An Algal Metabolite-Based PPAR-γ Agonist Displayed Anti-Inflammatory Effect via Inhibition of the NF-κB Pathway

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Compound (+)-(R,E)-6a1 Promoted PPAR-γ Translocation to Cell Nuclei
2.2. Cytotoxicity of (+)-(R,E)-6a1 to RAW264.7, Ac2F, and KB Cells
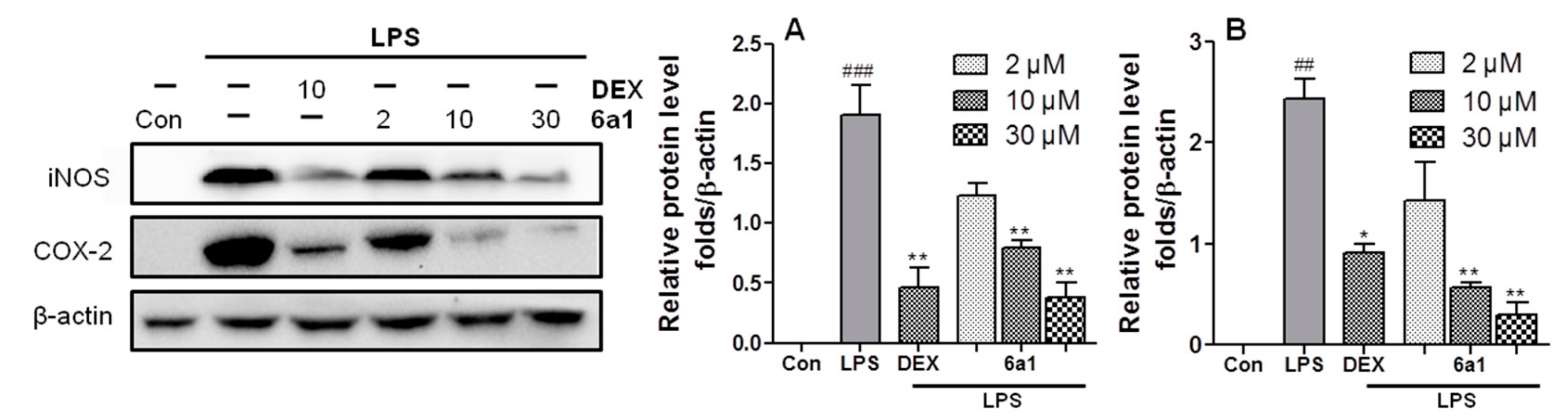
2.3. (+)-(R,E)-6a1 Inhibited LPS-Induced Expression of Proinflammatory Factors in RAW264.7 Cells
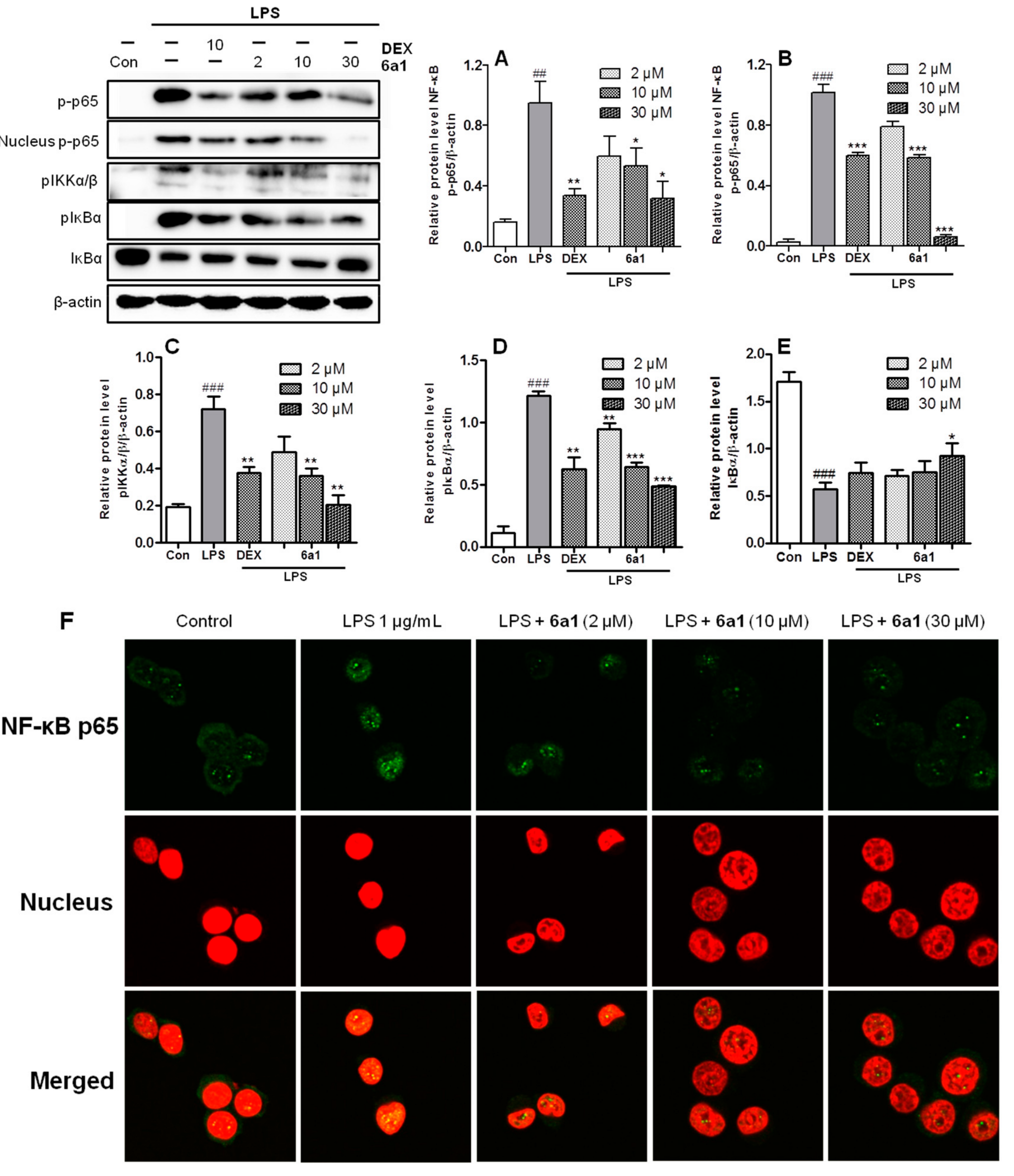
2.4. (+)-(R,E)-6a1 Inhibited LPS-Induced NF-Κb Signal Pathway in RAW264.7 Cells
3. Materials and Methods
3.1. Materials
3.2. Cell Culture and Cell Viability
3.3. Production Levels of NO and Cytokines Released into the Medium
3.4. Immunofluorescence Staining of NF-Κb P65 in RAW264.7 Cells
3.5. Western Blot Assay
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kliewer, S.A.; Xu, H.E.; Lambert, M.H.; Willson, T.M. Peroxisome proliferator-activated receptors: From genes to physiology. Recent Prog. Horm. Res. 2001, 56, 239–263. [Google Scholar] [CrossRef]
- Spiegelman, B.M. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes 1998, 47, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79. [Google Scholar] [CrossRef]
- Murphy, G.J.; Holder, J.C. PPAR-γ agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol. Sci. 2000, 21, 469–474. [Google Scholar] [CrossRef]
- Ono, M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008, 99, 1501–1506. [Google Scholar] [CrossRef]
- Moller, D.E.; Berger, J.P. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int. J. Obes. 2003, 27, S17–S21. [Google Scholar] [CrossRef] [Green Version]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Pillinger, M.H. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin. Immunol. 2005, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.S.; Pascual, G.; Li, M.; Welch, J.S.; Ricote, M.; Hsiang, C.H.; Sengchanthalangsy, L.L.; Ghosh, G.; Glass, C.K. 15-Deoxy-Δ12, 14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc. Nati. Acad. Sci. 2000, 97, 4844–4849. [Google Scholar] [CrossRef]
- Cernuda-Morollón, E.; Pineda-Molina, E.; Pérez-Sala, D. 15-deoxy-∆12,14-prostaglandin J2 Inhibition of NF-κB-DNA Binding through Covalent Modification of the p50 Subunit. J. Biol. Chem. 2001, 276, 35530–35536. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kapahi, P.; Natoli, G.; Takahashi, T.; Chen, Y.; Karin, M.; Santoro, M.G. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 2000, 403, 103. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Fiorucci, S. PPARs and other nuclear receptors in inflammation. Curr. Opin. Pharmacol. 2006, 6, 421–427. [Google Scholar] [CrossRef]
- Na, H.K.; Surh, Y.J. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol. Carcinogenesis. 2006, 45, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Pande, V.; Sousa, S.F.; Ramos, M.J. Direct covalent modification as a strategy to inhibit nuclear factor-kappa B. Curr. Med. Chem. 2009, 16, 4261–4273. [Google Scholar] [CrossRef]
- Dang, H.T.; Lee, H.J.; Yoo, E.S.; Shinde, P.B.; Lee, Y.M.; Hong, J.K.; Kim, D.K.; Jung, J.H. Anti-inflammatory constituents of the red alga Gracilaria verrucosa and their synthetic analogues. J. Nat. Prod. 2008, 71, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.T.; Lee, Y.M.; Kang, G.J.; Yoo, E.S.; Hong, J.K.; Lee, S.M.; Lee, S.K.; Pyee, Y.; Chung, H.J.; Moon, H.R.; et al. In vitro stability and in vivo anti-inflammatory efficacy of synthetic jasmonates. Bioorg. Med. Chem. 2012, 20, 4109–4116. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.T.; Lee, H.J.; Yoo, E.S.; Hong, J.K.; Bao, B.Q.; Choi, J.S.; Jung, J.H. New jasmonate analogues as potential anti-inflammatory agents. Bioorg. Med. Chem. 2008, 16, 10228–10235. [Google Scholar] [CrossRef]
- Ju, Z.R.; Su, M.Z.; Hong, J.K.; Ullah, S.; Kim, E.L.; Zhao, C.H.; Moon, H.R.; Kim, S.M. Design of PPAR-γ agonist based on algal metabolites and the endogenous ligand 15-deoxy-∆12,14-prostaglandin J2. Eur. J. Med. Chem. 2018, 157, 1192–1201. [Google Scholar] [CrossRef]
- Shiraki, T.; Kamiya, N.; Shiki, S.; Kodama, T.S.; Kakizuka, A.; Jingami, H. α, β-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor γ. J. Mol. Biol. 2005, 280, 14145–14153. [Google Scholar] [CrossRef]
- Soares, A.F.; Nosjean, O.; Cozzone, D.; D’Orazio, D.; Becchi, M.; Guichardant, M.; Ferry, G.; Boutin, J.A.; Lagarde, M.; Géloën, A. Covalent binding of 15-deoxy-∆12,14-prostaglandin J2 to PPARγ. Biochem. Biophys. Res. Commun. 2005, 337, 521–525. [Google Scholar] [CrossRef]
- Szanto, A.; Nagy, L. The many faces of PPARγ: anti-inflammatory by any means? Immunobiology 2008, 213, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Pisano, B.; Dugo, L.; Lanaro, A.; Maffia, P.; Patel, N.S.A.; Paola, R.D.; Latenti, A.; Genovese, T.; Chatterjee, P.K.; et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-γ, reduces acute inflammation. Eur. J. Pharmacol. 2004, 483, 79–93. [Google Scholar] [CrossRef]
- Su, M.Z.; Cao, J.F.; Huang, J.; Liu, S.; Im, D.S.; Yoo, J.W.; Jung, J.H. The in vitro and in vivo anti-inflammatory effects of a phthalimide PPAR-γ agonist. Mar. Drugs. 2017, 15, 7. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: a key role in inflammatory diseases. J. Clin. Invest. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, Z.; Su, M.; Li, D.; Hong, J.; Im, D.-S.; Kim, S.; Kim, E.L.; Jung, J.H. An Algal Metabolite-Based PPAR-γ Agonist Displayed Anti-Inflammatory Effect via Inhibition of the NF-κB Pathway. Mar. Drugs 2019, 17, 321. https://doi.org/10.3390/md17060321
Ju Z, Su M, Li D, Hong J, Im D-S, Kim S, Kim EL, Jung JH. An Algal Metabolite-Based PPAR-γ Agonist Displayed Anti-Inflammatory Effect via Inhibition of the NF-κB Pathway. Marine Drugs. 2019; 17(6):321. https://doi.org/10.3390/md17060321
Chicago/Turabian StyleJu, Zhiran, Mingzhi Su, Dandan Li, Jongki Hong, Dong-Soon Im, Suhkmann Kim, Eun La Kim, and Jee H. Jung. 2019. "An Algal Metabolite-Based PPAR-γ Agonist Displayed Anti-Inflammatory Effect via Inhibition of the NF-κB Pathway" Marine Drugs 17, no. 6: 321. https://doi.org/10.3390/md17060321