
Combination of Fish Oil and Selenium Enhances Anticancer Efficacy and Targets Multiple Signaling Pathways in Anti-VEGF Agent Treated-TNBC Tumor-Bearing Mice

Abstract
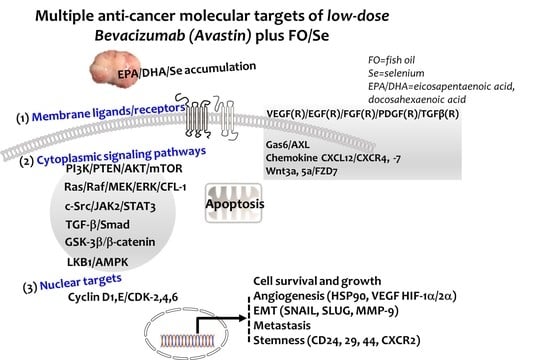
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
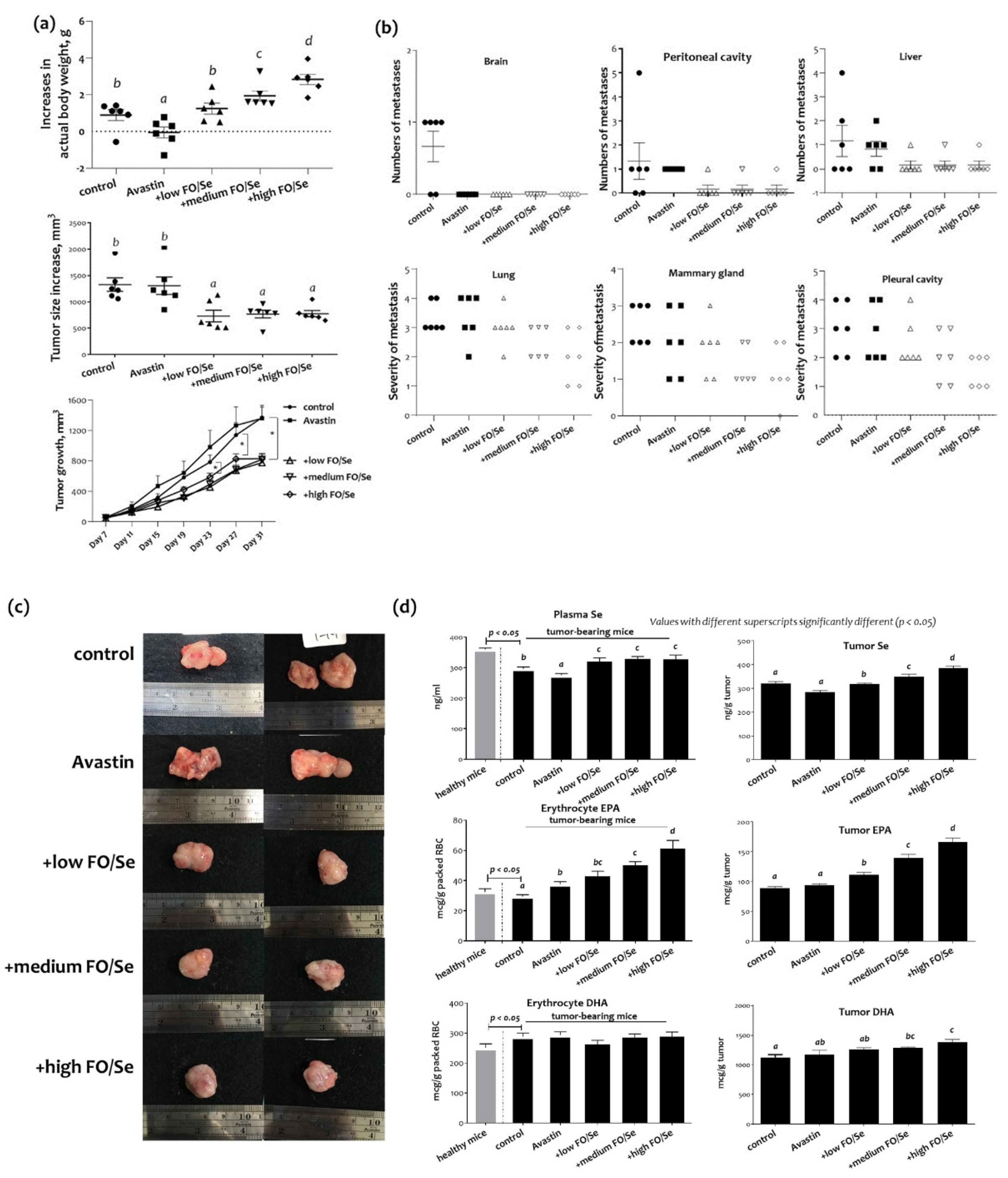
2.1. Body Weight, Tumor Weight, and Subcutaneous Tumor Size
2.2. Distant Metastatic Profiles
2.3. Tumor and Plasma Concentrations of Se and EPA/DHA
2.4. Protein Levels of Tumor Selenoproteins
2.5. HSP90/HIFs/COX-2/SOD-1 and MMP-9 in Tumors
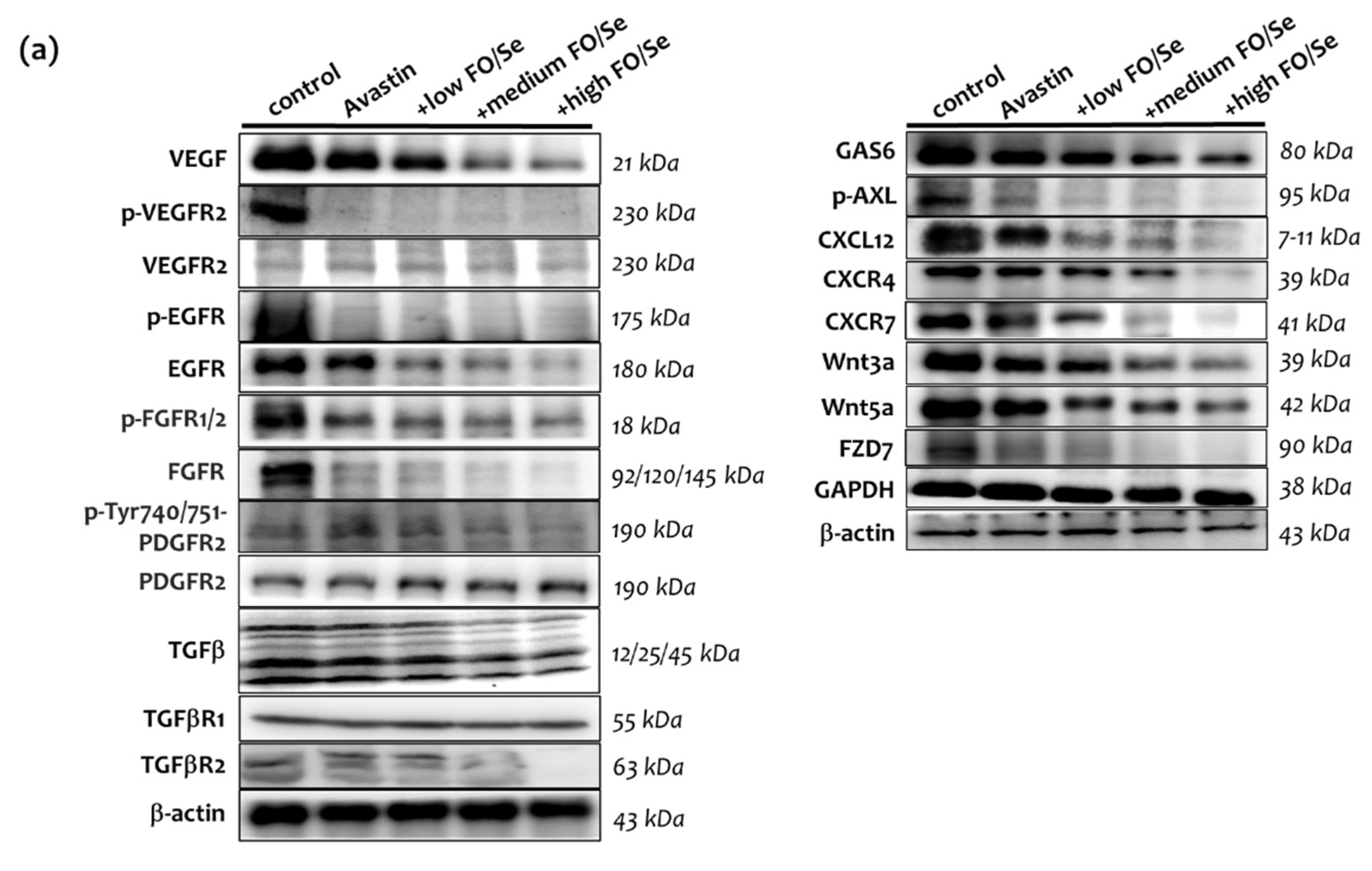
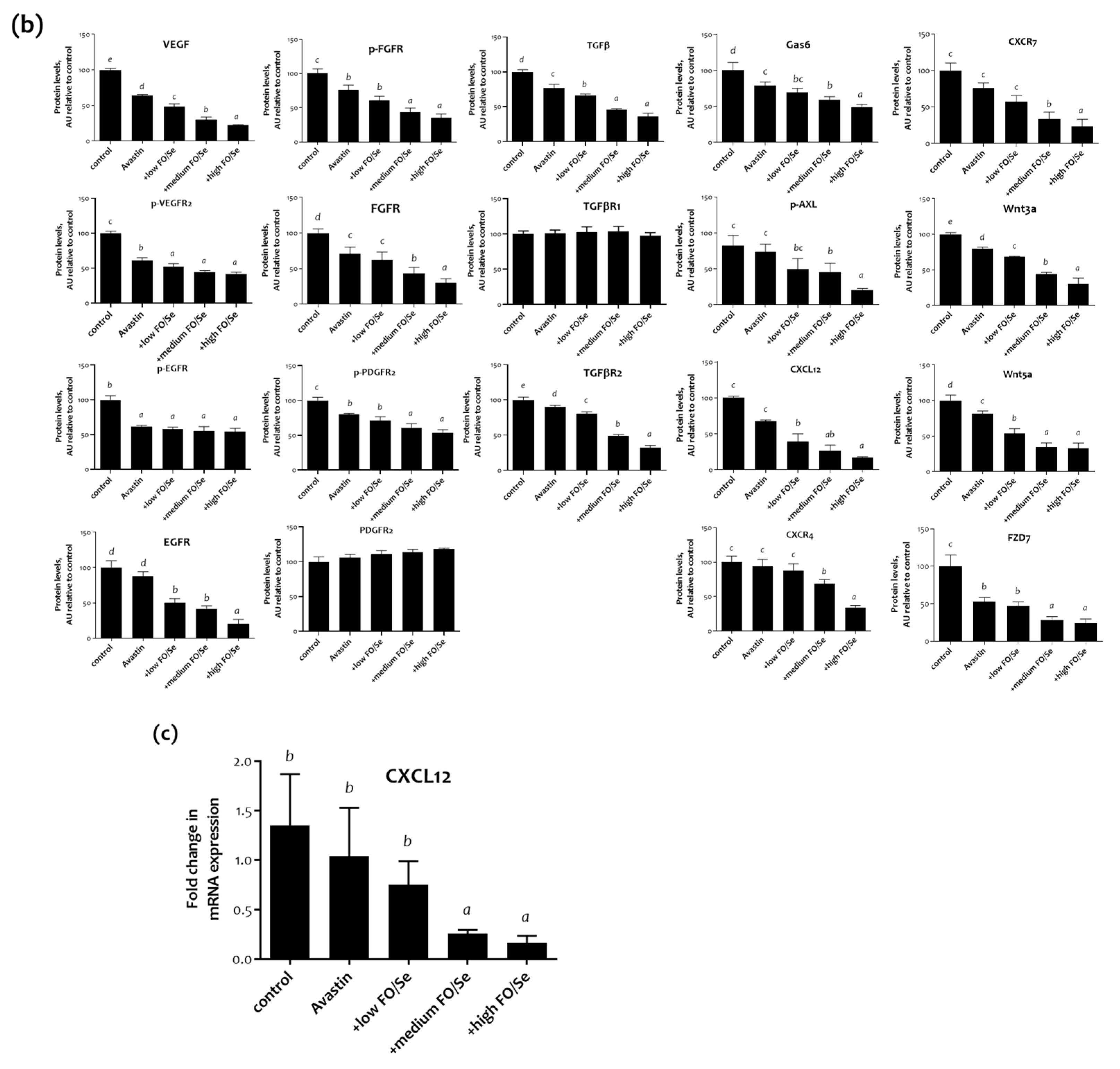
2.6. Pro-Angiogenic (Growth) Factors and Their Receptors
2.6.1. VEGF(R)/EGF(R)/FGF(R)/PDGF(R)/TGFβ(R)
2.6.2. Gas6/AXL Axis
2.6.3. Chemokine CXCL12/CXCR4, -7 Axis
2.6.4. Wnt3a, 5a/FZD7 Axis
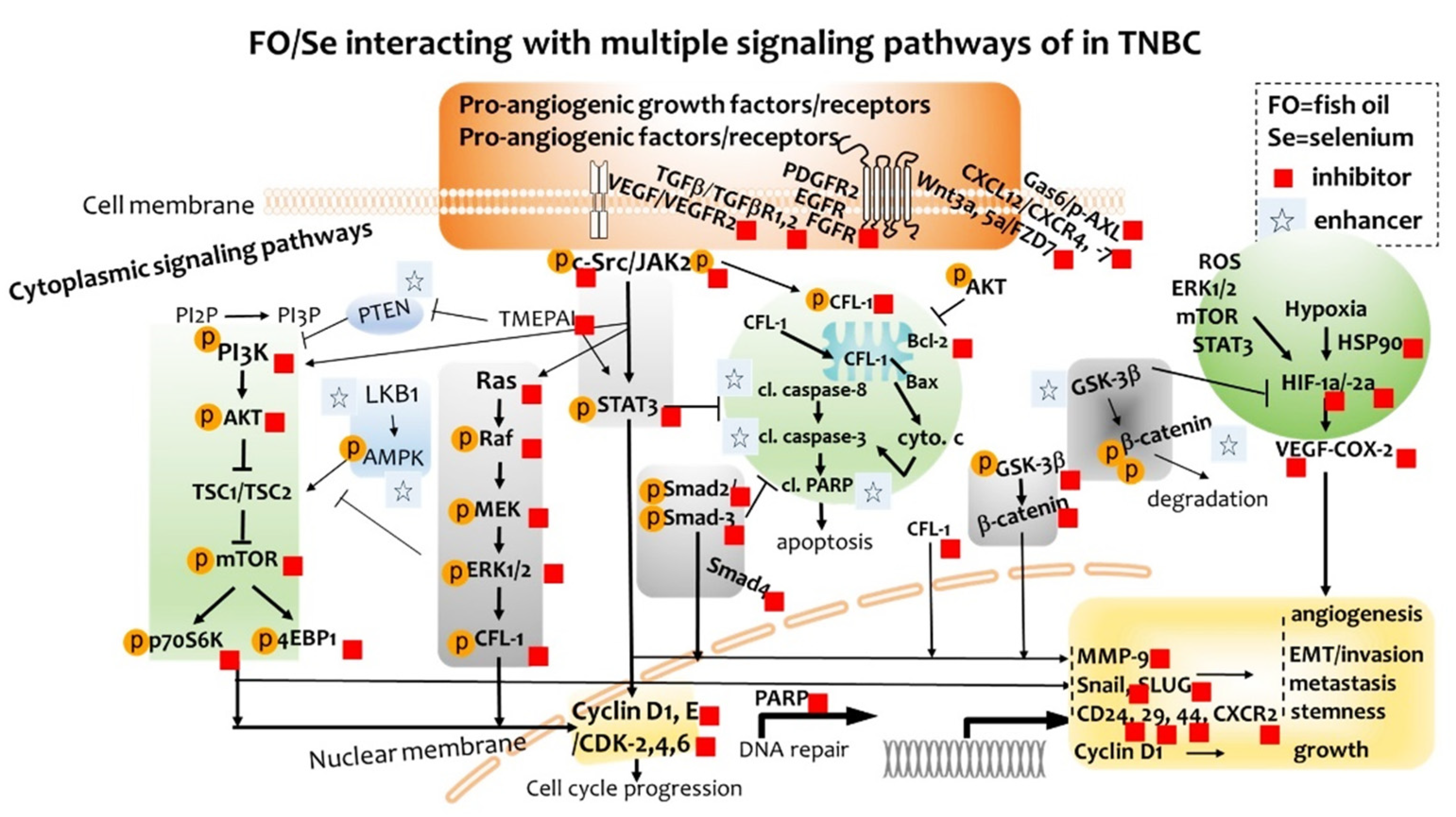
2.7. Cytoplasmic Signaling Pathways
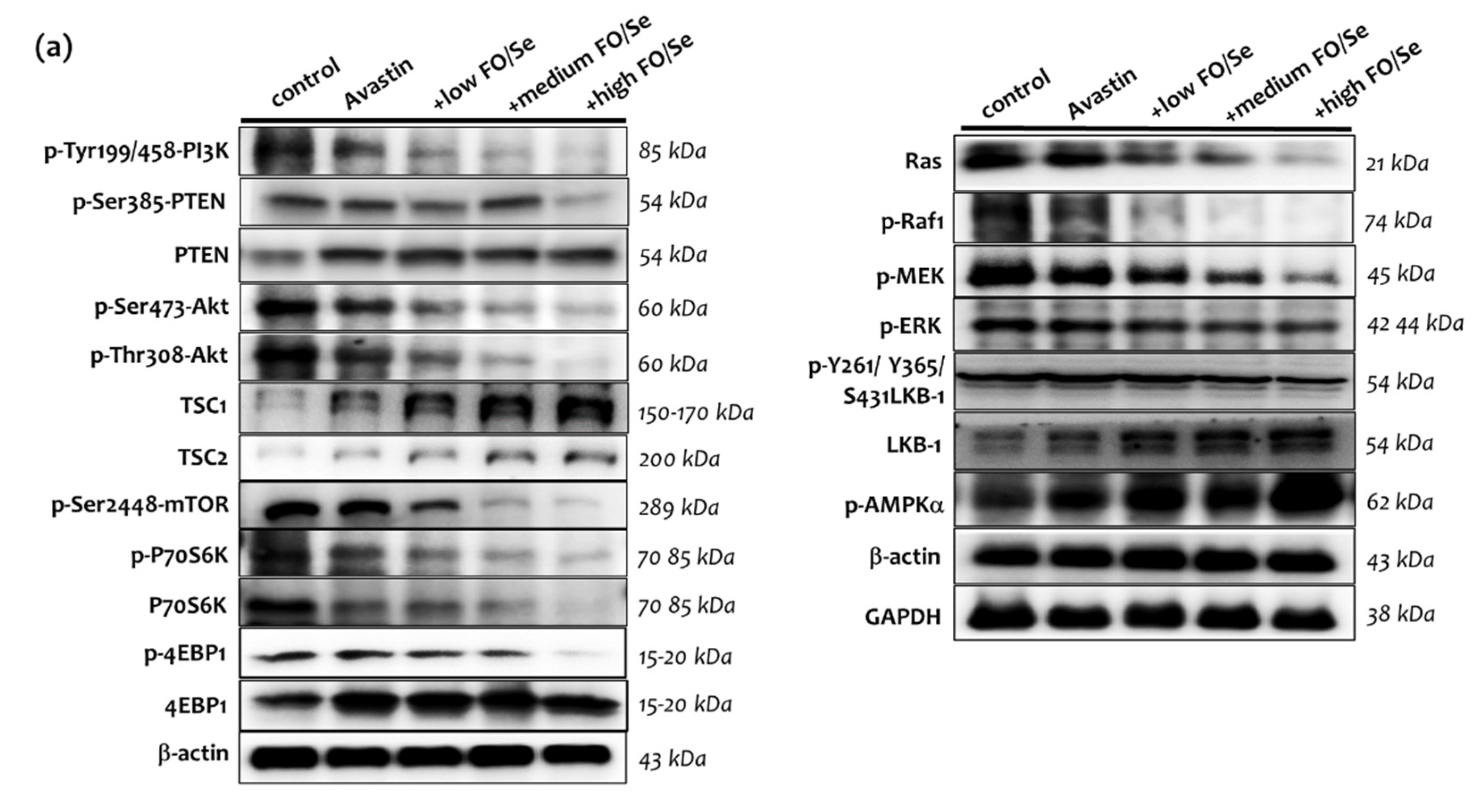
2.7.1. PI3K-PTEN-AKT-TSC1/TSC2-mTOR Axis
2.7.2. Ras-Raf-MEK-ERK, and LKB1-AMPK Pathway
2.7.3. TGFβ-Smad Pathway
2.7.4. c-Src-JAK2-STAT3, and GSK3β-β-Catenin Signaling Pathway
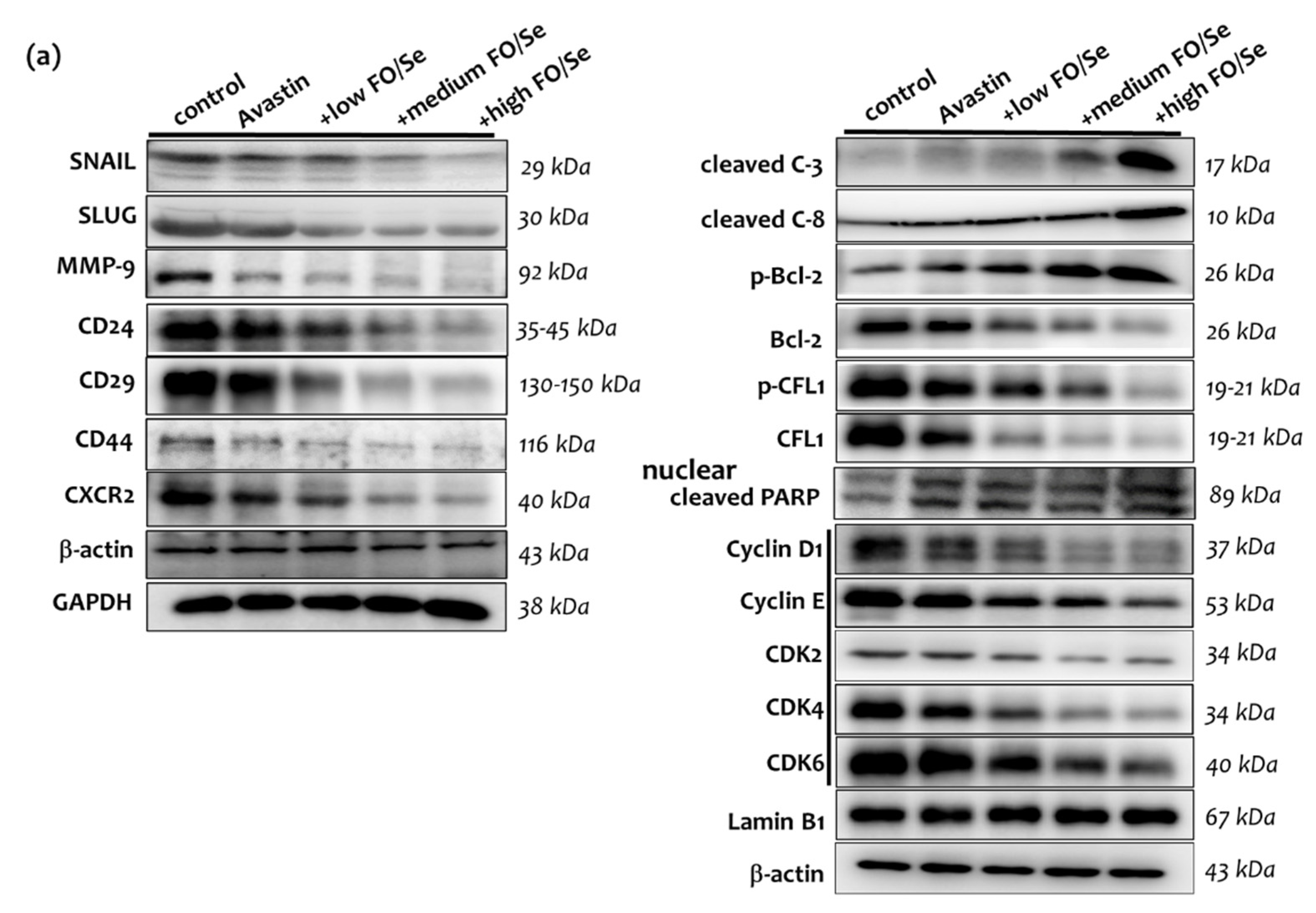
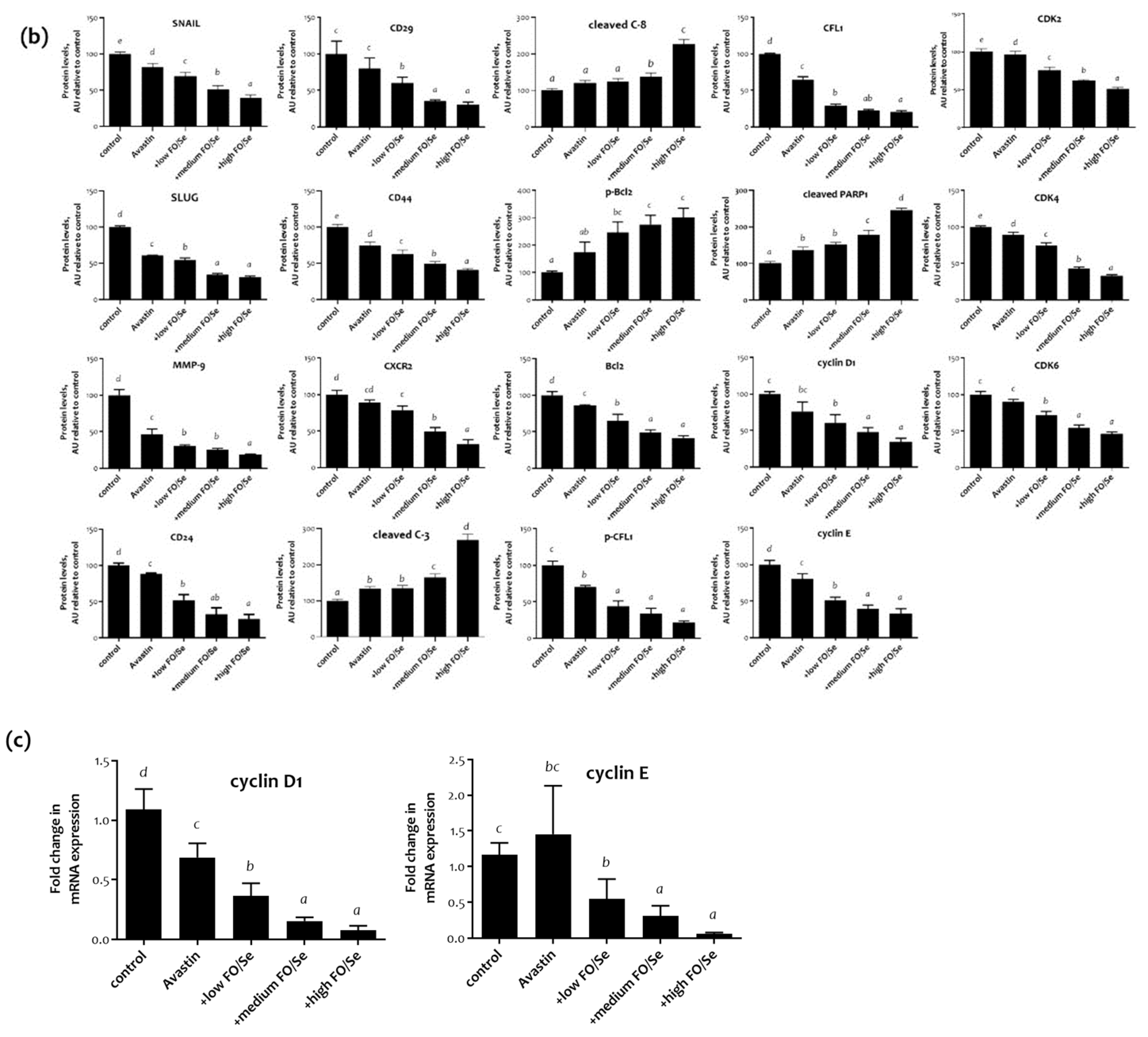
2.8. EMT-Activated Transcription Factors, and Nuclear Cyclin/Cyclin-Dependent Kinases
2.9. Tumor PARP-1, Caspases, Bcl-2, and CFL-1
2.10. CSCs Markers
3. Discussion
3.1. EPA/DHA Levels in TNBC
3.2. Se Accumulation and Selenoprotein Expression in TNBC
3.3. FO/Se and Pro-Angiogenic Growth Factors in TNBC
3.4. FO/Se and Pro-Angiogenic Factors in TNBC
3.5. FO/Se and PI3K-PTEN-AKT-mTOR Signaling in TNBC
3.6. FO/Se and Ras-Raf-MEK-ERK Signaling in TNBC
3.7. FO/Se and TGFβ-Smad2/3-TMEPA1 Signaling in TNBC
3.8. FO/Se and JAK2-STAT3 Signaling in TNBC
3.9. FO/Se and LKB1-AMPK Signaling in TNBC
3.10. FO/Se and GSK-3β-β-Catenin Signaling in TNBC
3.11. FO/Se and Apoptosis in TNBC
3.12. FO/Se and Cyclin-CDKs in TNBC
3.13. FO/Se and Hypoxia-HIF-COX-2 in TNBC
3.14. FO/Se and Self-Renewal in TNBC
3.15. FO/Se and EMT in TNBC
4. Materials and Methods
4.1. Anti-Angiogenic Agent and Nutritional Supplements
4.2. Antibodies
4.3. Cell Culture and Animal Experiment
4.4. Measurement of Omega-3 Fatty Acid EPA and DHA
4.5. Determination of Se Content
4.6. Western Blot Analysis
4.7. RNA Extraction and Real-Time PCR Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Aurantoside C targets and induces apoptosis in triple negative breast cancer cells. Mar. Drugs 2018, 16, 361. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Ahn, J.H.; Kim, S.B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3 (Suppl. 1), e000357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639. [Google Scholar] [CrossRef]
- Montero, A.J.; Escobar, M.; Lopes, G.; Glück, S.; Vogel, C. Bevacizumab in the treatment of metastatic breast cancer: Friend or foe? Curr. Oncol. Rep. 2012, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Mohsena, M.A.; Abdel-Malakb, C.A.; Nassef, S.M. Anti-angiogenic targeted therapy in triple negative breast cancer HCC-1806 cell line. Int. J. Sci. Eng. Res. 2018, 9, 93. [Google Scholar]
- Ribatti, D.; Nico, B.; Ruggieri, S.; Tamma, R.; Simone, G.; Mangia, A. Angiogenesis and anti-angiogenesis in triple-negative breast cancer. Transl. Oncol. 2016, 9, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stryker, Z.I.; Rajabi, M.; Davis, P.J.; Mousa, S.A. Evaluation of angiogenesis assays. Bio Med. 2019, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.; He, S.; Ye, T.; Shen, G.; Wan, Y.; Wang, Y. Antiangiogenic therapy using sunitinib combined with rapamycin retards tumor growth but promotes metastasis. Transl. Oncol. 2014, 7, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Liao, C.-H.; Tzeng, Y.-T.; Lai, G.-M.; Chang, C.-L.; Hu, M.-H.; Tsai, W.-L.; Liu, Y.-R.; Hsia, S.; Chuang, S.-E.; Chiou, T.-J.; et al. Omega-3 fatty acid-enriched fish oil and selenium combination modulates endoplasmic reticulum stress response elements and reverses acquired gefitinib resistance in HCC827 lung adenocarcinoma cells. Mar. Drugs. 2020, 18, 399. [Google Scholar] [CrossRef]
- Liu, J.; Ma, D.W. The role of n-3 polyunsaturated fatty acids in the prevention and treatment of breast cancer. Nutrients 2014, 6, 5184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander, S.L.; Mazurak, V.C.; Damaraju, S.; Field, C.J. Determination of the relative efficacy of eicosapentaenoic acid and docosahexaenoic acid for anti-cancer effects in human breast cancer models. Int. J. Mol. Sci. 2017, 18, 2607. [Google Scholar]
- Choi, R.; Kim, M.-J.; Sohn, I.; Kim, S.; Kim, I.; Ryu, J.M.; Choi, H.J.; Kim, J.-M.; Lee, S.K.; Yu, J.; et al. Serum trace elements and their associations with breast cancer subgroups in Korean breast cancer patients. Nutrients 2018, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.H.; Hsia, S.; Hsiung, D.Y.; Chen, P.C. Supplementation with selenium yeast on the prooxidant-antioxidant activities and anti-tumor effects in breast tumor xenograft-bearing mice. Int. J. Med. Sci. 2015, 12, 748. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.H.; Hsia, S.; Chung, C.H.; Lin, Y.C.; Shih, M.Y.; Chen, P.C.; Peng, C.L.; Henning, S.M.; Hsu, G.S.W.; Li, Z. Nutritional supplements in combination with chemotherapy or targeted therapy reduces tumor progression in mice bearing triple-negative breast cancer. J. Nutr. Biochem. 2021, 87, 108504. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.H.; Liu, P.J.; Lin, K.P.; Chen, P.C. Nutritional supplement therapy improves oxidative stress, immune response, pulmonary function, and quality of life in allergic asthma patients- An open-label pilot study. Altern. Med. Rev. 2012, 17, 42. [Google Scholar]
- Yu, H.M. Bypassing the D6-desaturase enzyme and directly providing n-3 and n-6 PUFA pathway intermediates reduces the survival of two human breast cancer cell lines. Eur. J. Lipid Sci. Technol. 2015, 117, 1378–1390. [Google Scholar] [CrossRef]
- Serini, S.; Fasano, E.; Piccioni, E.; Cittadini, A.R.; Calviello, G. Differential anti-cancer effects of purified EPA and DHA and possible mechanisms involved. Curr. Med. Chem. 2011, 18, 4065. [Google Scholar] [CrossRef]
- Short, S.P.; Williams, C.S. Selenoproteins in tumorigenesis and cancer progression. Adv. Cancer Res. 2017, 136, 49. [Google Scholar]
- Mumin, N.H.; Drobnitzky, N.; Patel, A.; Lourenco, L.M.; Cahill, F.F.; Jiang, Y.; Kong, A.; Ryan, A.J. Overcoming acquired resistance to HSP90 inhibition by targeting JAK-STAT signalling in triple-negative breast cancer. BMC Cancer. 2019, 19, 102. [Google Scholar] [CrossRef]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967. [Google Scholar] [CrossRef]
- Alsamri, H.; El Hasasna, H.; Al Dhaheri, Y.; Eid, A.H.; Attoub, S.; Iratni, R. Carnosol, a natural polyphenol, inhibits migration, metastasis, and tumor growth of breast cancer via a ROS-dependent proteasome degradation of STAT3. Front. Oncol. 2019, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Spencer, L.; Mann, C.; Metcalfe, M.; Webb, M.; Pollard, C.; Spencer, D.; Berry, D.; Steward, W.; Dennison, A. The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. Eur. J. Cancer. 2009, 45, 2077. [Google Scholar] [CrossRef] [PubMed]
- Darwito, D.; Dharmana, E.; Riwanto, I.; Budijitno, S.; Suwardjo, S.; Purnomo, J.; Widodo, I.; Ghozali, A.; Aryandono, T.; Anwar, S.L. Effects of omega-3 supplementation on Ki-67 and VEGF expression levels and clinical outcomes of locally advanced breast cancer patients treated with neoadjuvant CAF chemotherapy: A randomized controlled trial report. Asian Pac. J. Cancer Prev. 2019, 20, 911. [Google Scholar] [CrossRef]
- Sinha, I.; Null, K.; Wolter, W.; Suckow, M.A.; King, T.; Pinto, J.T.; Sinha, R. Methylseleninic acid downregulates hypoxia-inducible factor-1α in invasive prostate cancer. Int. J. Cancer. 2012, 130, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Aït-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic activity of anti-AXL antibody against triple-negative breast cancer patient-derived xenografts and metastasis. Clin. Cancer Res. 2017, 23, 2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaludin, S.Y.N.; Azimi, I.; Davis, F.M.; Peters, A.A.; Gonda, T.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Assessment of CXC ligand 12-mediated calcium signalling and its regulators in basal-like breast cancer cells. Oncol. Lett. 2018, 15, 4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, L.; Du, C.; Xie, X.; Liu, Y.; Wang, J.; Li, Z.; Jiang, M.; Cao, D.; Yan, X.; et al. CXCR2 is a novel cancer stem-like cell marker for triple-negative breast cancer. Onco Targets Ther. 2018, 11, 5559. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Liu, Z.; Yang, G.; Gao, D.; Niu, X. MicroRNA expression profiles in rats with selenium deficiency and the possible role of the Wnt/β-catenin signaling pathway in cardiac dysfunction. Int. J. Mol. Med. 2015, 35, 143. [Google Scholar] [CrossRef]
- Song, K.-S.; Jing, K.; Kim, J.-S.; Yun, E.-J.; Shin, S.; Seo, K.-S.; Park, J.-H.; Heo, J.-Y.; Kang, J.X.; Suh, K.-S.; et al. Omega-3-polyunsaturated fatty acids suppress pancreatic cancer cell growth in vitro and in vivo via downregulation of Wnt/Beta-catenin signaling. Pancreatology 2011, 11, 574. [Google Scholar] [CrossRef] [PubMed]
- Facompre, N.D.; Sinha, I.; El-Bayoumy, K.; Pinto, J.T.; Sinha, R. Remarkable inhibition of mTOR signaling by the combination of rapamycin and 1,4-phenylenebis (methylene) seleno-cyanate in human prostate cancer cells. Int. J. Cancer 2012, 131, 2134. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, S.Y.; Kim, I.S.; Park, O.J.; Kim, Y.M. The effect of combined treatment of selenium and curcumin on Akt and mTOR regulation in Hep3B hepato-carcinoma cells. Cancer Prev. Res. 2010, 15, 285–290. [Google Scholar]
- Nagaria, T.S.; Shi, C.; Leduc, C.; Hoskin, V.; Sikdar, S.; Sangrar, W.; Greer, P.A. Combined targeting of Raf and Mek synergistically inhibits tumorigenesis in triple negative breast cancer model systems. Oncotarget 2017, 8, 80804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosalpuria, K.; Hall, C.; Krishnamurthy, S.; Lodhi, A.; Hallman, D.M.; Baraniuk, M.S.; Bhattacharyya, A.; Lucci, A. Cyclooxygenase-2 expression in non-metastatic triple-negative breast cancer patients. Mol. Clin. Oncol. 2014, 2, 845. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Zhang, J.; Zhao, J.; Mu, K.; Zhang, J.; Jin, Z.; Yu, J.; Liu, J. Precision medicine based on tumorigenic signaling pathways for triple-negative breast cancer. Oncol. Lett. 2018, 16, 4984. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Lu, G.; Li, Q.; Zhang, J.; Huang, Z.; Gao, X. Selenium modulates MMP2 expression through the TGFβ1/Smad signalling pathway in human umbilical vein endothelial cells and rabbits following lipid disturbance. J. Trace Elem. Med. Biol. 2017, 4, 59. [Google Scholar] [CrossRef]
- Andrade-Vieira, R.; Han, J.H.; Marignani, P.A. Omega-3 polyunsaturated fatty acid promotes the inhibition of glycolytic enzymes and mTOR signaling by regulating the tumor suppressor LKB1. Cancer Biol. Ther. 2013, 14, 1050. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Choi, J.H.; Nam, J.S. Targeting cancer stem cells in triple-negative breast cancer. Cancers 2019, 11, 965. [Google Scholar] [CrossRef] [Green Version]
- Singha, P.K.; Pandeswara, S.; Geng, H.; Lan, R.; Venkatachalam, M.A.; Dobi, A.; Srivastava, S.; Saikumar, P. Increased Smad3 and reduced Smad2 levels mediate the functional switch of TGF-β from growth suppressor to growth and metastasis promoter through TMEPAI/PMEPA1 in triple negative breast cancer. Genes Cancer 2019, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.P.; Lee, W.L.; Tang, Y.Q.; Yap, W.H. Honokiol: A review of its anticancer potential and mechanisms. Cancers 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Li, J.; Hao, Q.; Vadgama, J.V.; Wu, Y. AMP-activated protein kinase: A potential therapeutic target for triple-negative breast cancer. Breast Cancer Res. 2019, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.T.; Kim, Y.M.; Surh, Y.J.; Baik, H.W.; Lee, S.K.; Ha, J.; Park, O.J. Selenium regulates cyclooxygenase-2 and extracellular signal-regulated kinase signaling pathways by activating AMP-activated protein kinase in colon cancer cells. Cancer Res. 2006, 66, 10057–10063. [Google Scholar] [CrossRef] [Green Version]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöning, J.P.; Monteiro, M.; Gu, W. Drug resistance and cancer stem cells: The shared but distinct roles of hypoxia-inducible factors HIF1α and HIF2α. Clin. Exp. Pharmacol. Physiol. 2017, 44, 153. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Han, C.; Dai, Y.; Shen, M.; Wu, T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclo-oxygenase-2. Mol. Cancer Ther. 2009, 8, 3046. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jiang, S.; Tian, X.; Jiang, Y. Expression of cleaved caspase-3 predicts good chemo-therapy response but poor survival for patients with advanced primary triple-negative breast cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 4363. [Google Scholar]
- Niu, Y.; Xu, J.; Sun, T. Cyclin-dependent kinases 4/6 inhibitors in breast cancer: Current status, resistance, and combination strategies. J. Cancer 2019, 10, 5504–5517. [Google Scholar] [CrossRef]
- Alkan, Z.; Duong, F.L.; Hawkes, W.C. Selenoprotein W controls epidermal growth factor receptor surface expression, activation and degradation via receptor ubiquitination. Biochim. Biophys. Acta. 2015, 1853, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation, and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidi, A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006, 66, 6683–6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Ganther, H.; Lu, J. Monomethyl selenium--specific inhibition of MMP-2 and VEGF expression: Implications for angiogenic switch regulation. Mol. Carcinog. 2000, 29, 236. [Google Scholar] [CrossRef]
- An, S.J.; Huang, Y.S.; Chen, Z.H.; Han, J.F.; Yang, J.J.; Zhou, Q.; Xie, Z.; Yang, Y.; Yan, H.H.; Wu, Y.-L. Lower Ras expression as an independent predictor of patient outcomes in lung cancer treated with bevacizumab plus chemotherapy. Cancer Gene Ther. 2014, 21, 110. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.-C.; Deng, X.; Chen, L.; Kim, C.C.H.; Lau, S.; et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437. [Google Scholar] [CrossRef]
- O’Conor, C.J.; Chen, T.; González, I.; Cao, D.; Peng, Y. Cancer stem cells in triple-negative breast cancer: A potential target and prognostic marker. Biomark. Med. 2018, 12, 813. [Google Scholar] [CrossRef]
- Jamdade, V.S.; Sethi, N.; Mundhe, N.A.; Kumar, P.; Lahkar, M.; Sinha, N. Therapeutic targets of triple-negative breast cancer: A review. Br. J. Pharmacol. 2015, 172, 4228–4237. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.B.; Gupta, M.; Hoang, V.T.; Chakrabarty, S.; Wright, T.D.; Elliot, S.; Chopra, I.K.; Monlish, D.; Anna, K.; Burow, M.E.; et al. Novel diphenylamine analogs induce mesenchymal to epithelial transition in triple negative breast cancer. Front Oncol. 2019, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia. 2010, 15, 201. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, C.-H.; Hsia, S.; Chung, C.-H.; Lin, Y.-C.; Shih, M.-Y.; Chen, P.-C.; Hsu, G.-S.W.; Fan, C.-T.; Peng, C.-L. Combination of Fish Oil and Selenium Enhances Anticancer Efficacy and Targets Multiple Signaling Pathways in Anti-VEGF Agent Treated-TNBC Tumor-Bearing Mice. Mar. Drugs 2021, 19, 193. https://doi.org/10.3390/md19040193
Guo C-H, Hsia S, Chung C-H, Lin Y-C, Shih M-Y, Chen P-C, Hsu G-SW, Fan C-T, Peng C-L. Combination of Fish Oil and Selenium Enhances Anticancer Efficacy and Targets Multiple Signaling Pathways in Anti-VEGF Agent Treated-TNBC Tumor-Bearing Mice. Marine Drugs. 2021; 19(4):193. https://doi.org/10.3390/md19040193
Chicago/Turabian StyleGuo, Chih-Hung, Simon Hsia, Chieh-Han Chung, Yi-Chun Lin, Min-Yi Shih, Pei-Chung Chen, Guoo-Shyng W. Hsu, Ciou-Ting Fan, and Chia-Lin Peng. 2021. "Combination of Fish Oil and Selenium Enhances Anticancer Efficacy and Targets Multiple Signaling Pathways in Anti-VEGF Agent Treated-TNBC Tumor-Bearing Mice" Marine Drugs 19, no. 4: 193. https://doi.org/10.3390/md19040193