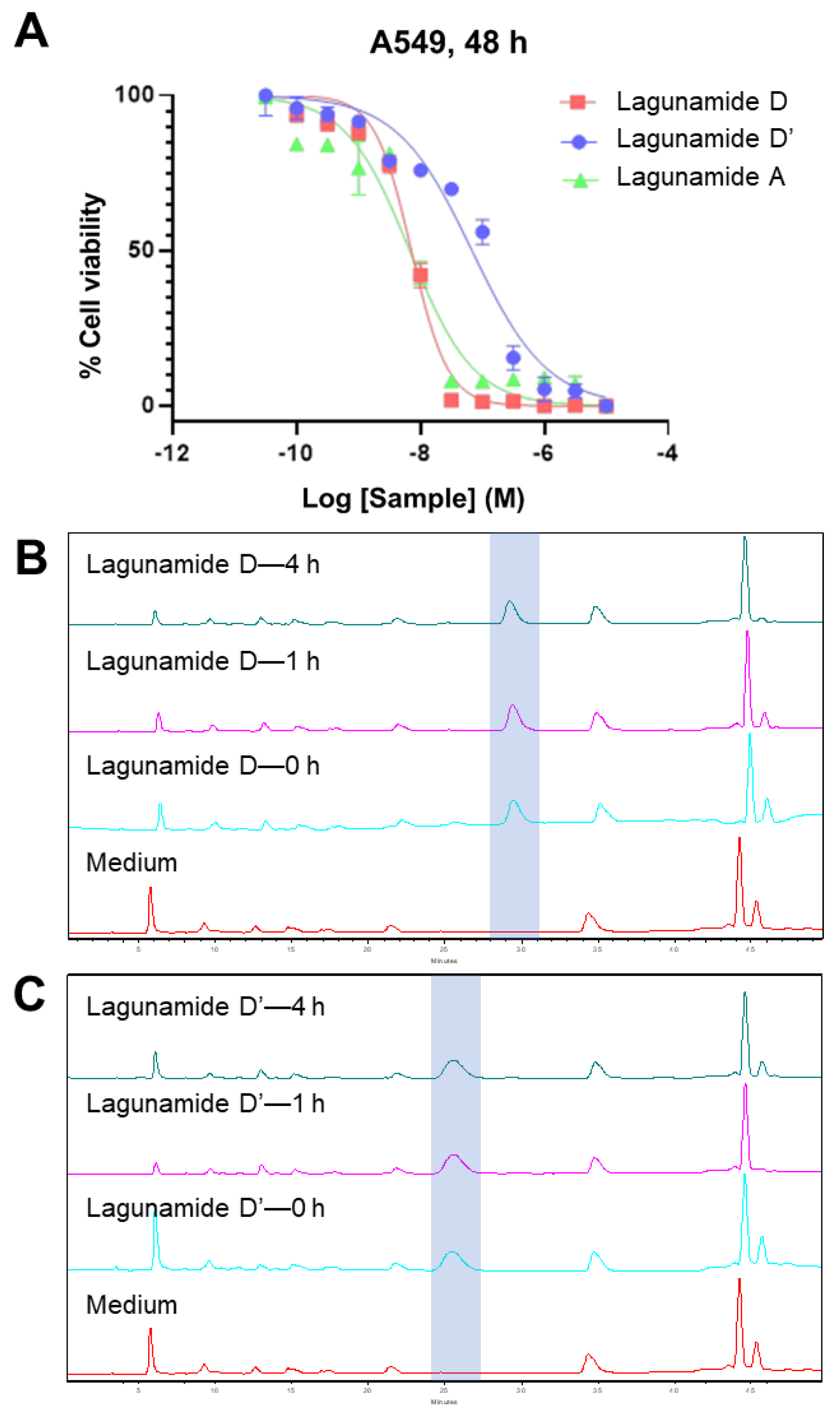
2.2. In Vitro Biological Evaluation
3. Materials and Methods
3.1. General
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Stability Assessment
3.5. Preparation of 2-Hydroxy-3-methylpentanoic Acid (Hmpa)
3.6. Acid Hydrolysis and Enantioselective Analysis
3.7. Modified Mosher’s Analysis
3.8. Kishi NMR Database
3.9. General Cell Culture Procedure
3.10. Cell Viability Assay (MTT)
3.11. Stability in Cell Culture Medium
3.12. Caspase Activity Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salvador-Reyes, L.A.; Luesch, H. Biological targets and mechanisms of action of natural products from marine cyanobacteria. Nat. Prod. Rep. 2015, 32, 478–503. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, M.; Maruthanayagam, V.; Sundararaman, M. A review of pharmacological and toxicological potentials of marine cyanobacterial metabolites. J. Appl. Toxicol. 2012, 32, 153–185. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Reyes, L.A.; Engene, N.; Paul, V.J.; Luesch, H. Targeted natural products discovery from marine cyanobacteria using combined phylogenetic and mass spectrometric evaluation. J. Nat. Prod. 2015, 78, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef]
- Jaspars, M.; De Pascale, D.; Andersen, J.H.; Reyes, F.; Crawford, A.D.; Ianora, A. The marine biodiscovery pipeline and ocean medicines of tomorrow. J. Mar. Biol. Assoc. UK 2016, 96, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M.; Branch, N.P. Drugs and drug candidates from marine sources: An assessment of the current “state of play”. Planta Med. 2015, 82, 775–789. [Google Scholar] [CrossRef]
- Suenaga, K.; Mutou, T.; Shibata, T.; Itoh, T.; Fujita, T.; Takada, N.; Hayamizu, K.; Takagi, M.; Irifune, T.; Kigoshi, H.; et al. Aurilide, a cytotoxic depsipeptide from the sea hare Dolabella auricularia: Isolation, structure determination, synthesis, and biological activity. Tetrahedron 2004, 60, 8509–8527. [Google Scholar] [CrossRef]
- Han, B.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.K.; Tan, L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Yoshida, W.Y.; Takada, Y.; Kimura, J.; Yang, L.; Mooberry, S.L.; Scheuer, P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004, 67, 1332–1340. [Google Scholar] [CrossRef]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Williams, P.G.; Yoshida, W.Y.; Quon, M.K.; Moore, R.E.; Paul, V.J. The structure of palau’amide, a potent cytotoxin from a species of the marine cyanobacterium Lyngbya. J. Nat. Prod. 2003, 66, 1545–1549. [Google Scholar] [CrossRef]
- Sato, S.I.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine natural product aurilide activates the OPA1-mediated apoptosis by binding to prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tan, C.H.; Kishi, Y. Toward creation of a universal NMR database for stereochemical assignment: The case of 1,3,5-trisubstituted acyclic systems. Helv. Chim. Acta 2000, 83, 2562–2571. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tan, C.-H.; Kishi, Y. Toward creation of a universal NMR database for stereochemical assignment: Complete structure of the desertomycin/oasomycin class of natural products structural properties of fatty acids and related compounds are inherent to the specific stereochemical. J. Am. Chem. Soc. 2001, 123, 2076–2078. [Google Scholar] [CrossRef] [PubMed]
- Umehara, M.; Takada, Y.; Nakao, Y.; Kimura, J. Intramolecular ester exchange of potent cytotoxic kulokekahilide-2. Tetrahedron Lett. 2009, 50, 840–843. [Google Scholar] [CrossRef]
- Kaneda, M.; Kawaguchi, S.; Fujii, N.; Ohno, H.; Oishi, S. Structure−activity relationship study on odoamide: Insights into the bioactivities of aurilide-family hybrid peptide−polyketides. ACS Med. Chem. Lett. 2018, 9, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Kurokawa, R.; Arai, D.; Kanto, K.K.; Okino, T.; Nakao, Y.; Kushiro, T.; Yoshida, M.; Matsumoto, K. A quantitative shRNA screen identifies ATP1A1 as a gene that regulates cytotoxicity by aurilide B. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Huang, W.; Li, L.; Sun, X.; Song, S.; Xu, Q.; Zhang, L.; Wei, B.-G.; Deng, X. Structure determinants of lagunamide A for anticancer activity and its molecular mechanism of mitochondrial apoptosis. Mol. Pharm. 2016, 13, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | C/H No. | δCa | δH (J in Hz) b | COSY b | HMBC b d |
|---|---|---|---|---|---|
| N-Me-Ala | 1 | 170.17 | |||
| 2 | 57.93 | 3.91, q (6.8) | 3 | 1, 3, 4 | |
| 3 | 12.74 | 1.26, d (6.8) | 2 | 1 | |
| 4 | 36.95 | 3.21, s | 2, 5 | ||
| Ile | 5 | 170.88 | |||
| 6 | 51.07 | 4.92, dd (9.3, 7.7) | 7, NH (1) | 5, 7, 8, 10, 11 | |
| 7 | 37.20 | 1.69, ddd (10.5, 7.9, 3.1) | 6, 8a, 8b, 10 | ||
| 8a | 23.00 | 1.51, m | 7, 8b, 9 | ||
| 8b | 1.36, m | 7, 8a, 9 | |||
| 9 | 10.13 | 0.84, m | 8a, 8b | 8 | |
| 10 | 14.55 | 0.89, d (6.9) | 7 | 6, 7, 8 | |
| NH (1) | 7.21, m | 6 | 11 | ||
| N-Me-Gly | 11 | 168.18 | |||
| 12a | 50.38 | 3.98, d (−18.4) | 12b | 11, 13 | |
| 12b | 3.31 c | 12a | 11, 13, 14 | ||
| 13 | 35.65 | 2.73, s | 12, 14 | ||
| N-Me-Phe | 14 | 170.18 | |||
| 15 | 52.33 | 5.28, dd (10.2, 5.3) | 16a, 16b | 14, 16, 17, 23 | |
| 16a | 34.25 | 2.92, dd (−14.4, 10.3) | 15, 16b | 14, 15, 17, 18/22 | |
| 16b | 2.77, dd (−15.0, 5.8) | 15, 16a | 15, 17, 18/22 | ||
| 17 | 137.16 | ||||
| 18/22 | 129.30 | 7.10, m | 16, 18/22, 20 | ||
| 19/21 | 127.51 | 7.15, m | 17, 19/21 | ||
| 20 | 125.93 | 7.13, m | 18/22 | ||
| 23 | 29.24 | 2.85, s | 15, 24 | ||
| Ala | 24 | 172.45 | |||
| 25 | 44.51 | 4.31, qd (6.9, 6.9) | 26, NH (2) | 24, 26 | |
| 26 | 14.68 | 0.69, d (7.0) | 25 | 24, 25 | |
| NH (2) | 8.49, d (6.5) | 25 | 25, 26, 27 | ||
| Hmpa | 27 | 169.49 | |||
| 28 | 75.30 | 4.86, d (3.2) | 29 | 29, 32 | |
| 29 | 36.21 | 1.81, m | 28, 30a, 30b, 32 | ||
| 30a | 25.65 | 1.38, m | 29, 30b, 31 | 28, 29, 31, 32 | |
| 30b | 1.25, m | 29, 30a, 31 | 28, 29, 31, 32 | ||
| 31 | 11.52 | 0.87, m | 30a, 30b | 29, 30 | |
| 32 | 14.00 | 0.83, m | 29 | 28, 29, 30 | |
| Dihydroxy acid | 33 | 168.34 | |||
| 34 | 126.83 | ||||
| 35 | 144.45 | 7.11, m | 36a, 36b, 43 | ||
| 36a | 29.56 | 2.11, ddd (−14.4, 9.8, 9.8) | 35, 36b, 37 | 34, 35, 37 | |
| 36b | 1.89, m | 35, 36a, 37 | |||
| 37 | 69.24 | 3.53, m | 36a, 36b, 38, OH | ||
| OH | 4.13, br d (4.4) | 37 | |||
| 38 | 40.85 | 1.89, m | 37, 39, 44 | ||
| 39 | 74.56 | 4.76, m | 38, 40a, 40b | ||
| 40a | 33.62 | 1.56, m | 40b, 39 | ||
| 40b | 1.40, m | 40a, 39 | |||
| 41 | 16.84 | 1.23, m | 42 | ||
| 42 | 14.12 | 0.86, m | 41 | 40, 41 | |
| 43 | 11.84 | 1.81, br s | 35 | 33, 34, 35 | |
| 44 | 9.40 | 0.79, d (6.9) | 38 | 37, 38, 39 |
| Unit | C/H No. | δCa | δH (J in Hz) b | COSY b | HMBC b d e |
|---|---|---|---|---|---|
| N-Me-Ala | 1 | 170.48 | |||
| 2 | 58.48 | 3.88, q (6.7) | 3 | 1, 3, 4 | |
| 3 | 13.25 | 1.30, d (6.8) | 2 | 1, 2 | |
| 4 | 37.46 | 3.22, s | 2, 5 | ||
| Ile | 5 | 170.58 | |||
| 6 | 51.11 | 4.77, dd (10.1, 10.1) | 7, NH (1) | 5, 7, 8, 10, 11 | |
| 7 | 36.12 | 1.78, m | 6, 8a, 8b, 10 | ||
| 8a | 23.21 | 1.63, m | 7, 8b, 9 | 7, 9 | |
| 8b | 1.40, m | 7, 8a, 9 | 7, 9 | ||
| 9 | 11.35 | 0.87, m | 8a, 8b | 7, 8 | |
| 10 | 14.28 | 0.78, d (6.8) | 7 | 6, 7, 8 | |
| NH (1) | 7.72, d (9.8) | 6 | 6, 11 | ||
| N-Me-Gly | 11 | 167.99 | |||
| 12a | 50.57 | 4.02, d (−18.7) | 12b | 11, 13 | |
| 12b | 2.93, d (−18.7) | 12a | 11, 13, 14 | ||
| 13 | 35.56 | 2.73, s | 12, 14 | ||
| N-Me-Phe | 14 | 170.37 | |||
| 15 | 52.60 | 5.28, dd (10.0, 5.5) | 16a, 16b | 14, 16, 17, 23, 24 | |
| 16a | 34.37 | 2.91, dd (−14.5, 9.9) | 15, 16b | 15, 17, 18/22 | |
| 16b | 2.81, dd (−14.4, 5.4) | 15, 16a | 15, 17, 18/22 | ||
| 17 | 137.44 | ||||
| 18/22 | 129.21 | 7.11, m | 16, 20 | ||
| 19/21 | 127.61 | 7.16, m | 17, 19/21 | ||
| 20 | 125.99 | 7.13, m | 18/22 | ||
| 23 | 29.26 | 2.86, s | 15, 24 | ||
| Ala | 24 | 173.11 | |||
| 25 | 44.56 | 4.34, qd (7.0, 7.0) | 26, NH (2) | 24, 26, 27 | |
| 26 | 14.98 | 0.72, d (7.1) | 25 | 24, 25 | |
| NH (2) | 8.56, d (6.7) | 25 | 25, 26, 27 | ||
| Hmpa | 27 | 169.87 | |||
| 28 | 75.66 | 4.83, d (2.9) | 29 | 27, 29, 30, 32, 33 e | |
| 29 | 36.31 | 1.83, m | 28, 30a, 30b, 32 | 30, 32 | |
| 30a | 26.12 | 1.39, m | 29, 30b, 31 | 28, 29, 32 | |
| 30b | 1.28, m | 29, 30a, 31 | 28, 29, 32 | ||
| 31 | 9.52 | 0.86, m | 30a, 30b | 29, 30 | |
| 32 | 13.63 | 0.82, m | 29 | 28, 29, 30 | |
| Dihydroxy acid | 33 | 168.17 | |||
| 34 | 127.51 | ||||
| 35 | 140.68 | 6.75, ddd (6.8, 6.8, 1.2) | 36a, 36b, 43 | ||
| 36a | 27.98 | 2.66, m | 35, 36b, 37 | ||
| 36b | 2.36, ddd (−15.6, 7.8, 7.8) | 35, 36a, 37 | 34, 35, 37 | ||
| 37 | 73.79 | 4.85, ddd (5.0, 5.0, 5.0) | 36a, 36b, 38 | 1 e, 35, 38, 44 | |
| 38 | 41.07 | 1.66, m | 37, 39, 44 | 36, 37, 39, 40, 44 | |
| 39 | 71.00 | 3.31 c | 38, 40, OH | ||
| OH | 4.52, d (5.5) | 39 | 38, 39, 40 | ||
| 40 | 36.28 | 1.27, m | 39 | ||
| 41a | 18.47 | 1.42, m | 41b, 42 | 39, 40, 42 | |
| 41b | 1.25, m | 41a, 42 | 39, 40, 42 | ||
| 42 | 13.93 | 0.84, m | 41 | 41 | |
| 43 | 12.03 | 1.85, br s | 35 | 33, 34, 35 | |
| 44 | 11.12 | 0.76, d (7.0) | 38 | 37, 38, 39 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, D.; Putra, M.Y.; Ye, T.; Paul, V.J.; Luesch, H. Isolation, Structure Elucidation and Biological Evaluation of Lagunamide D: A New Cytotoxic Macrocyclic Depsipeptide from Marine Cyanobacteria. Mar. Drugs 2019, 17, 83. https://doi.org/10.3390/md17020083
Luo D, Putra MY, Ye T, Paul VJ, Luesch H. Isolation, Structure Elucidation and Biological Evaluation of Lagunamide D: A New Cytotoxic Macrocyclic Depsipeptide from Marine Cyanobacteria. Marine Drugs. 2019; 17(2):83. https://doi.org/10.3390/md17020083
Chicago/Turabian StyleLuo, Danmeng, Masteria Y. Putra, Tao Ye, Valerie J. Paul, and Hendrik Luesch. 2019. "Isolation, Structure Elucidation and Biological Evaluation of Lagunamide D: A New Cytotoxic Macrocyclic Depsipeptide from Marine Cyanobacteria" Marine Drugs 17, no. 2: 83. https://doi.org/10.3390/md17020083