Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products


College of Pharmacy and Research Institute of Pharmaceutical Sciences, Gyeongsang National University, Jinju Daero 501, Jinju 52828, Gyeongnam, Korea
*
Author to whom correspondence should be addressed.
Mar. Drugs 2019, 17(9), 500; https://doi.org/10.3390/md17090500
Submission received: 23 July 2019
/
Revised: 7 August 2019
/
Accepted: 16 August 2019
/
Published: 27 August 2019
(This article belongs to the Collection Marine Compounds and Cancer)
Abstract
:In this paper, the chemical conjugation of marine natural products with other bioactive molecules for developing an advanced anti-cancer agent is described. Structural complexity and the extraordinary biological features of marine natural products have led to tremendous research in isolation, structural elucidation, synthesis, and pharmacological evaluation. In addition, this basic scientific achievement has made it possible to hybridize two or more biologically important skeletons into a single compound. The hybridization strategy has been used to identify further opportunities to overcome certain limitations, such as structural complexity, scarcity problems, poor solubility, severe toxicity, and weak potency of marine natural products for advanced development in drug discovery. Further, well-designed marine chimera molecules can function as a platform for target discovery or degradation. In this review, the design, synthesis, and biological evaluation of recent marine chimera molecules are presented.
1. Introduction
The struggle for existence in a natural environment occasionally makes a species develop its own unique weapons such as speed, power, or even toxins. As these toxins possess biologically potent activity and unique modes of actions, these natural products have been regarded as a robust platform for further medicinal research [1,2]. Thus far, marine natural products have been particularly highlighted for their extraordinary bioactivity under highly diluted conditions [3,4,5]. Therefore, it is plausible to utilize marine natural products as a hit or lead compound in drug discovery and its further development [6,7].
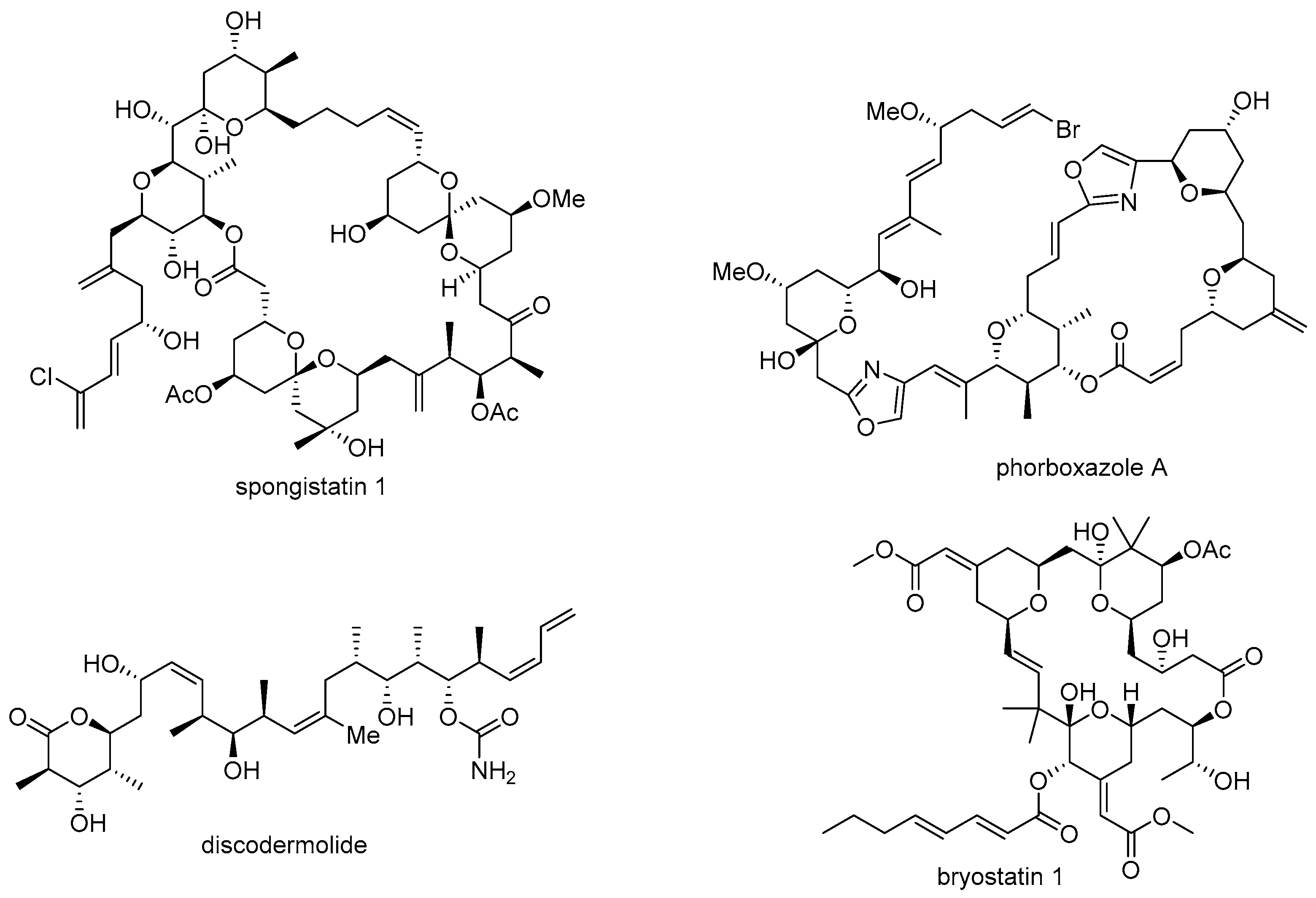
Although marine natural products could have the powerful potential for drug discovery, there are also a few obstacles associated with them. First, it is rather difficult to secure a sufficient amount of these products for further study [8]. In most cases, the medicinal study of natural products requires a substantial number of test samples for elucidation of target protein/receptor and following signaling pathway. However, harsh conditions, difficulty in access, and scarcity of the target organism are hindrances to large scalable synthesis of important marine natural products, such as spongistatin 1 (13.8 mg from 400 kg of marine sponge) [9], phorboxazole A (95 mg from 236 g of Phorbas sp.) [10], discodermolide (7 mg from 434 g of Discodermia dissoluta) [11], bryostatin 1 (0.9~1.8 g from 10,000 gallons of Bugula neritina), [12,13] etc. The unmet needs from natural resources occasionally leads to samples being obtained via chemical synthesis, and it usually remains difficult to satisfy substantial supply requirements [14,15,16] (Figure 1).
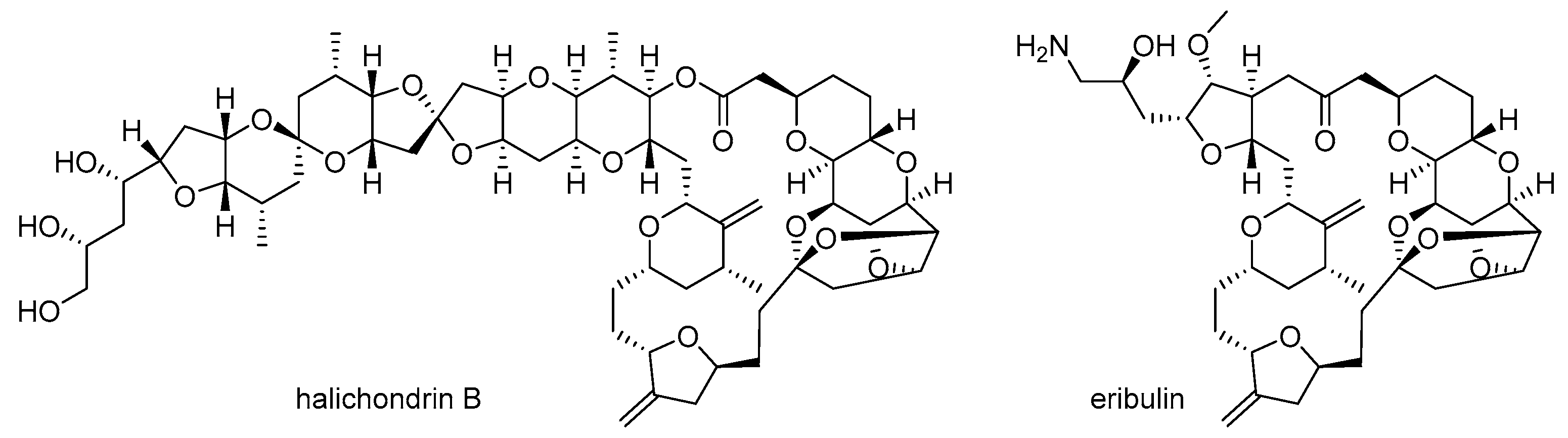
Second, structural complexity is another hurdle. The highly complex structure of marine natural products frequently makes it very difficult to modify or synthesize them on a large scale [17,18]. In order to improve the biological activity of these products, both chemical modification and related structure-activity relationship (SAR) study of marine natural products are necessary [19]. However, their highly complex structure hampers efficient modifications and any subsequent systematic research [20]. In order to address this structural complexity, a more simplified analog of marine natural products can be a breakthrough in drug discovery, as evident in the case of halichondrin B [21] and its simplified analog eribulin [22] (Figure 2). Although halichondrin B possesses extraordinary cytotoxicity against B-16 melanoma cells (IC50 0.093 ng/mL) and other tumor cell lines, its structure is too complex to be an advanced drug or drug candidate for related cancer therapy. Consequently, the truncated ketone analog eribulin of halichondrin B was developed through an entirely synthetic approach [23] and approved for treatment of metastatic breast cancer as a mesylate salt.
The third hurdle is biological activity and its selectivity. Since marine natural products are derived from marine organisms and not humans, their application to the human body may trigger undesired biological processes [24]. In order to solve this selectivity issue, various analogs are made and tested [25,26]. In addition, systematic study for mechanism of action or ligand-target binding usually follows valuable analogs [27]. Nonetheless, marine natural products continue to serve as a versatile starting point for drug discovery because of their unique structural framework and biological activity. Simultaneously, the strategy for the efficient modification of this complex molecule has also been evolving.
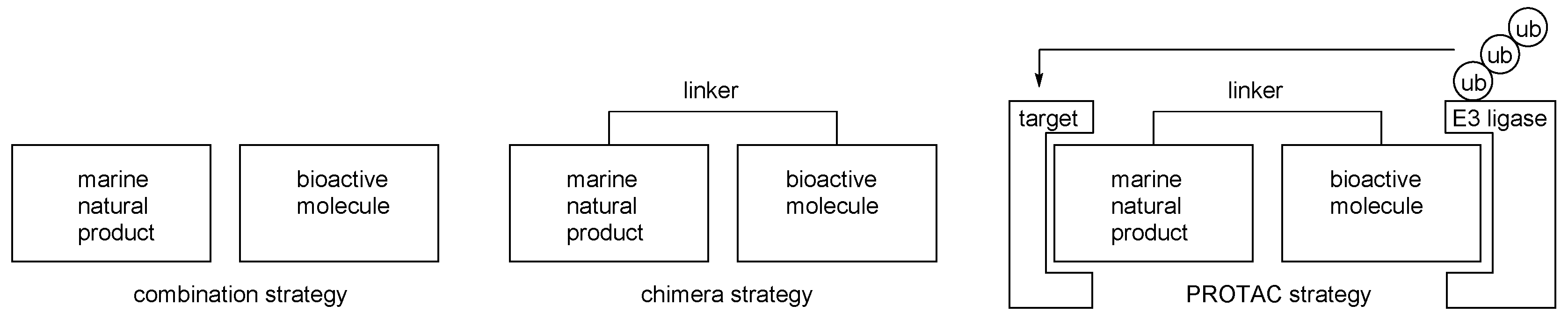
Molecular hybridization can be a good strategy for advanced marine natural products [28,29,30,31]. Certain drawbacks of marine natural products such as structural complexity or non-selective biological activities were overcome through a combination with other bioactive molecules. This combination strategy was evolved to develop chimera molecules, pursuing not merely a synergistic sequence but a systematically operating biological sequence. For example, the hybridization of a ligand and ubiquitin recruiting probe enables the degradation of target biomolecules via cellular sequence, such as ubiquitination and following the proteasome pathway. This proteolysis targeting chimera (PROTAC) approach showcases a well-designed chimera strategy from natural products [32,33,34,35,36,37]. (Figure 3) In addition, biotinylation of active ligand provides affinity column chromatography to enable target protein isolation [38]. In addition to these strategies, an improvement of basic activity can be anticipated. In this regard, advanced chimeric molecules from marine natural products with anticancer activity is reviewed here.
2. Results
2.1. Conjugation with Other Active Molecules
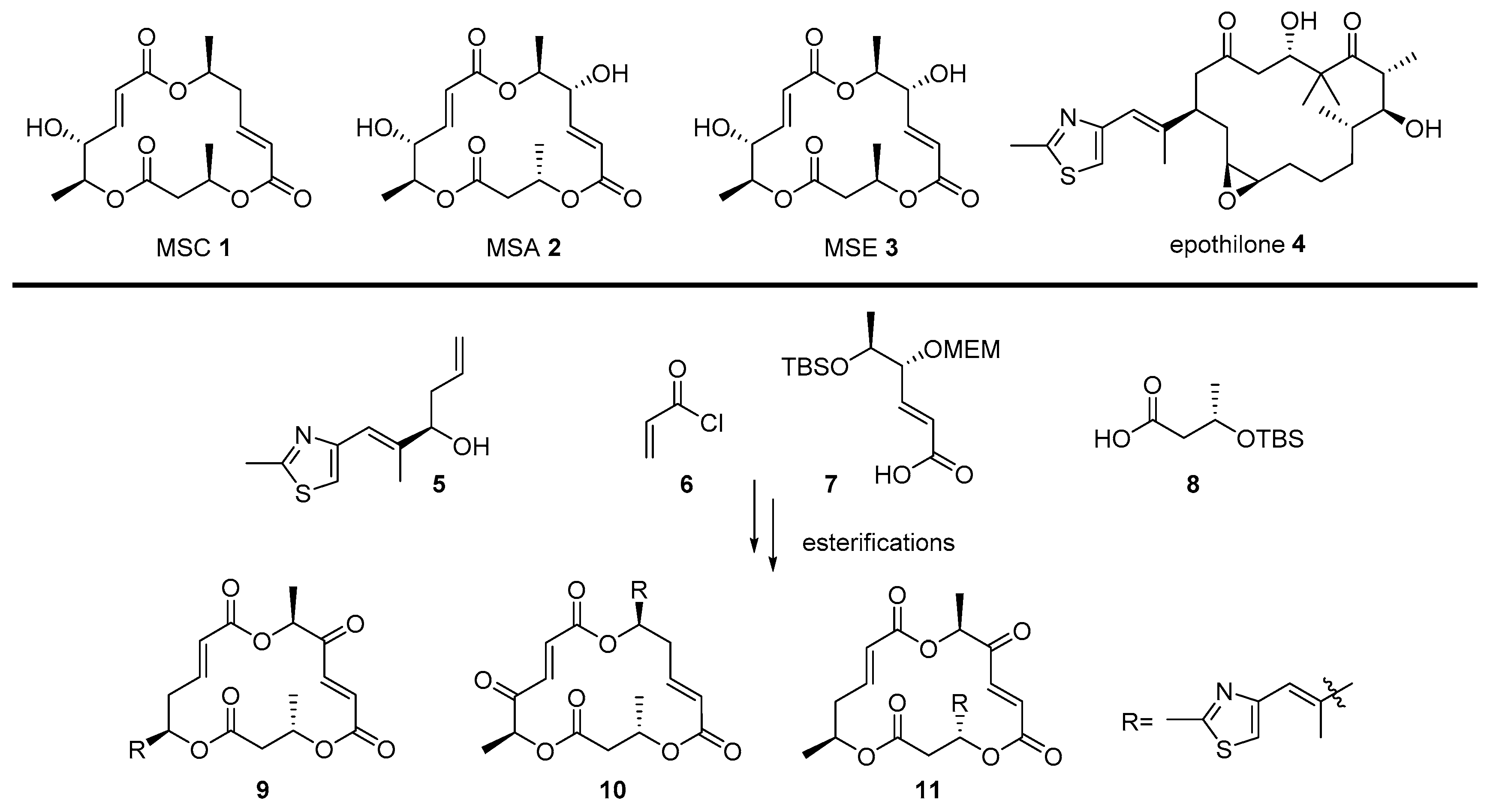
The macrosphelide (MS) family [39], comprising 13 natural isomers, are 15- or 16-membered macrolide antitumor agents. They are derived from marine sponge Periconia byssoides [40] (MSC 1, MSE-H, and MSL) and soil fungi Microsphaeropsis sp. FO-5050 [41] (MSA-D, MSJ, and MSK) and show cell adhesion inhibitory activity or immunosuppressive activity [42]. In addition, a derivatization of macrosphelide skeleton based on MSA 2 or MSE 3 enables the activation of apoptosis in human lymphoma U937 cells, although their activity is slightly weak [43]. In order to increase this apoptosis-inducing activity in cancer cells, preparation of a chimera compound with another 16-membered anticancer agent, epothilone 4—which shows not only tubulin-disrupting profiles and anticancer activity, similar to the paclitaxel, but also an apoptosis-inducing property in cancer cells to inhibit tumor cell growth—was pursued [44]. Based on their similar structural features and biological properties, hybridization of MS and epothilone 4 was performed, pursuing an advanced anticancer agent.
Scheme 1 illustrates the synthetic plan of the MS-epothilone chimera. Known intermediate 5 [45] for total synthesis of epothilone was prepared and linked to fragments 6, 7, [42] and 8 of MS via iterative esterification and deprotection of secondary alcohol moiety. For an elucidation of the structure-activity relationship, a thiazole side chain of epothilone was introduced in each CH3 group in the MS skeleton to provide the desired MS-epothilone chimera 9–11.
Screening of chimeras 9–11 exhibited an increase in their desired potencies, which is attributed to this hybridization strategy. In particular, chimera 9 showed more potent apoptosis-inducing activity in the U937 cell line as compared to other analogs or parent compounds MSA 2 or MSE 3. It is noteworthy that other chimeras also showed an increased apoptotic property, while parent MSA 2 or MSE 3 did not have the same activity at the same concentration (1 μM, 12 h incubation). Further, successful hybridization of MS with epothilone 4 presents that the chimeric molecule strategy can be a powerful solution to the limitation of marine natural products themselves.
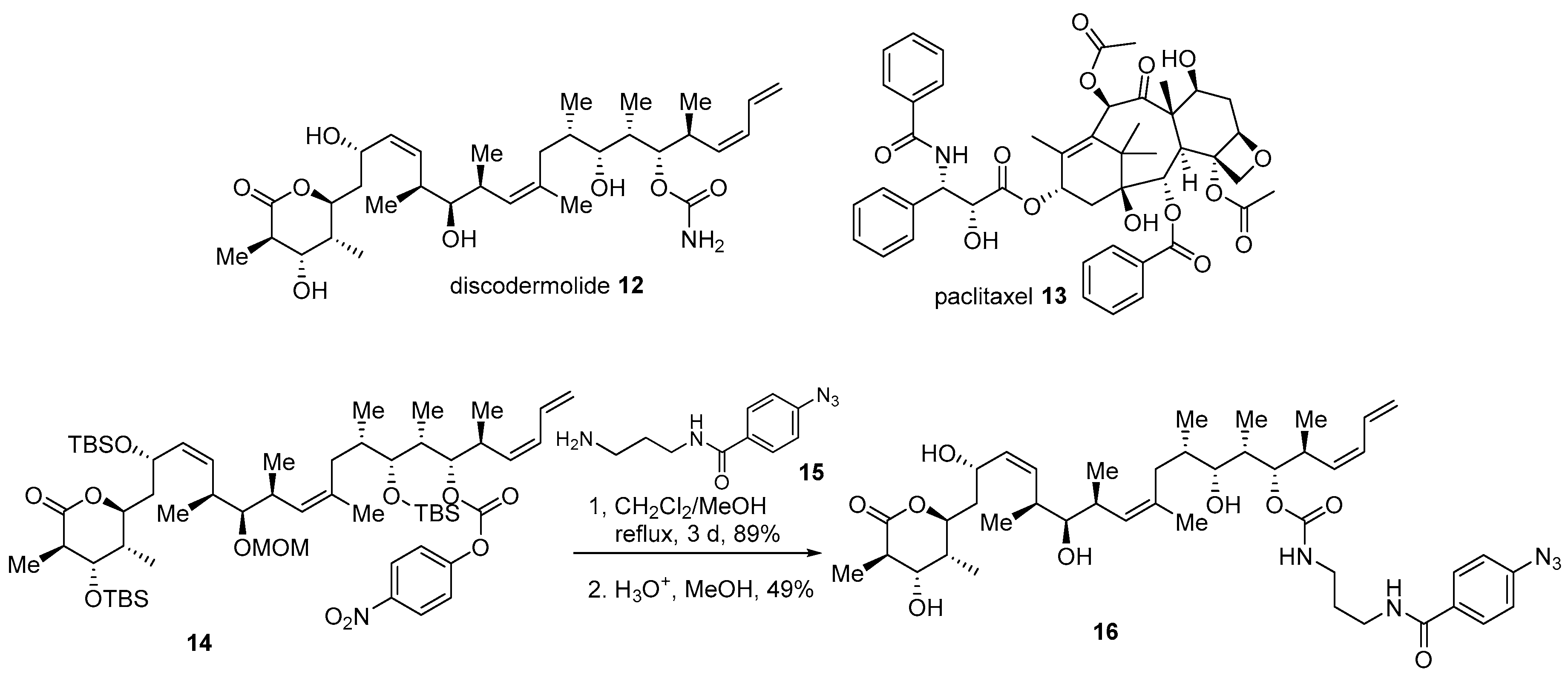
The complex structure of marine products can hamper efficient design or synthesis of advanced chimera molecules. In such cases, in silico study may offer another solution. Discodermolide 12, a marine polyketide product from Discodermia dissolute, possesses potent antiproliferative activity against various human cancer cell lines [11]. Mechanically, it stabilizes microtubules and finally arrests cells in the mitosis status, just as paclitaxel does [46]. However, unlike paclitaxel 13, a previous SAR and docking study of discodermolide at the paclitaxel binding site of tubulin revealed that free carbamate of discodermolide could be modified [47]. Based on the following study, conjugated diene in discodermolide resides in an aromatic pocket of tubulin. Moreover, the attachment of a simple aromatic group did not decrease its own anticancer activity. With this early study, the introduction of an aromatic side chain to discodermolide was pursued [47]. Synthetic intermediate 14 was coupled with various amine side chains, such as 15 with photolabile functionality, to finally produce carbamate 16 in good yield (Scheme 2). It was gratifying to note that the biological evaluation of this hybrid 16 showed an improved antiproliferative profile against human cancer cell lines. When it was treated to the lung cancer cell line A549, inhibition of cell growth at IC50 1.21 ± 0.35 nM was demonstrated, while discodermolide 12 showed a value of IC50 9.34 ± 0.56 nM (paclitaxel IC50 3.14 ± 0.09 nM). This pharmacological advance implies that the hybridization strategy based on the docking study could be another option for structurally complex molecules.
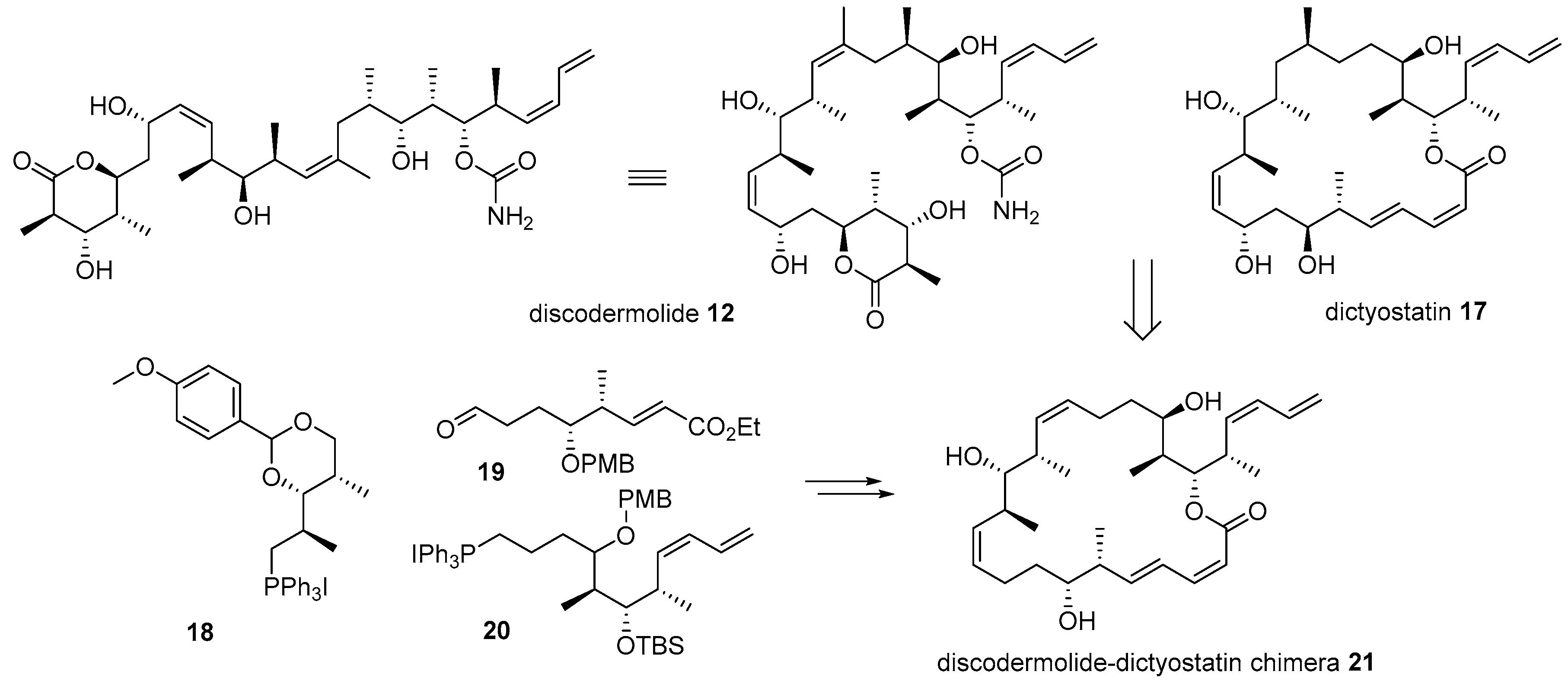
Further, hybridization of discodermolide 12 and dictyostatin 17 was also performed based on their similar structural features and anticancer activities, as depicted in Scheme 3 [48]. Dictyostatin 17 is a 22-membered macrolide natural product from the Indian [49] or Caribbean ocean sponge [50]. Dictyostatin has also garnered substantial attention due to its extraordinary antiproliferative effect (ED50 0.38 nM for P388 leukemia cell) [51]. A comparison of discodermolide and dictyostatin makes it plausible to say that they have similar backbones and substituents, except for the 22-membered lactone of dictyostatin. Based on this observation, hybridization of these two powerful anticancer natural products was performed [48]. During the synthesis and SAR study of discodermoilde 12, a practical synthetic route to it was developed and applied to chimera preparation. Para-methoxybenzyl (PMP)-acetal 18 was coupled with aldehyde 19 using Wittig olefination to construct pivotal Z-alkene of target chimera. After some functional group interconversion and another Wittig olefination with diene 20, discodermolide-dictyostatin chimera 21 was efficiently prepared. In addition, the biological activity of the chimera was also examined. Although chimera 21 possesses a relatively simple structure compared to discodermolide 12, it displayed one-third the potency of discodermolide in a displacement test using [3H]-paclitaxel bound to microtubules. This simplified structure with moderate activity might open new possibilities for further development of related natural products.
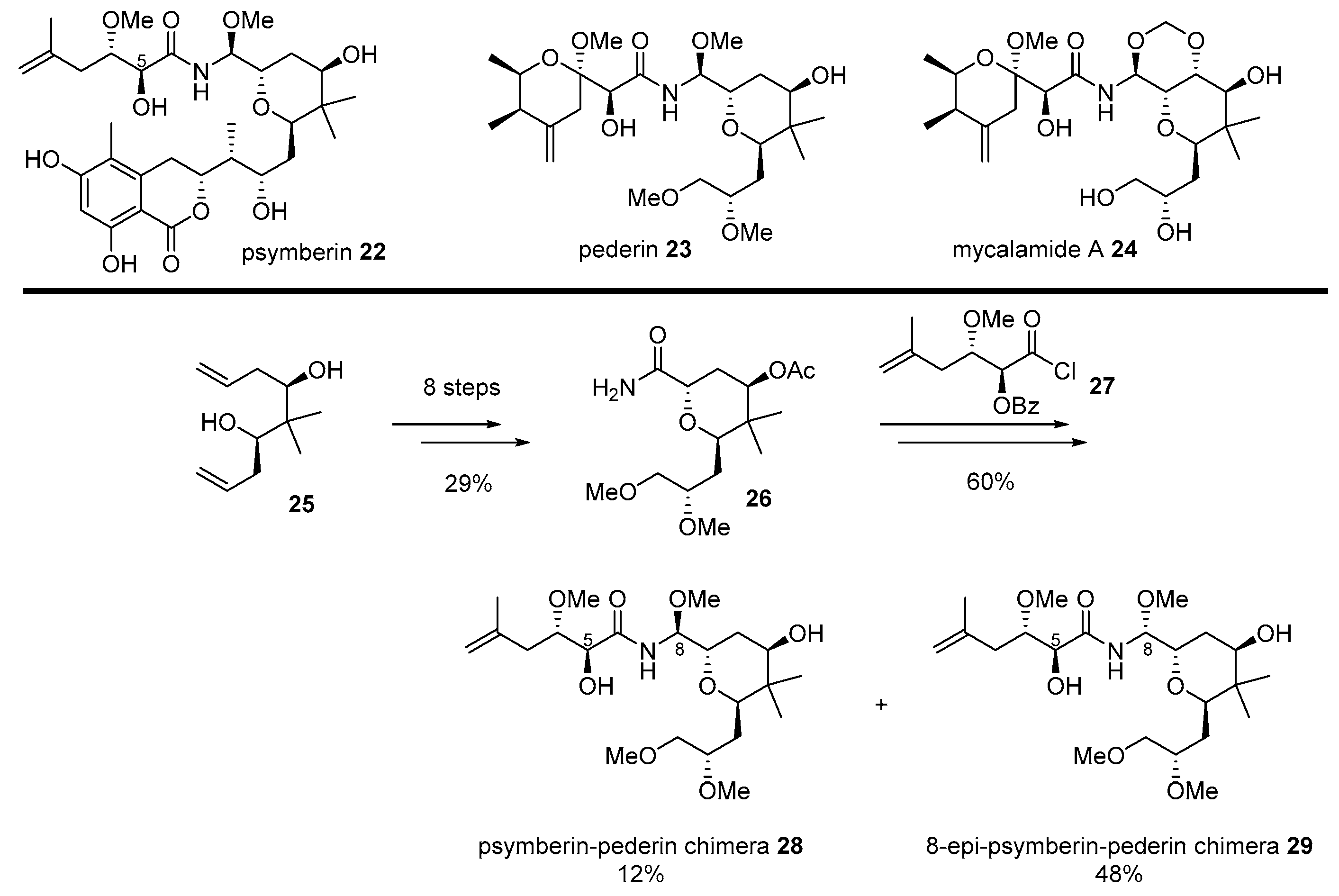
Since great cytotoxicity for solid tumors was observed using Psammocinia extracts from the waters of Papua New Guinea [52], significant effort has been made to isolate active ingredients produced from moderated cytotoxic marine natural products, such as cyclocinamide A [53], swinholide A [54], and furanosesterpenes [55]. However, these compounds did not explain the extraordinary cytotoxic properties of crude extract. The active ingredient was re-examined only in the early 2000s. Finally, highly anti-proliferative marine natural product psymberin 22 was isolated [52,56], but its absolute configuration and C4-stereochemistry were not confirmed. An additional research program elucidated its mysterious structure through total synthesis by the De Brabander group [57].
After confirmation of the correct structure, psymberin 22 was compared to classic natural compounds pederin 23 from rove beetles or mycalamide A 24 from marine invertibrates [58]. Although their structure possesses unique aminal-amide and trans-substituted tetrahydropyran skeletons, psymberin 22 has a characteristic dihydroisocoumarin moiety, while pederin/mycalamide A has another tetrahydropyran with the exomethylene group. As pederin/mycalamide A has been well reported as a powerful anticancer natural product based on eukaryotic protein synthesis inhibitors [52,56], a similar skeleton with a different side chain inspired the hybridization of the two natural products. It was anticipated that applying well-known biological properties of pederin to the psymberin-based would lead discovery [59] (Scheme 4).
Bishomoallylic alcohol 25, a synthetic intermediate for total synthesis of psymberin [57], was transformed into tetrahydropyranyl amide 26 in eight steps, with a 29% yield. It lacks a unique dihydroisocoumarin chain in psymberin, while it possesses a dimethoxy group in pederin. In order to introduce a side chain of psymberin, acyl halide 27 was attached and reduced with NaBH4. Final deprotection using LiOH/MeOH condition afforded separable C8-diastereomeric mixture of 28 and 29. These two psymberin-pederin chimeras were used to unveil the SAR of this natural product family. It is interesting to note that the absence of dihydroisocoumarin moiety gave rise to a significant loss of psymberin’s own cytotoxic property, while C8-epimer of the natural product caused a slight loss of this property. When they were treated to colon cancer cell line KM12, psymberin 25 or its C8-epimer showed a powerful or moderate toxicity (IC50 0.45 nM and 37 nM, respectively). However, its truncated analog 28 or C8-epi analog 29 showed little or no toxicity to the same cell line (IC50 710 nM and >1000 nM, respectively). These chimeras could be compared to pederin/mycalamide A as well. With a substituted tetrahydropyranyl side chain, which is rather common in natural pederin isomers, mycalamide A showed potent cytotoxicity (IC50 0.95 nM) as well. It was proven that the common substituted side chain, or dihydroisocoumarin unit, plays a pivotal role in its own anticancer activity, employing a direct comparison of mother natural products and daughter chimeras. This case is a good indication of the effectiveness of a chimera strategy for drug development.
Topsentin 30 is a bisindolyl marine alkaloid isolated from several marine sponges in Mediterranean Topsentia genitrix or Korean Spongosorites sp [60]. As this alkaloid exhibits excellent cytotoxicity to cancer cells and features a rather simple structure when compared to the other active marine natural products [61], numerous researchers have attempted to develop more potent and drug-like analogs [62]. One of the indole side chains in topsentin was changed to an aminothiazole unit, as a thiadiazole in dendrodoine 31, another marine alkaloid from Dendroda grossular [63]. With the first chimera diaminoindolylthiazole (DIT) 32 in hand, additional hybridization with curcumin 33 was performed to yield diaminocinnamoylthiazole (DCT) 34 and its analogs [64] (Scheme 5). Moreover, DITs and DCTs were screened to the cancer cell line. It is interesting that DITs are very effective in the induction of apoptosis in HeLa cells (IC50 1~45 μM), while DCTs play an active role in downregulating TNF-induced NF-κB activation. This research indicates that the hybridization of a simple functional group may also offer an opportunity to improve biological activity.
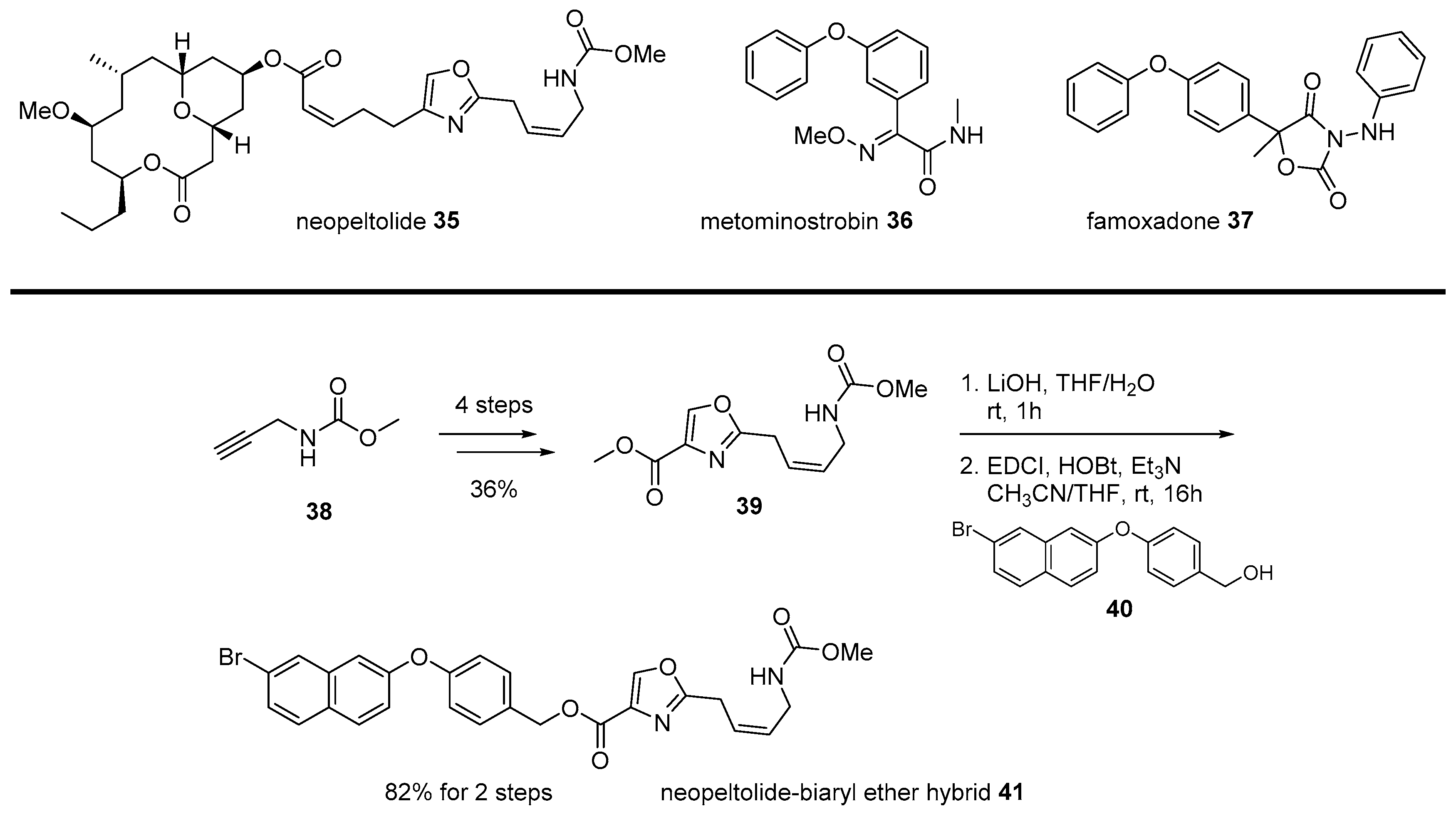
Neopeltolide 35 is another example of the chimera strategy of marine natural products. Since its isolation from marine sponge Daedalopelta Sollas in 2007 [65], urgent synthetic efforts were made to elucidate its correct structure [66]. As this marine macrolide showed various cytotoxic or cytostatic activity for numerous cancer cell lines, substantial research was conducted, including asymmetric synthesis, mechanism study, and analog synthesis. In addition, its antifungal activity was also focused upon. Contrary to its varied anticancer activity, its inhibitory activity to Candida albicans was highly potent at the minimum inhibitory concentration (MIC) 0.625 μg/mL [67]. This antifungal inhibition could be used for patients infected with the acquired immune deficiency syndrome (AIDS) or related fungal diseases. Further, the mechanism of action of neopeltolide was also studied to reveal that the agent works as the cytochrome bc1 complex inhibitor and inhibits the adenosine triphosphate (ATP) synthesis in mitochondria [68].
Although systemic research on neopeltolide was undertaken, its application in drug discovery was hampered by its complex structure. Neopeltolide 35 features a cis-substituted tetrahydropyran skeleton with an ansacyclic macrolactone framework. This formidable structure made it difficult to prepare neopeltolide on a large scale or related analogs in a varied manner. An interesting strategy was the chimeric application of neopeltolide with biaryl ether active molecules [69]. An SAR study reported that the oxazolyl carbamate skeleton plays an important role in its activity profile, while a complex macrolide skeleton has little effect on it [70]. Instead of this complex and less important left chain in neopeltolide, simple biaryl moiety was inspired by other similar fungicide metominostrobin 36, famoxadone 37, or other biaryl ethers, as illustrated in Scheme 6 [71].
Methyl propargyl carbamate 38 was converted into oxazolyl ester 39 in four steps and with high yield. Ester 39 was hydrolyzed and esterified with various primary alcohol 40 with biaryl moiety. Among synthesized chimeras, naphthyl benzyl ether 41 showed the most potent inhibitory activity (IC50 12 nM) for porcine succinate cytochrome c reductase (SCR). As this chimera simplified its mother structure, additional development for improved antifungal agent was expected.
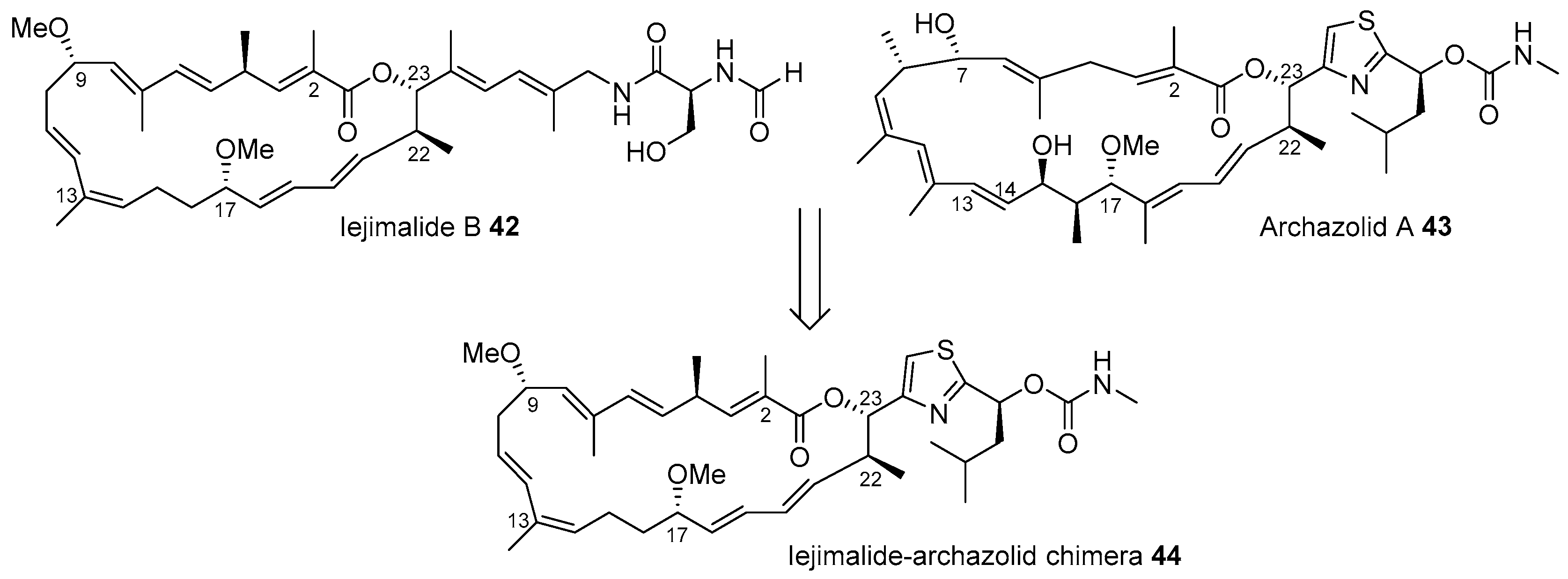
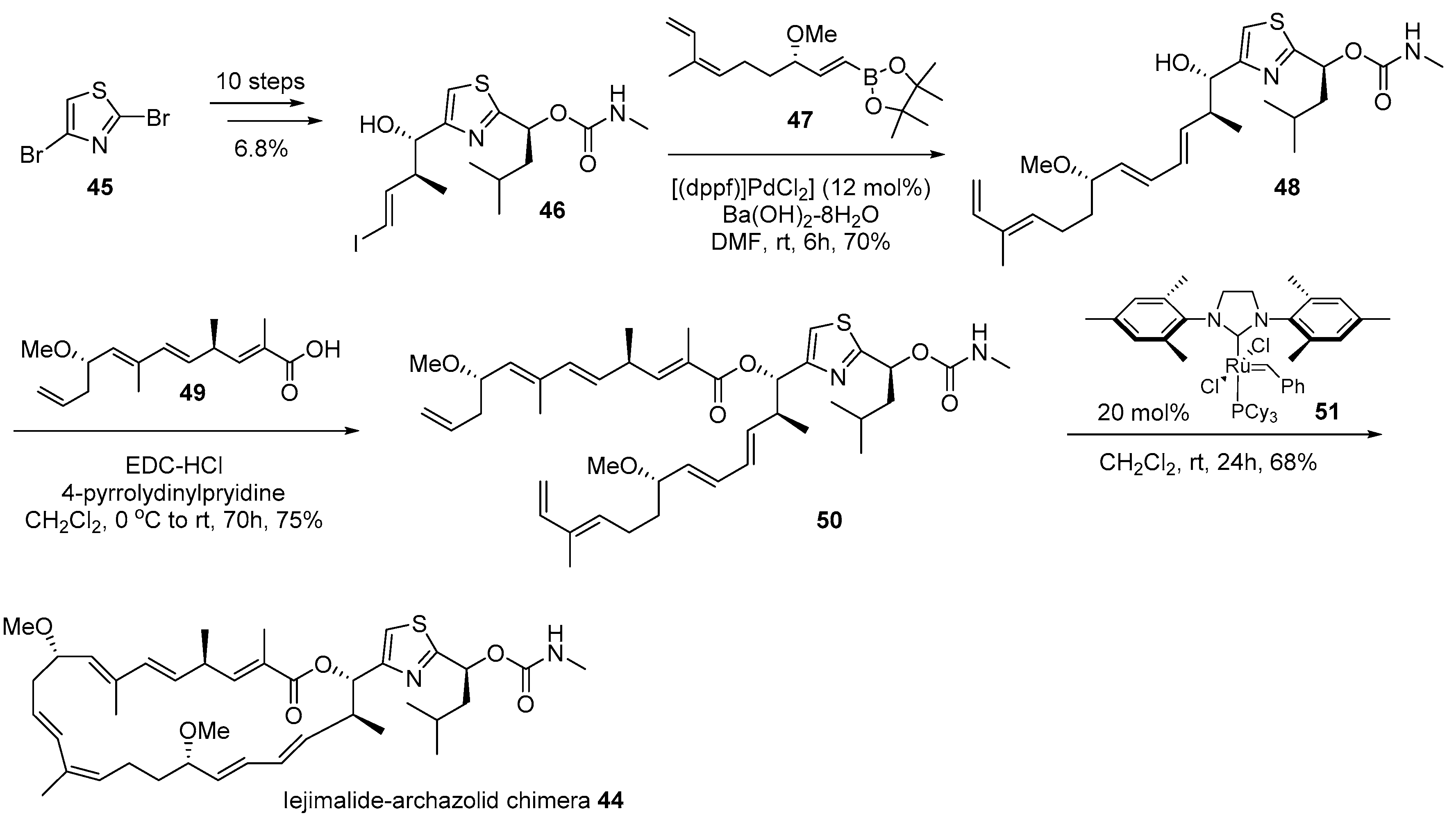
Figure 4 presents the hybridization of iejimalide and archazolid skeletons [72]. Iejimalide B 42 and its family compounds were isolated from Eudistoma cf. rigida or Cystodytes sp., which were collected from an island in Japan [73]. Due to its remarkable anticancer activity and selectivity to NCI 60 cell lines, this polyunsaturated macrolide has been focused upon for its promising role in drug discovery [74]. However, its scarcity in natural resources led to not only total synthesis [75,76] but also structural simplification based on a hybridization strategy [72]. Archazolid A 43, isolated from terrestrial myxobacteria [77], possesses a very similar structure as that of iejimalide B 42; however, archazolid A 43 features a relatively simple side chain with thiazole and methyl carbamate moiety. In addition, because archazolid 43 showed potent inhibitory activity to vacuolar-type ATPases as well [77], iejimalide-archazolid chimera 44 was designed and synthesized.
Dibromothiazole 45 was converted to vinyliodide 46 using a 10-step sequence containing the Corey-Bakshi-Shibata (CBS) reduction [78], Marshall alkylation [79], and lithium-halogen exchange reactions as shown in Scheme 7. It was coupled with known catechol borane [76] 47, using the Suzuki coupling condition [80] to afford tetraene 48. Finally, the esterification of tetraene 48 with known carboxylic acid [76] 49 and ring closing metathesis [81] produced the desired chimera 44 in good yield. Biological evaluation of chimera 44 against various cancer cell lines was performed and it was proven that this hybridization yielded reduced toxicity to cancer cell lines, although certain cell lines such as lung adeno (LXFA 629L, IC50 0.32 μM) or colorectal (CSF HT29, IC50 0.49 μM)) were slightly sensitive to chimera 44.
2.2. Conjugation with Other Functional Compounds
For drug discovery and sequential development, marine natural products require more detailed studies, such as on-target protein isolation. The chemical conjugation of marine natural products with other functional compounds provides special opportunities in this regard. Biotin is a good example. Biotin, well known for its strong interaction with streptavidin [82,83,84], can be utilized to identify the target protein of natural products [85,86,87,88]. As this hybridization strategy may lead to the loss of its own binding affinity to natural products, it usually requires preliminary study to identify where to ligate biotin and what to use as a linker between biotin and the natural product. This pre-research requires additional efforts, thereby making it difficult to apply this method to a general natural product. However, in certain cases, well-designed chimeras showcase the usefulness of this hybridization protocol.
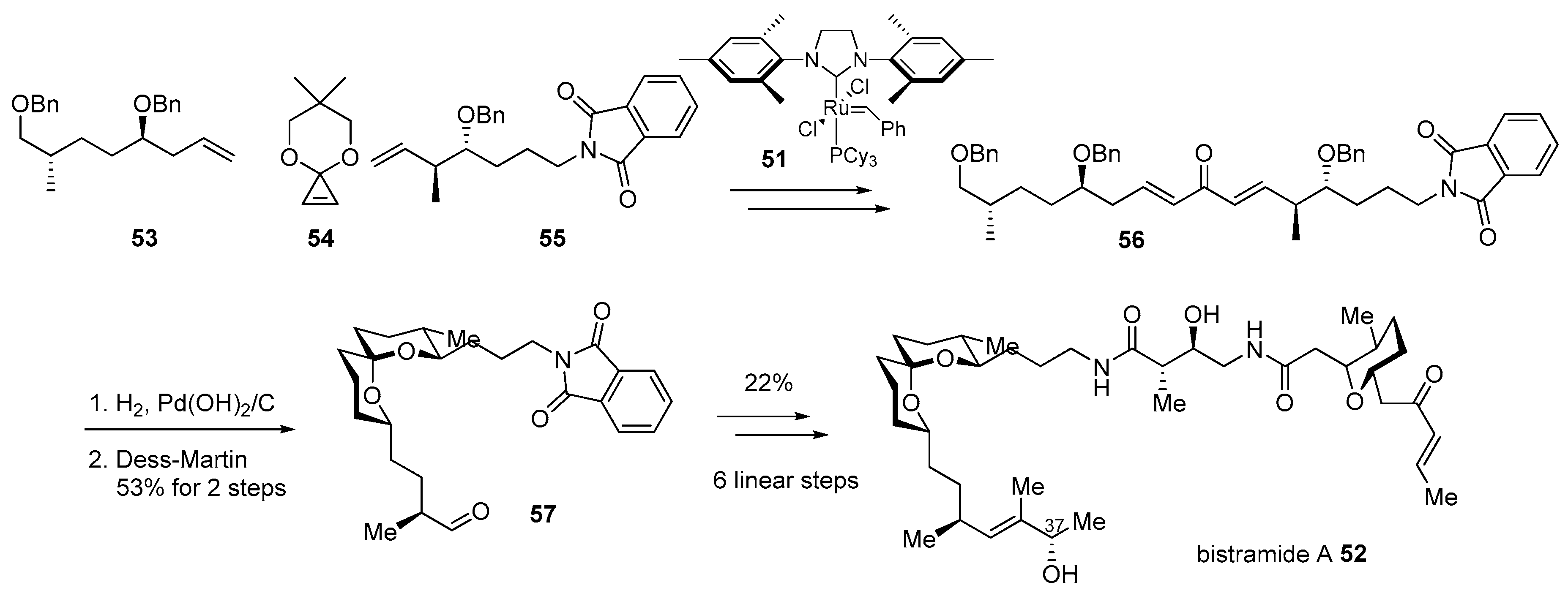
Bistramide A 52, isolated from marine metabolite of Lissoclinum bistratum [89,90,91], is a good example of this hybridization. As the bistramide family has shown potent cytotoxic properties on various cancer cell lines, such as non-small cell broncho-pulmonary carcinoma or HL60 cells as well as unique spiroketal skeleton [92], this marine product has been a captivating target for synthetic chemists. After pioneering research on structural elucidation and synthesis of skeletons or other isomers [93], enantioselective total synthesis of bistramide A was accomplished in 2004 [94]. This synthetic route features iterative cross metathesis of terminal alkenes 53 and 55 with the geometrically strained cyclopropene 54 [95] in the presence of Grubbs second-generation catalyst 55 to construct bisunsaturated ketone 56. The treatment of H2 with Pd(OH)2/C to this ketone induced hydrogenation/hydrogenolysis and spontaneous cyclization to afford the desired spiroketal 57 in 53% yield after the Dess–Martin oxidation sequence. Ketal 57 was converted to bistramide A 52 after employing amidation with other building blocks in six linear steps. This efficient and convergent synthesis made it possible to not only elucidate the C37 chiral center [96] but also provide further opportunity for hybridization with other active molecules (Scheme 8).
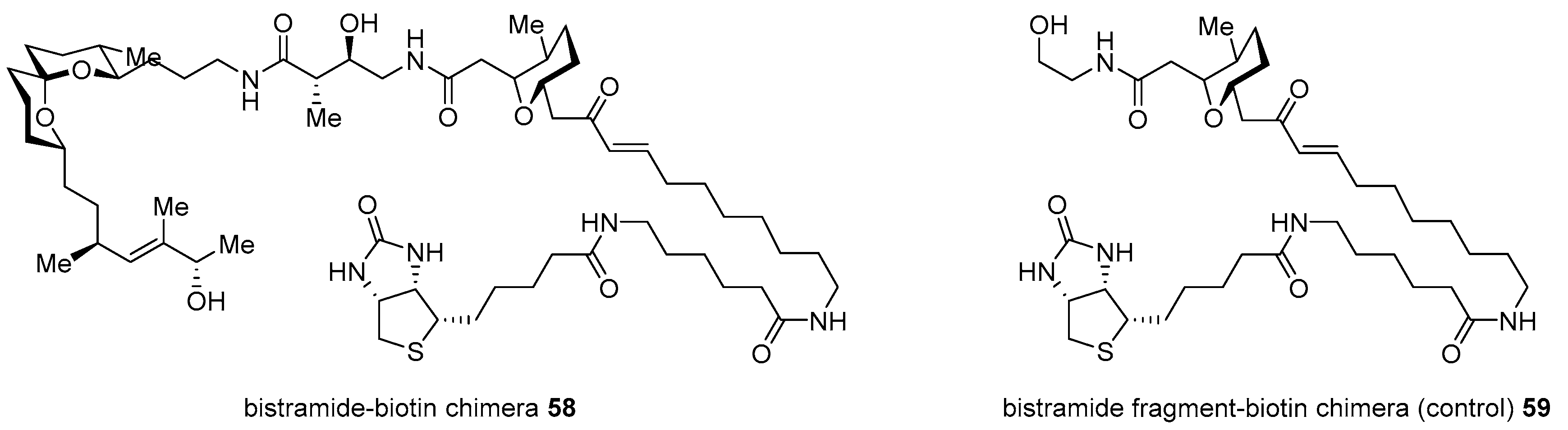
Although potent and active, bistramide A 52 has been reported since its first isolation [97]; its mechanism of action was unknown until the preparation of bistramide-biotin chimera, as illustrated in Scheme 8 [98]. Its protein kinase C(PKC) δ inhibitory features caused PKCδ to be a cellular target in HL-60 cells. However, an in vitro study with real-time fluorogenic kinase assay system [99] revealed that bistramide A did not have strong affinity to PKCδ as its inhibitory activity. In order to identify an early-stage target, target protein fishing was planned. Based on the synthetic strategy previously described [94], homologation with carbon linker and final attachment of biotin was accomplished in order to produce desired chimera 58 as well as fragment-biotin chimera 59 for a controlled experiment (Figure 5). The treatment of these two chimeras into whole-cell lysate from A549 cell revealed direct binding of monomeric G-actin (Kd 7nM) as a primary target protein. Using this target fishing study with biotin-chimera enabled the study of a more detailed mechanism of action.
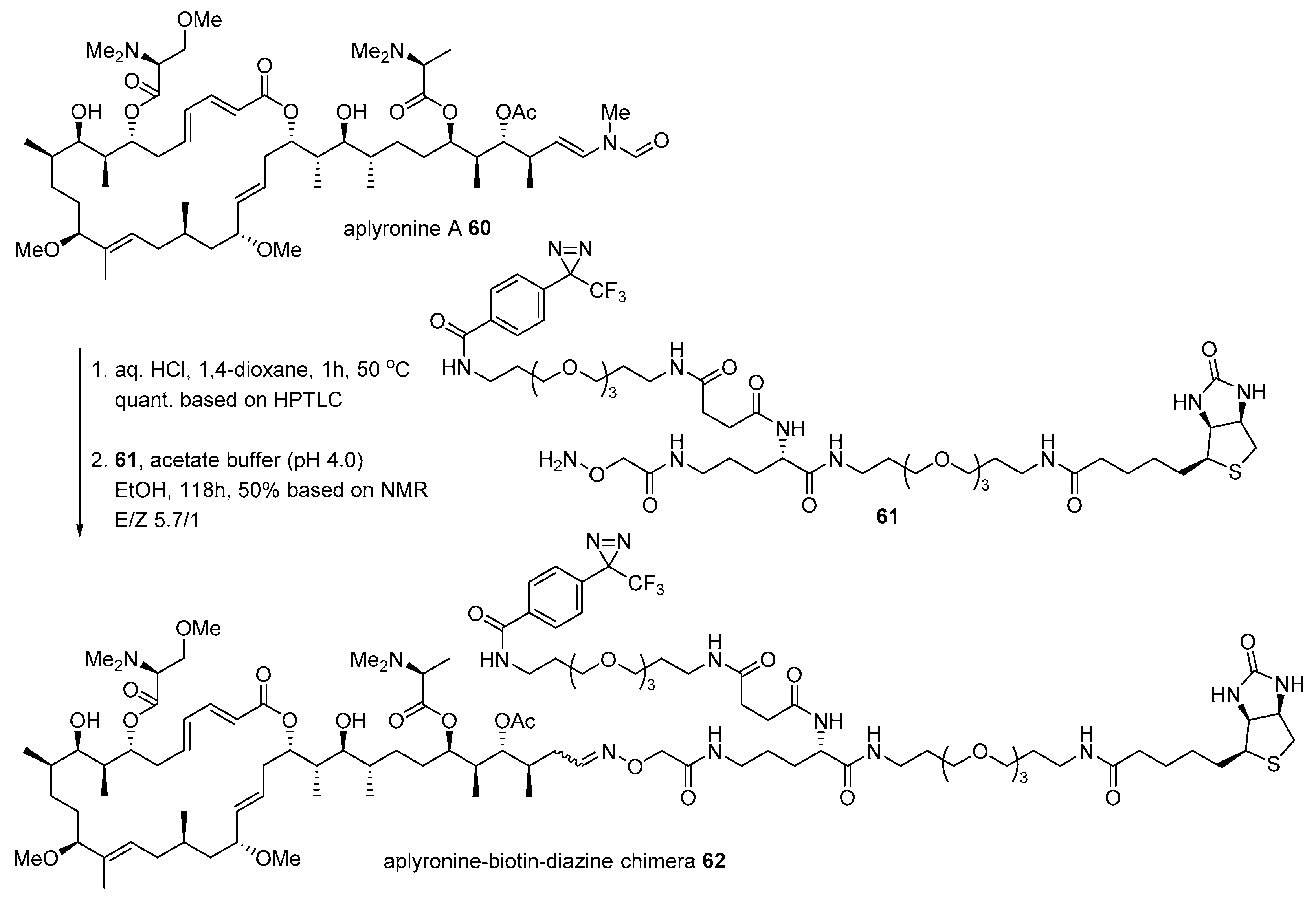
Aplyronine A 60, isolated from Japanese sea hare Aplysia kurodai [100], is another case for the conjugation of marine natural product with biotin to identify the interaction of actin. While bistramide A 52 binds to actin, aplyronine A binds to the actin/tubulin complex, which is a 1:1:1 trimeric complex [101]. The Kigoshi group treated aplyronine A with aqueous HCl to produce the corresponding aldehyde, which was condensed with hydroxylamine 61 to form an oxime bond as an inseparable mixture of aplyronine-biotin-diazine chimera 62 (Scheme 9). When it was treated to HeLa S3 cells, α/β tubulins appeared in SDS-PAGE with some other nonspecific binding proteins. In order to validate this Western blot result, purified actin (from rabbit) or tubulin (from porcine brain) was treated with chimera 62. This validation study examined that unusual aplyronine/actin/tubulin heterotrimeric complex is responsible for its extraordinary antitumor effect through cell arrest at mitosis in HeLa S3 cells (IC50 0.45 nM) [102].
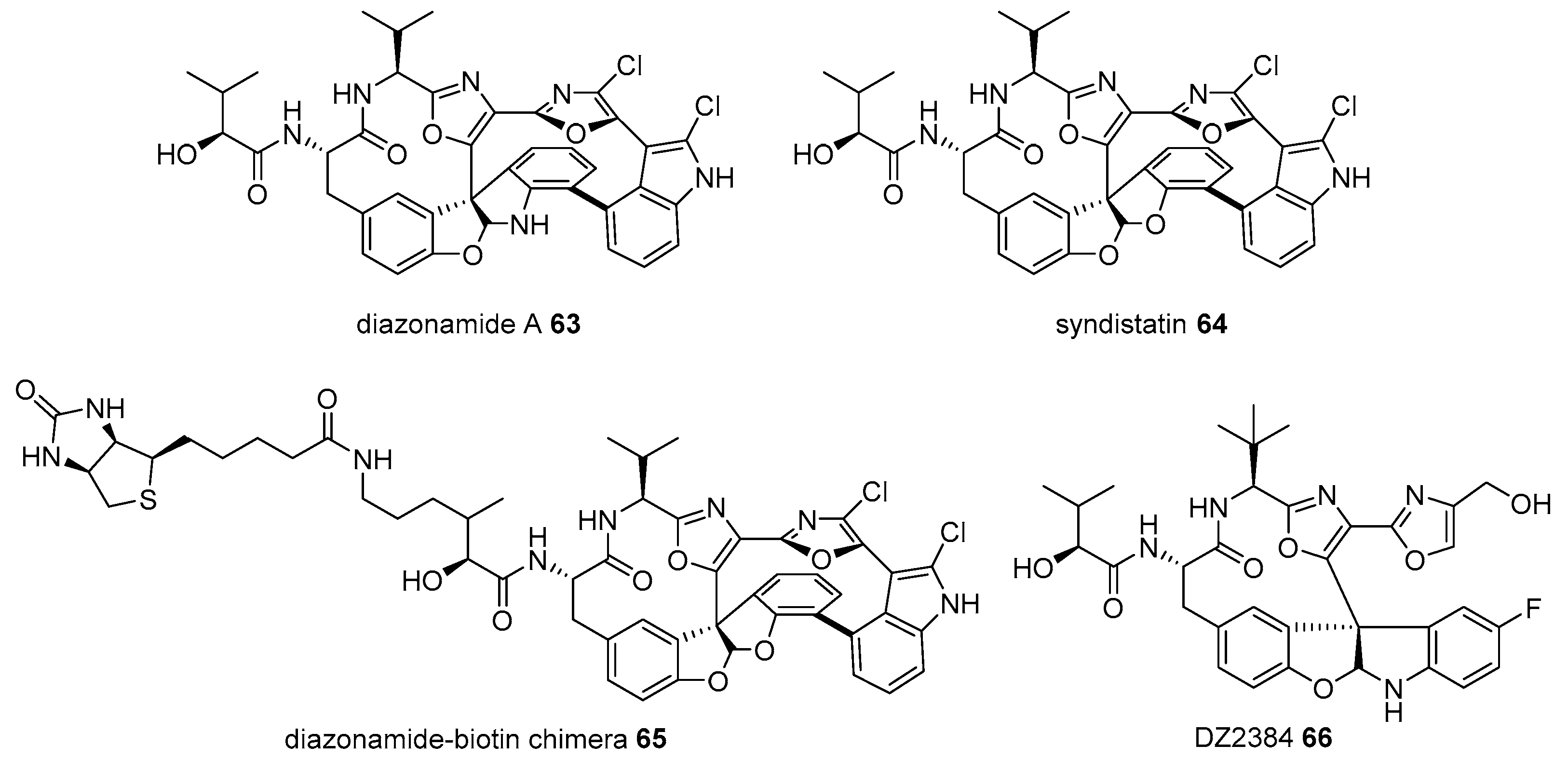
Diazonamide A 63 is a marine natural product from Diazona chinensis, located on the ceilings of small caves on the Siquijor Islands in the Philippines [103]. Despite its scarcity in natural sources (54 mg from 256 g of D. chinensis), its extraordinary antimitotic property in human colon carcinoma and murine melanoma cancer cell lines has garnered substantial interest from chemists and biologists [104,105,106]. This tremendous study led to structural elucidation, efficient chemical synthesis, and related analog preparation. Its target protein study is interesting. When diazonamide (syndistatin 64)-biotin chimera 65 was prepared and treated with HeLa extract, two additional bands of an affinity matrix were observed on an SDS/PAGE compared to those in the control experiment. Mass identification indicated that these two bands were ornithine δ-amino transferase, although why the matrix separates is unknown [107]. As ornithine δ-amino transferase is well known as a mitochondrial enzyme in the TCA cycle [108], it was slightly surprising. However, further study using the RNAi-mediated knockdown method and additional investigation proved that ornithine δ-amino transferase is essential for mitotic cell division. Thus, target protein and its corresponding pathways of diazonamide were unveiled to lead to the discovery of advanced diazonamide analog DZ2384 66 [109]. This process shows a 5–50 times higher efficacy without neurotoxicity at an effective dose (Figure 6).
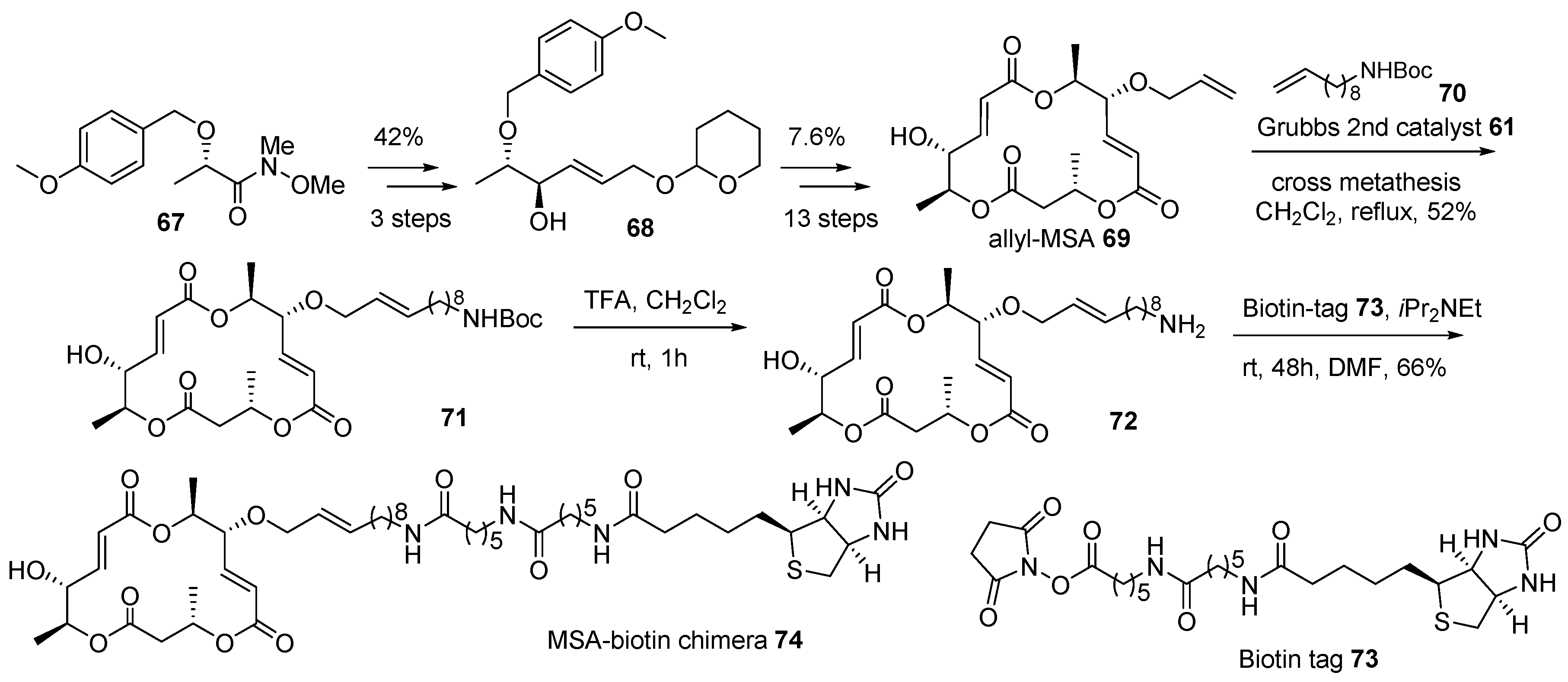
Further, macrosphelide (MS) skeleton was also utilized in this chimeric research, as depicted in Scheme 10 [110]. Although it has a structural framework and biological profiles, its mechanism of action is not well studied in the MS family. In order to identify the target protein of MSA 2 and elucidate the following pathway, MSA-biotin hybridization was planned. The protected Weinreb-amide of (S)-lactic acid 67 [111] was transformed into allylic alcohol 68 using a three-step protocol—alkylnylatoin, carbonyl reduction, and alkyne reduction—in 42% yield. This key intermediate 68 in hand, allylation, iterative esterification, and deprotection sequence finally afforded allyl-MSA 69 in 7.6% yield and 13 steps. Biotin was linked to allyl-MSA 69 after additional manipulation. For efficient target fishing, a long carbon chain was selected as a linker. Terminal alkene in allyl-MSA was utilized in cross metathesis to introduce a long carbon linker and protect the amine functional group in amino MSA 71. Finally, acidic deprotection and following amidation with biotin tag 73 produced the desired MSA-biotin chimera 74. Target fishing studies, employing this chimera, on potent cell-cell adhesion inhibitor are ongoing.
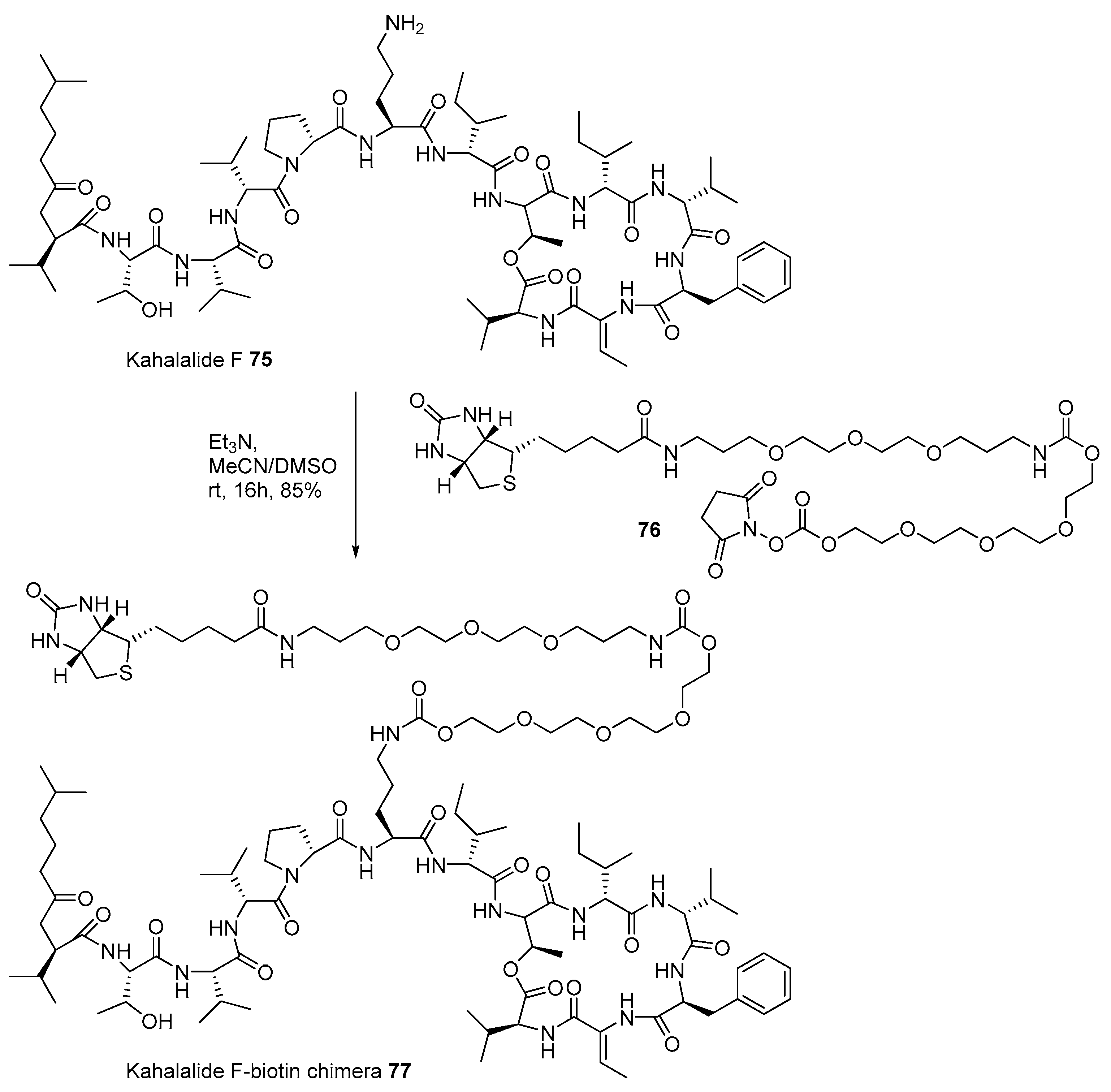
The marine natural product-biotin chimera strategy was also applied to identify the target protein of kahalalide F 75. Ever since the isolation of kahalalide F from marine mollusk, Elysia rufescens, in 1993 [112], this polypeptide marine product has been used in clinical trials because of its powerful anticancer activity and low cellular toxicity [113]. For advanced development, identification of the target protein using biotinylation of kahalalide F was also attempted, as presented in Scheme 11. Kahalalide F was coupled with biotinylated linker 76 [114], prepared from biotin and tetraethyleneglycol, under a weak basic condition to produce desired chimera 77 in 85% yield. When chimera 77 was treated to T7 cDNA phage for reverse chemical proteomics, human ribosomal protein S25 was found to be responsible for its potent anticancer property in a dose-dependent manner.
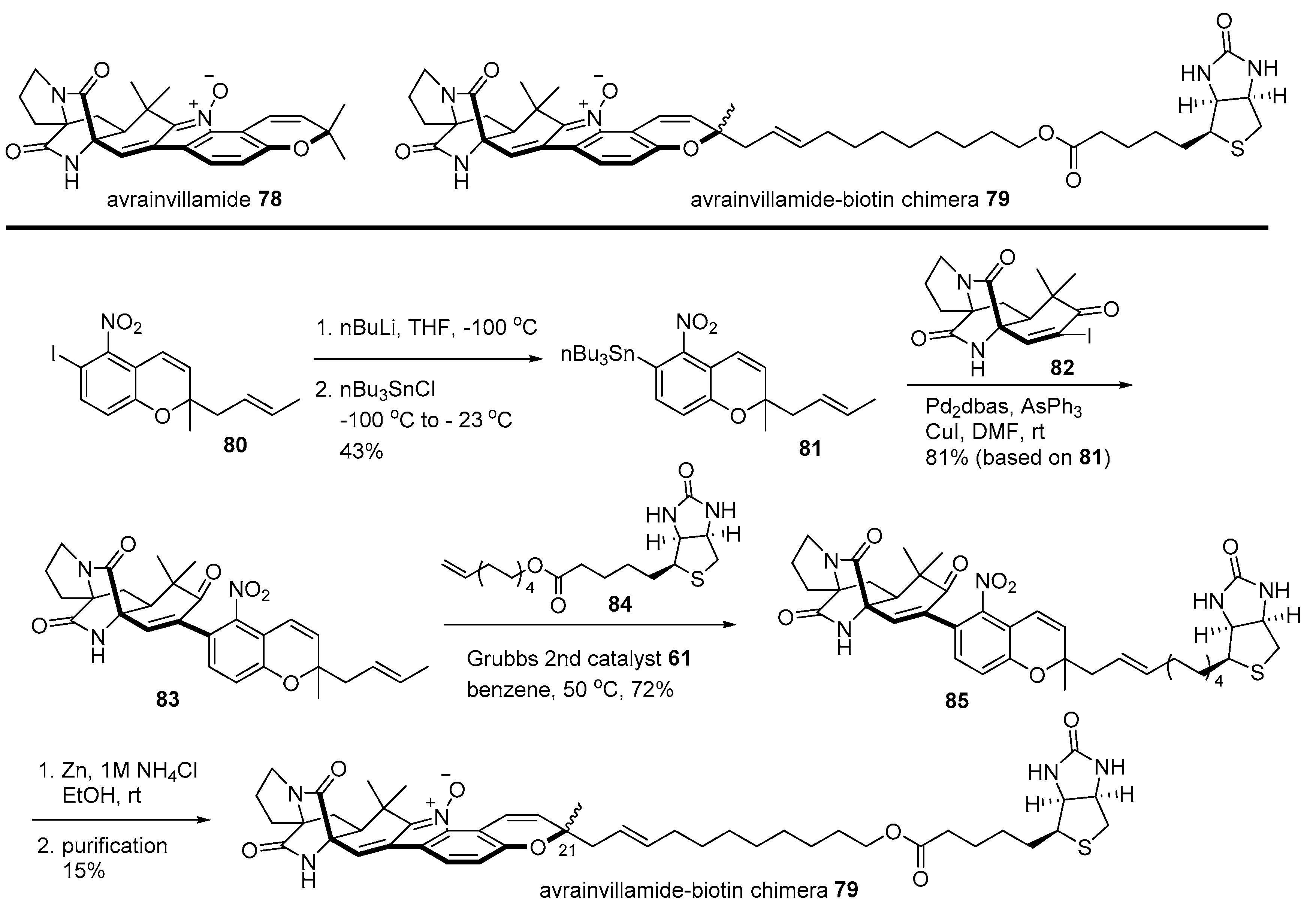
Avrainvillamide is another good example of the biotin chimera strategy [115]. In order to unveil the mysterious target protein of powerful avrainvillamide, isolated from Aspergillus sp. CNC358 [116], a biotinylated natural product 79 was designed. Synthesis of chimera 79 is depicted in Scheme 12. Iodoarene 80 [116] was stannylated to yield aromatic stannane 81 via metal/halogen exchange and a substitution reaction. Then, stannane 81 was coupled with vinyl iodide 82 [117] to produce nitroarene 83, which was converted into biotinylated arene 85 employing cross metathesis with terminal alkene 84 [118]. Reductive cyclization of 85 and HPLC purification afforded the desired chimera 79 (1:1 mixture of diastereomers at the C21 chiral center). Finally, Western blotting and MS/MS sequencing of these chimera-treated T-47 D cells revealed that nucleophosmin was a target protein of avrainvillamide. As nucleophosmin is a multifunctional protein in numerous tumors [119], a more detailed study was subsequently conducted to disclose the mode of action of avrainvillamide and its related natural product stephacidin [120].
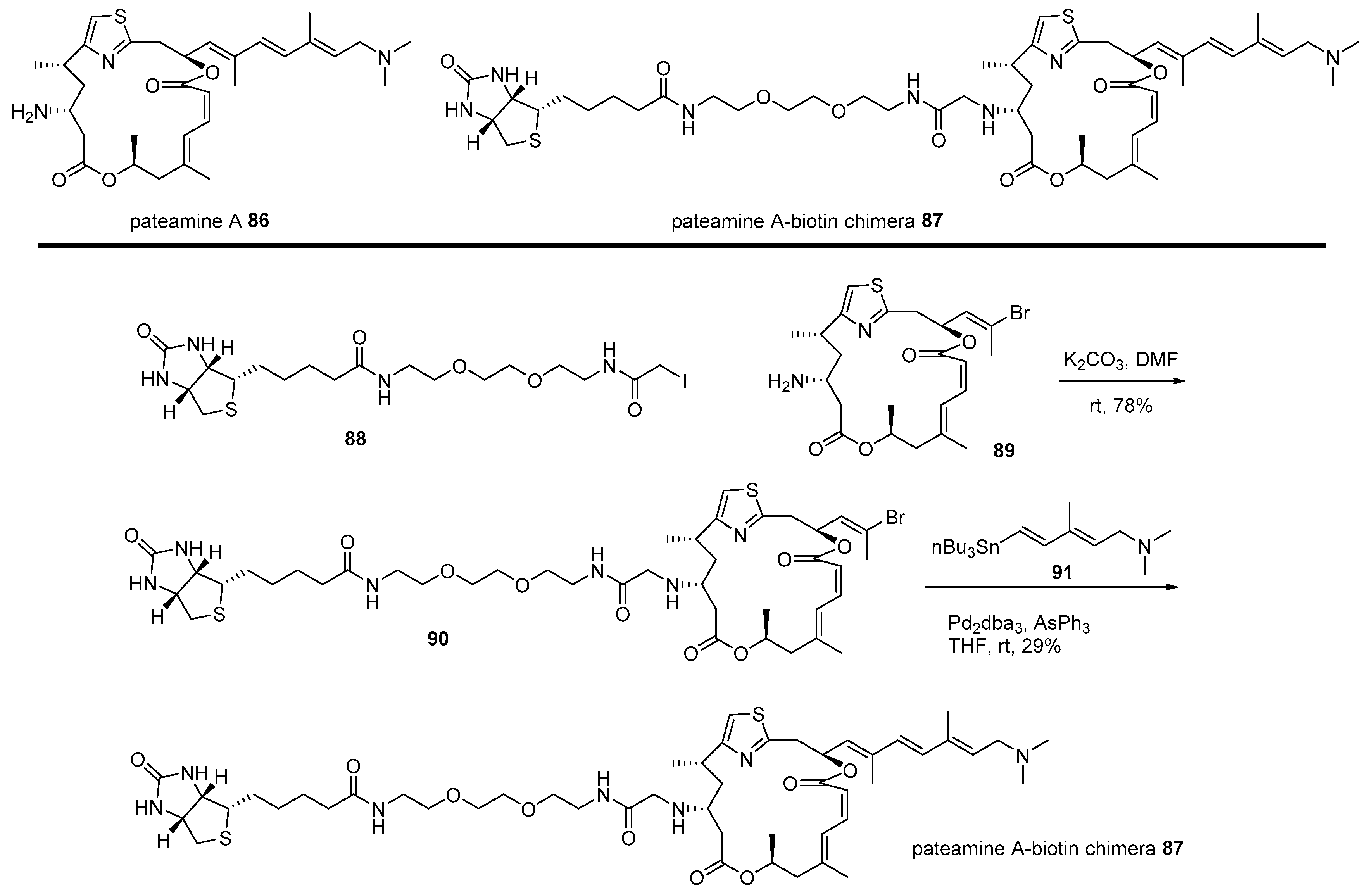
The hybridization strategy was also utilized to identify the target protein of pateamine, which is isolated from the New Zealand marine sponge [121]. As this marine natural product showed not only remarkable cytotoxicity to tumor cell line P338 [121] but also extraordinary immunosuppressive activity [122], target protein identification was pursued, as illustrated in Scheme 13 [123]. Primary iodide 88 [124] was alkylated with truncated pateamine 89 [123], which is more stable than pateamine 86. Further, corresponding vinyl bromide 90 [124] was coupled with vinyl stannane 91 [122] to produce the desired pateamine-biotin chimera 87 in moderate yield. The pull-down method of this affinity-matrix 87 showed that it bound to and inhibited the association of eIF4A and eIF4B, finally inhibiting the cap-dependent eukaryotic translation sequence [124]. This fundamental study has also opened the way for more detailed research and further applications of pateamine analogs [125].
3. Conclusions
As an endless resource of bioactive molecules, marine natural products stand a reliable chance in the world of drug discovery. However, certain limitations of these products, such as complex structure, toxicity, selectivity, and even potency have driven researchers to identify additional strategies for these natural compounds. Chimeras yielded by the hybridization of two active molecules are one such solution. Changing the active skeleton of the mother framework also added more opportunities, but this process also comes with its own set of unique drawbacks, such as loss of function, scarcity of supply, and complex structure for chemical modification. In order to overcome these limitations, SAR, structural simplification, development of the synthetic route, and the medicinal chemistry approach have been studied thus far. Based on this fundamental progress, currently, hybridization of active natural products is being utilized to improve their own biological properties and elucidate veiled mechanism of actions. The efficient application of this chimera strategy to other active molecules remains a direction for groundbreaking research in the future.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (NRF-2016R1C1B2006699).
Acknowledgments
We are grateful to the two reviewers for their constructive input.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, J.M.; Goncalves, B.D.; Gomez, M.V.; Vieira, L.B.; Ribeiro, F.M. Animal toxins as therapeutic tools to treat neurodegenerative diseases. Front. Pharmacol. 2018, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, P.; D’Auria, M.V.; Muller, C.D.; Tammela, P.; Vuorela, H.; Yli-Kauhaluoma, J. Exploring marine resources for bioactive compounds. Planta Med. 2014, 80, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lou, H.-X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Chicacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooper, J.N. Antineoplastic agents. 257. Isolation and structure of spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] [CrossRef]
- Smith, A.B.; Razler, T.M.; Meis, R.M.; Pettit, G.R. Synthesis and biological evaluation of phorboxazole congeners leading to the discovery and preparative-scale synthesis of (+)-chlorophorboxazole a possessing picomolar human solid tumor cell growth inhibitory activity. J. Org. Chem. 2008, 73, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Schulte, G.K. Discodermolide: A new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissoluta. J. Org. Chem. 1990, 55, 4912–4915. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Schaufelberger, D.E.; Koleck, M.P.; Beutler, J.A.; Vatakis, A.M.; Alvarado, A.B.; Andrews, P.; Marzo, L.; Muschik, G.; Roach, J.; Ross, J.T. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 1991, 54, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B., III; Tomioka, T.; Risatti, C.A.; Sperry, J.B.; Sfouggatakis, C. Gram-scale synthesis of (+)-spongistatin 1: Development of an improved, scalable synthesis of the F-ring subunit, fragment union, and final elaboration. Org. Lett. 2008, 10, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Beauchamp, T.J.; LaMarche, M.J.; Kaufman, M.D.; Qiu, Y.; Arimoto, H.; Jones, D.R.; Kobayashi, K. Evolution of a gram-scale synthesis of (+)-discodermolide. J. Am. Chem. Soc. 2000, 122, 8654–8664. [Google Scholar] [CrossRef]
- Mickel, S.J.; Niederer, D.; Daeffler, R.; Osmani, A.; Kuesters, E.; Schmid, E.; Schaer, K.; Gamboni, R.; Chen, W.; Loeser, E. Large-scale synthesis of the anti-cancer marine natural product (+)-Discodermolide. Part 5: Linkage of fragments C1-6 and C7-24 and finale. Org. Process. Res. Dev. 2004, 8, 122–130. [Google Scholar] [CrossRef]
- Choudhary, A.; Naughton, L.; Montánchez, I.; Dobson, A.; Rai, D. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Lear, M.J.; Hirai, K.; Ogawa, K.; Yamashita, S.; Hirama, M. A convergent total synthesis of the kedarcidin chromophore: 20-years in the making. J. Antibiot. 2019, 72, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.-H. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “state of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- McBride, A.; Butler, S.K. Eribulin mesylate: A novel halichondrin B analogue for the treatment of metastatic breast cancer. Am. J. Health Syst. Pharm. 2012, 69, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total synthesis of halichondrin B and norhalichondrin B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-derived pharmaceuticals—Challenges and opportunities. Biomol. Ther. 2016, 24, 561. [Google Scholar] [CrossRef] [PubMed]
- Bebbington, M.W. Natural product analogues: Towards a blueprint for analogue-focused synthesis. Chem. Soc. Rev. 2017, 46, 5059–5109. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015, 13, 5302–5343. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Edit. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Edit. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Mehta, G.; Singh, V. Hybrid systems through natural product leads: An approach towards new molecular entities. Chem. Soc. Rev. 2002, 31, 324–334. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101. [Google Scholar] [CrossRef]
- Salami, J.; Crews, C.M. Waste disposal-An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Olaáh, J.; Ovaádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2017, 61, 482–491. [Google Scholar] [CrossRef]
- Leslie, B.J.; Hergenrother, P.J. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 2008, 37, 1347–1360. [Google Scholar] [CrossRef]
- Paek, S. Synthetic advances in macrosphelides: Natural anticancer agents. Molecules 2014, 19, 15982–16000. [Google Scholar] [CrossRef]
- Yamada, T.; Iritani, M.; Doi, M.; Minoura, K.; Ito, T.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, macrosphelides C, E–G and I, produced by a Periconia species separated from an Aplysia sea hare. J. Chem. Soc. Perkin Trans. 1 2001, 3046–3053. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, Y.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar] [CrossRef]
- Sunazuka, T.; Hirose, T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Ōmura, S.; Sprengeler, P.A.; Smith, A.B. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell—Cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar] [CrossRef]
- Paek, S. Development of advanced macrosphelides: Potent anticancer agents. Molecules 2015, 20, 4430–4449. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Ishihara, K.; Ahmed, K.; Zhao, Q.; Kondo, T.; Nemoto, H. Synthesis of macrosphelides with a thiazole side chain: New antitumor candidates having apoptosis-inducing property. Org. Lett. 2006, 8, 4609–4612. [Google Scholar] [CrossRef]
- Meng, D.; Bertinato, P.; Balog, A.; Su, D.; Kamenecka, T.; Sorensen, E.J.; Danishefsky, S.J. Total syntheses of epothilones A and B. J. Am. Chem. Soc. 1997, 119, 10073–10092. [Google Scholar] [CrossRef]
- Xia, S.; Kenesky, C.S.; Rucker, P.V.; Smith, A.B.; Orr, G.A.; Horwitz, S.B. A photoaffinity analogue of discodermolide specifically labels a peptide in β-tubulin. Biochemistry 2006, 45, 11762–11775. [Google Scholar] [CrossRef]
- Smith, A.B., III; Sugasawa, K.; Atasoylu, O.; Yang, C.H.; Horwitz, S.B. Design and synthesis of (+)-discodermolide–paclitaxel hybrids leading to enhanced biological activity. J. Med. Chem. 2011, 54, 6319–6327. [Google Scholar] [CrossRef]
- Shin, Y.; Choy, N.; Balachandran, R.; Madiraju, C.; Day, B.W.; Curran, D.P. Discodermolide/dictyostatin hybrids: Synthesis and biological evaluation. Org. Lett. 2002, 4, 4443–4446. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Boyd, M.R.; Schmidt, J.M. Isolation and structure of the cancer cell growth inhibitor dictyostatin 1. J. Chem. Soc. Chem. Commun. 1994, 1111–1112. [Google Scholar] [CrossRef]
- Isbrucker, R.A.; Cummins, J.; Pomponi, S.A.; Longley, R.E.; Wright, A.E. Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem. Pharmacol. 2003, 66, 75–82. [Google Scholar] [CrossRef]
- Zanato, C.; Pignataro, L.; Hao, Z.; Gennari, C. A practical synthesis of the C1–C9 fragment of dictyostatin. Synthesis 2008, 2008, 2158–2162. [Google Scholar]
- Cichewicz, R.H.; Valeriote, F.A.; Crews, P. Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the pederin family. Org. Lett. 2004, 6, 1951–1954. [Google Scholar] [CrossRef]
- Clark, W.D.; Corbett, T.; Valeriote, F.; Crews, P. Cyclocinamide A. An unusual cytotoxic halogenated hexapeptide from the marine sponge Psammocinia. J. Am. Chem. Soc. 1997, 119, 9285–9286. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tanaka, J.I.; Katori, T.; Matsuura, M.; Kitagawa, I. Structure of swinholide A, a potent cytotoxic macrolide from the Okinawan marine sponge Tehonella Swinhoei. Tetrahedron Lett. 1989, 30, 2963–2966. [Google Scholar] [CrossRef]
- Faulkner, D.J. Variabilin, an antibiotic from the sponge, Ircinia variabilis. Tetrahedron Lett. 1973, 39, 3821–3822. [Google Scholar] [CrossRef]
- Pettit, G.R.; Xu, J.; Chapuis, J.; Pettit, R.K.; Tackett, L.P.; Doubek, D.L.; Hooper, J.N.; Schmidt, J.M. Antineoplastic Agents. 520. Isolation and Structure of Irciniastatins A and B from the Indo-Pacific Marine Sponge Ircinia r amosa. J. Med. Chem. 2004, 47, 1149–1152. [Google Scholar] [CrossRef]
- Jiang, X.; García-Fortanet, J.; De Brabander, J.K. Synthesis and complete stereochemical assignment of psymberin/irciniastatin A. J. Am. Chem. Soc. 2005, 127, 11254–11255. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.; Pannell, L.K. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar] [CrossRef]
- Jiang, X.; Williams, N.; De Brabander, J.K. Synthesis of psymberin analogues: Probing a functional correlation with the pederin/mycalamide family of natural products. Org. Lett. 2007, 9, 227–230. [Google Scholar] [CrossRef]
- Bartik, K.; Braekman, J.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentins, new toxic bis-indole alkaloids from the marine sponge Topsentia genitrix. Can. J. Chem. 1987, 65, 2118–2121. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis (indolyl) imidazoles from Caribbean deep-sea sponges of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Gu, X.; Wan, X.; Jiang, B. Syntheses and biological activities of bis (3-indolyl) thiazoles, analogues of marine bis (indole) alkaloid nortopsentins. Bioorg. Med. Chem. Lett. 1999, 9, 569–572. [Google Scholar] [CrossRef]
- Hogan, I.; Sainsbury, M. The synthesis of dendrodoine, 5-[3-(N, N-dimethylamino-1, 2, 4-thiadiazolyl]-3-indolylmethanone, a metabolite of the marine tunicate dendroda grossular. Tetrahedron 1984, 40, 681–682. [Google Scholar] [CrossRef]
- Juneja, M.; Vanam, U.; Paranthaman, S.; Bharathan, A.; Keerthi, V.S.; Reena, J.K.; Rajaram, R.; Rajasekharan, K.N.; Karunagaran, D. 4-amino-2-arylamino-5-indoloyl/cinnamoythiazoles, analogs of topsentin-class of marine alkaloids, induce apoptosis in hela cells. Eur. J. Med. Chem. 2013, 63, 474–483. [Google Scholar] [CrossRef]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Scheidt, K.A. Total synthesis and structural revision of the marine macrolide neopeltolide. J. Am. Chem. Soc. 2008, 130, 804–805. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Hines, J.; Crews, C.M.; Scheidt, K.A. Total synthesis and structure—Activity investigation of the marine natural product neopeltolide. J. Am. Chem. Soc. 2009, 131, 12406–12414. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Janjic, J.; Suzuki, M.; Sabharwal, S.S.; Schumacker, P.T.; Kron, S.J.; Kozmin, S.A. Synthesis enables identification of the cellular target of leucascandrolide A and neopeltolide. Nat. Chem. Biol. 2008, 4, 418. [Google Scholar] [CrossRef]
- Zhu, X.L.; Z’hang, R.; Wu, Q.; Song, Y.; Wang, Y.; Yang, J.; Yang, G. Natural Product Neopeltolide as A Cytochrome bc1 Complex Inhibitor: Mechanism of Action and Structural Modification. J. Agric. Food Chem. 2019, 67, 2774–2781. [Google Scholar] [CrossRef]
- Vintonyak, V.V.; Kunze, B.; Sasse, F.; Maier, M.E. Total synthesis and biological activity of neopeltolide and analogues. Chem. Eur. J. 2008, 14, 11132–11140. [Google Scholar] [CrossRef]
- Xiong, L.; Li, H.; Jiang, L.; Ge, J.; Yang, W.; Zhu, X.L.; Yang, G. Structure-based discovery of potential fungicides as succinate ubiquinone oxidoreductase inhibitors. J. Agric. Food Chem. 2017, 65, 1021–1029. [Google Scholar] [CrossRef]
- Moulin, E.; Nevado, C.; Gagnepain, J.; Kelter, G.; Fiebig, H.; Fürstner, A. Synthesis and evaluation of an Iejimalide-archazolid chimera. Tetrahedron 2010, 66, 6421–6428. [Google Scholar] [CrossRef]
- Kobayashi, J.; Cheng, J.; Ohta, T.; Nakamura, H.; Nozoe, S.; Hirata, Y.; Ohizumi, Y.; Sasaki, T. Iejimalides A and B, novel 24-membered macrolides with potent antileukemic activity from the Okinawan tunicate Eudistoma cf. rigida. J. Org. Chem. 1988, 53, 6147–6150. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Ishibashi, M.; Sasaki, T.; Kobayashi, J. Iejimalides C and D, new antineoplastic 24-membered macrolide sulfates from the Okinawan marine tunicate Eudistoma cf. rigida. Tetrahedron Lett. 1991, 32, 797–798. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Waser, M. Total synthesis of iejimalide B. Angew. Chem. Int. Edit. 2006, 45, 5837–5842. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Waser, M.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Moulin, E.; Müller, O. Total Synthesis of Iejimalide A–D and Assessment of the Remarkable Actin-Depolymerizing Capacity of These Polyene Macrolides. J. Am. Chem. Soc. 2007, 129, 9150–9161. [Google Scholar] [CrossRef]
- Sasse, F.; Steinmetz, H.; Hoefle, G.; Reichenbach, H. Archazolids, New Cytotoxic Macrolactones from Archangium gephyra (Myxobacteria). J. Antibiot. 2003, 56, 520–525. [Google Scholar] [CrossRef]
- Corey, E.J.; Helal, C.J. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: A new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Edit. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
- Marshall, J.A. Chiral allylic and allenic metal reagents for organic synthesis. J. Org. Chem. 2007, 72, 8153–8166. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Ogba, O.; Warner, N.; O’Leary, D.; Grubbs, R. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, O.H.; Nordlund, H.R.; Hytönen, V.P.; Kulomaa, M.S. Brave new (strept) avidins in biotechnology. Trends Biotechnol. 2007, 25, 269–277. [Google Scholar] [CrossRef]
- Dundas, C.M.; Demonte, D.; Park, S. Streptavidin–biotin technology: Improvements and innovations in chemical and biological applications. Appl. Microbiol. Biotechnol. 2013, 97, 9343–9353. [Google Scholar] [CrossRef]
- Kim, J.; Cantor, A.B.; Orkin, S.H.; Wang, J. Use of in vivo biotinylation to study protein–protein and protein–DNA interactions in mouse embryonic stem cells. Nat. Protoc. 2009, 4, 506–517. [Google Scholar] [CrossRef]
- Manz, B.; Heubner, A.; Kohler, I.; Grill, H.; Pollow, K. Synthesis of Biotin-Labelled Dexamethasone Derivatives: Novel Hormone-Affinity Probes. Eur. J. Biochem. 1983, 131, 333–338. [Google Scholar] [CrossRef]
- Wright, M.; Sieber, S. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708. [Google Scholar] [CrossRef] [Green Version]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of direct protein targets of small molecules. ACS Chem. Biol. 2010, 6, 34–46. [Google Scholar] [CrossRef]
- Sato, S.; Murata, A.; Shirakawa, T.; Uesugi, M. Biochemical target isolation for novices: Affinity-based strategies. Chem. Biol. 2010, 17, 616–623. [Google Scholar] [CrossRef]
- Gouiffes, D.; Moreau, S.; Helbecque, N.; Bernier, J.; Henichart, J.; Barbin, Y.; Laurent, D.; Verbist, J. Proton nuclear magnetic study of bistramide A, a new cytotoxic drug isolated from Lissoclinum bistratum Sluiter. Tetrahedron 1988, 44, 451–459. [Google Scholar] [CrossRef]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; McCaffrey, E.J.; Parry, D.L.; Watters, D.J. Novel cytotoxic compounds from the ascidian Lissoclinum bistratum. J. Med. Chem. 1989, 32, 1354–1359. [Google Scholar] [CrossRef]
- Biard, J.; Roussakis, C.; Kornprobst, J.; Gouiffes-Barbin, D.; Verbist, J.; Cotelle, P.; Foster, M.P.; Ireland, C.M.; Debitus, C. Bistramides A, B, C, D, and K: A new class of bioactive cyclic polyethers from Lissoclinum bistratum. J. Nat. Prod. 1994, 57, 1336–1345. [Google Scholar] [CrossRef]
- Riou, D.; Roussakis, C.; Biard, J.F.; Verbist, J.F. Comparative study of the antitumor activity of bistramides A, D and K against a non-small cell broncho-pulmonary carcinoma. Anticancer Res. 1993, 13, 2331–2334. [Google Scholar]
- Wipf, P.; Uto, Y.; Yoshimura, S. Total synthesis of a stereoisomer of bistramide C and assignment of configuration of the natural product. Chem. Eur. J. 2002, 8, 1670–1681. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Liu, D.; Kozmin, S.A. Synthesis of bistramide A. J. Am. Chem. Soc. 2004, 126, 9546–9547. [Google Scholar] [CrossRef]
- Gagne, M.R.; Grubbs, R.H.; Feldman, J.; Ziller, J.W. Catalytic activity of a well-defined binuclear ruthenium alkylidene complex. Organometallics 1992, 11, 3933–3935. [Google Scholar] [CrossRef]
- Foster, M.P.; Mayne, C.L.; Dunkel, R.; Pugmire, R.J.; Grant, D.M.; Kornprobst, J.M.; Verbist, J.F.; Biard, J.F.; Ireland, C.M. Revised structure of bistramide A (bistratene A): Application of a new program for the automated analysis of 2D INADEQUATE spectra. J. Am. Chem. Soc. 1992, 114, 1110–1111. [Google Scholar] [CrossRef]
- Johnson, W.E.B.; Watters, D.J.; Suniara, R.K.; Brown, G.; Bunce, C.M. Bistratene A induces a microtubule-dependent block in cytokinesis and altered stathmin expression in HL60 cells. Biochem. Biophys. Res. Commun. 1999, 260, 80–88. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin is the primary cellular receptor of bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef]
- Sun, H.; Low, K.E.; Woo, S.; Noble, R.L.; Graham, R.J.; Connaughton, S.S.; Gee, M.A.; Lee, L.G. Real-time protein kinase assay. Anal. Chem. 2005, 77, 2043–2049. [Google Scholar] [CrossRef]
- Yamada, K.; Ojika, M.; Ishigaki, T.; Yoshida, Y.; Ekimoto, H.; Arakawa, M. Aplyronine A, a potent antitumor substance and the congeners aplyronines B and C isolated from the sea hare Aplysia kurodai. J. Am. Chem. Soc. 1993, 115, 11020–11021. [Google Scholar] [CrossRef]
- Kita, M.; Hirayama, Y.; Yoneda, K.; Yamagishi, K.; Chinen, T.; Usui, T.; Sumiya, E.; Uesugi, M.; Kigoshi, H. Inhibition of microtubule assembly by a complex of actin and antitumor macrolide aplyronine A. J. Am. Chem. Soc. 2013, 135, 18089–18095. [Google Scholar] [CrossRef]
- Paterson, I.; Fink, S.J.; Lee, L.Y.; Atkinson, S.J.; Blakey, S.B. Total synthesis of aplyronine C. Org. Lett. 2013, 15, 3118–3121. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; Van Duyne, G.D.; Clardy, J. Isolation and structure determination of diazonamides A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991, 113, 2303–2304. [Google Scholar] [CrossRef]
- Li, J.; Jeong, S.; Esser, L.; Harran, P.G. Total synthesis of nominal Diazonamides—Part 1: Convergent preparation of the structure proposed for (−)-Diazonamide A. Angew. Chem. Int. Edit. 2001, 40, 4765–4769. [Google Scholar] [CrossRef]
- Li, J.; Burgett, A.W.; Esser, L.; Amezcua, C.; Harran, P.G. Total synthesis of nominal diazonamides—Part 2: On the true structure and origin of natural isolates. Angew. Chem. Int. Edit. 2001, 40, 4770–4773. [Google Scholar] [CrossRef]
- Nicolaou, K.; Bella, M.; Chen, D.Y.; Huang, X.; Ling, T.; Snyder, S.A. Total synthesis of diazonamide A. Angew. Chem. Int. Edit. 2002, 41, 3495–3499. [Google Scholar] [CrossRef]
- Wang, G.; Shang, L.; Burgett, A.W.; Harran, P.G.; Wang, X. Diazonamide toxins reveal an unexpected function for ornithine δ-amino transferase in mitotic cell division. Proc. Natl. Acad. Sci. USA 2007, 104, 2068–2073. [Google Scholar] [CrossRef]
- Seiler, N. Ornithine aminotransferase, a potential target for the treatment of hyperammonemias. Curr. Drug Targets 2000, 1, 119–154. [Google Scholar] [CrossRef]
- Wieczorek, M.; Tcherkezian, J.; Bernier, C.; Prota, A.E.; Chaaban, S.; Rolland, Y.; Godbout, C.; Hancock, M.A.; Arezzo, J.C.; Ocal, O. The synthetic diazonamide DZ-2384 has distinct effects on microtubule curvature and dynamics without neurotoxicity. Sci. Transl. Med. 2016, 8, 365ra159. [Google Scholar] [CrossRef]
- Yun, H.; Sim, J.; An, H.; Lee, J.; Lee, H.S.; Shin, Y.K.; Paek, S.; Suh, Y. Design and synthesis of a macrosphelide A-biotin chimera. Org. Biomol. Chem. 2014, 12, 7127–7135. [Google Scholar] [CrossRef]
- Paek, S.; Seo, S.; Kim, S.; Jung, J.; Lee, Y.; Jung, J.; Suh, Y. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Faircloth, G.; Marchante, M.d.C.C. Kahalalide F and ES285: Potent anticancer agents from marine molluscs. In Molluscs; Springer: Berlin/Heidelberg, Germany, 2006; pp. 363–379. [Google Scholar]
- Piggott, A.M.; Karuso, P. Rapid identification of a protein binding partner for the marine natural product kahalalide F by using reverse chemical proteomics. ChemBioChem 2008, 9, 524–530. [Google Scholar] [CrossRef]
- Wulff, J.E.; Siegrist, R.; Myers, A.G. The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J. Am. Chem. Soc. 2007, 129, 14444–14451. [Google Scholar] [CrossRef]
- Sugie, Y.; Hirai, H.; Inagaki, T.; Ishiguro, M.; Kim, Y.; Kojima, Y.; Sakakibara, T.; Sakemi, S.; Sugiura, A.; Suzuki, Y. A new antibiotic CJ-17, 665 from Aspergillus ochraceus. J. Antibiot. 2001, 54, 911–916. [Google Scholar] [CrossRef]
- Herzon, S.B.; Myers, A.G. Enantioselective synthesis of stephacidin B. J. Am. Chem. Soc. 2005, 127, 5342–5344. [Google Scholar] [CrossRef]
- Wulff, J.E.; Herzon, S.B.; Siegrist, R.; Myers, A.G. Evidence for the rapid conversion of stephacidin B into the electrophilic monomer avrainvillamide in cell culture. J. Am. Chem. Soc. 2007, 129, 4898–4899. [Google Scholar] [CrossRef]
- Chan, W.Y.; Liu, Q.R.; Borjigin, J.; Busch, H.; Rennert, O.M.; Tease, L.A.; Chan, P.K. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 1989, 28, 1033–1039. [Google Scholar] [CrossRef]
- Myers, A.G.; Herzon, S.B.; Wulff, J.E.; Siegrist, R.; Svenda, J.; Zajac, M.A. Synthesis of Avrainvillamide, Stephacidin B, and Analogues Thereof. U.S. Patent 20110166170A1, 1 July 2011. [Google Scholar]
- Northcote, P.T.; Blunt, J.W.; Munro, M.H. Pateamine: A potent cytotoxin from the New Zealand marine sponge, Mycale sp. Tetrahedron Lett. 1991, 32, 6411–6414. [Google Scholar] [CrossRef]
- Romo, D.; Rzasa, R.M.; Shea, H.A.; Park, K.; Langenhan, J.M.; Sun, L.; Akhiezer, A.; Liu, J.O. Total synthesis and immunosuppressive activity of (−)-pateamine A and related compounds: Implementation of a β-lactam-based macrocyclization. J. Am. Chem. Soc. 1998, 120, 12237–12254. [Google Scholar] [CrossRef]
- Low, W.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef]
- Low, W.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Rzasa, R.M.; Shea, H.A.; Li, S.; Park, K.; Ma, G. Isolation and identification of eukaryotic initiation factor 4A as a molecular target for the marine natural product Pateamine A. Meth. Enzymol. 2007, 431, 303–324. [Google Scholar]
- Low, W.; Dang, Y.; Bhat, S.; Romo, D.; Liu, J.O. Substrate-dependent targeting of eukaryotic translation initiation factor 4A by pateamine A: Negation of domain-linker regulation of activity. Chem. Biol. 2007, 14, 715–727. [Google Scholar] [CrossRef]
Figure 1.
Structure of certain bioactive marine natural products.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 3.
Comparison of the combination and chimera strategies.

Scheme 1.
Hybridization of macrosphelide (MS) skeleton and epothilone side chain [44].
Scheme 1.
Hybridization of macrosphelide (MS) skeleton and epothilone side chain [44].

Scheme 2.
Hybridization of discodermolide 12 with side chain moiety of paclitaxel 13 [47].
Scheme 2.
Hybridization of discodermolide 12 with side chain moiety of paclitaxel 13 [47].

Scheme 3.
Structural comparison and hybridization of discodermolide 12 with dictyostatin 17 [48].
Scheme 3.
Structural comparison and hybridization of discodermolide 12 with dictyostatin 17 [48].

Scheme 4.
Hybridization of psymberin 22 with pederin 23 [59].
Scheme 4.
Hybridization of psymberin 22 with pederin 23 [59].

Scheme 5.
Hybridization from topsentin 30, dendrodoine 31, and curcumin 1 33 [64].
Scheme 5.
Hybridization from topsentin 30, dendrodoine 31, and curcumin 1 33 [64].

Scheme 6.
Hybridization from neopeltolide and biaryl ether hybrid [71].
Scheme 6.
Hybridization from neopeltolide and biaryl ether hybrid [71].

Figure 4.
Design of iejimalide B and archazolid chimeras [72].
Figure 4.
Design of iejimalide B and archazolid chimeras [72].

Scheme 7.
Synthesis of iejimalide-archazolid chimera 44 [72].
Scheme 7.
Synthesis of iejimalide-archazolid chimera 44 [72].

Scheme 8.
Total synthesis of bistramide A 52 [94].
Scheme 8.
Total synthesis of bistramide A 52 [94].

Figure 5.
Biotin chimera based on bistramide A [98].
Figure 5.
Biotin chimera based on bistramide A [98].

Scheme 9.
Preparation of aplyronine-biotin-diazine chimera 62 [101].
Scheme 9.
Preparation of aplyronine-biotin-diazine chimera 62 [101].

Figure 6.
Preparation of biotinylated diazonamide and its advanced analog DZ2384 66 [107].
Figure 6.
Preparation of biotinylated diazonamide and its advanced analog DZ2384 66 [107].

Scheme 10.
Preparation of biotinylated marcosphelide chimera 74 [110].
Scheme 10.
Preparation of biotinylated marcosphelide chimera 74 [110].

Scheme 11.
Preparation of Kahalalide F-biotin chimera 77 [114].
Scheme 11.
Preparation of Kahalalide F-biotin chimera 77 [114].

Scheme 12.
Preparation of avrainvillamide-biotin chimera 79 [115].
Scheme 12.
Preparation of avrainvillamide-biotin chimera 79 [115].

Scheme 13.
Preparation of pateamine A-biotin chimera 87 [124].
Scheme 13.
Preparation of pateamine A-biotin chimera 87 [124].

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ha, M.W.; Song, B.R.; Chung, H.J.; Paek, S.-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Mar. Drugs 2019, 17, 500. https://doi.org/10.3390/md17090500
AMA Style
Ha MW, Song BR, Chung HJ, Paek S-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Marine Drugs. 2019; 17(9):500. https://doi.org/10.3390/md17090500
Chicago/Turabian StyleHa, Min Woo, Bo Reum Song, Hye Jin Chung, and Seung-Mann Paek. 2019. "Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products" Marine Drugs 17, no. 9: 500. https://doi.org/10.3390/md17090500
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.