
Secondary Metabolites with α-Glucosidase Inhibitory Activity from Mangrove Endophytic Fungus Talaromyces sp. CY-3

 , , ,
, , ,
Abstract
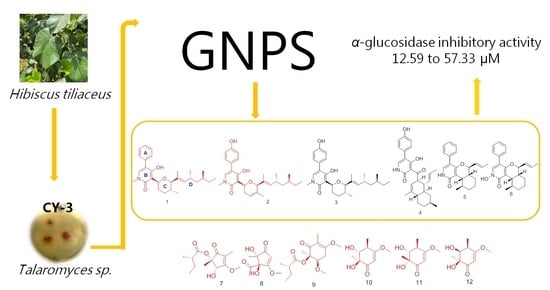
:
1. Introduction
2. Results
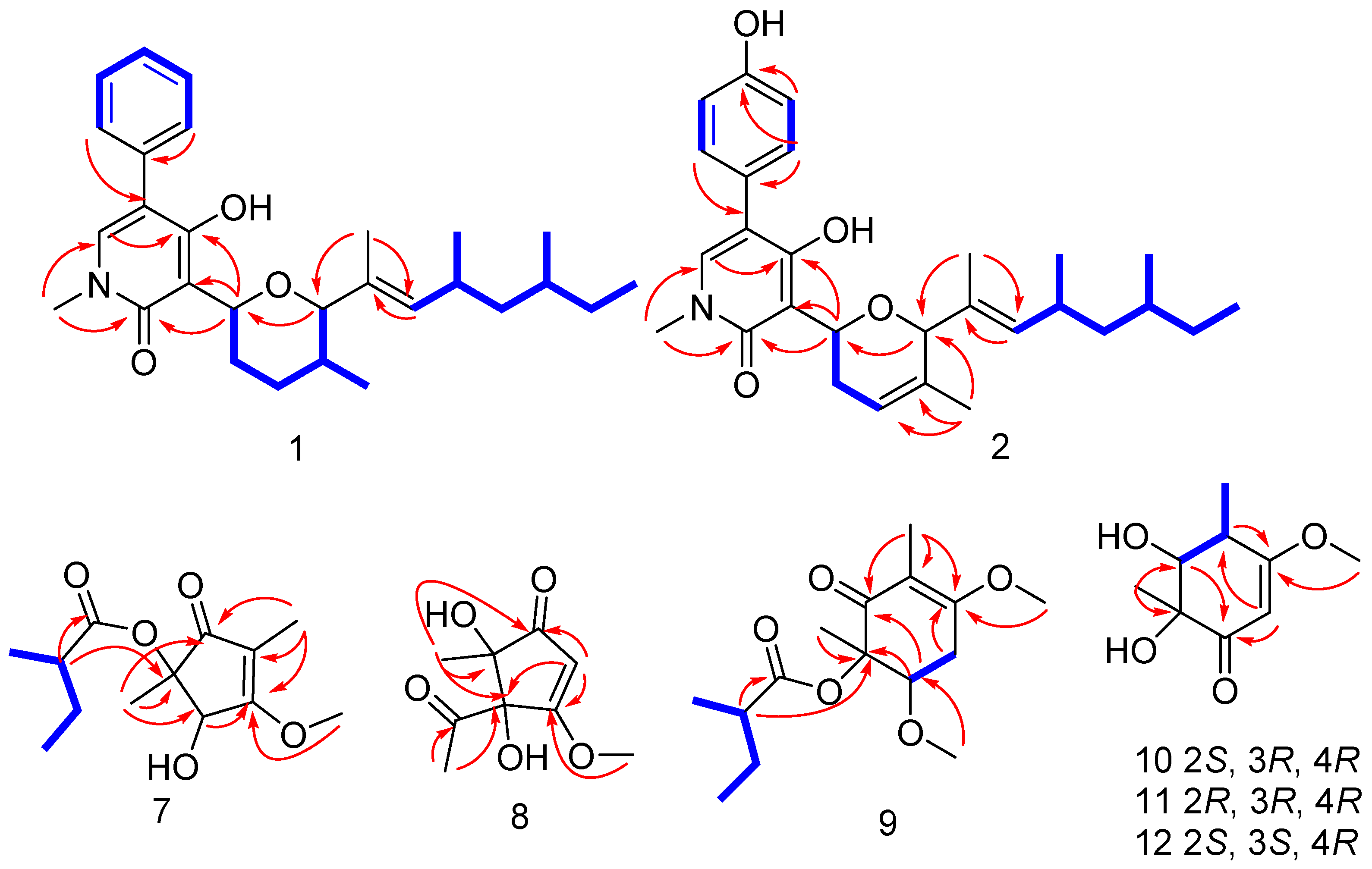
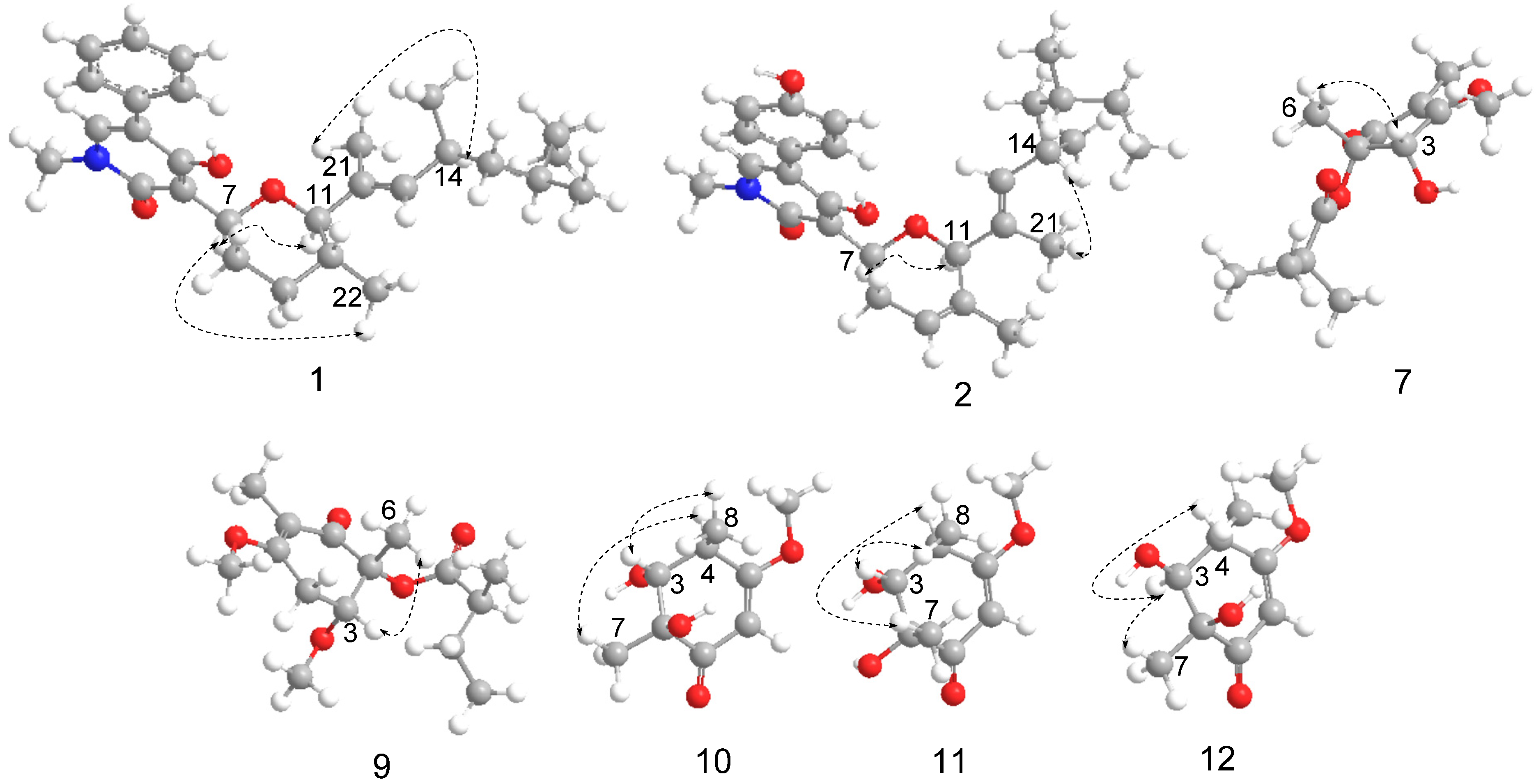
2.1. Structure Identification
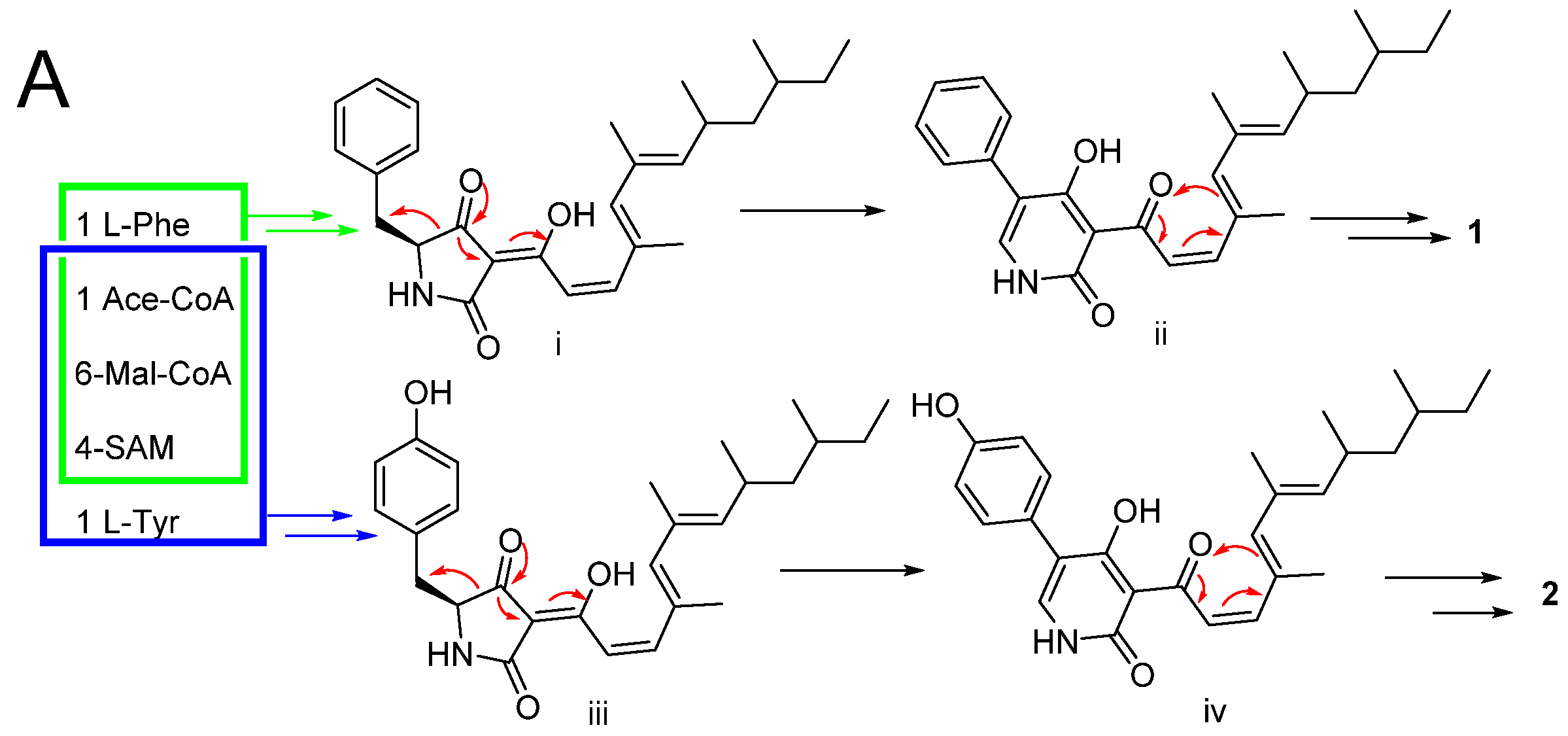
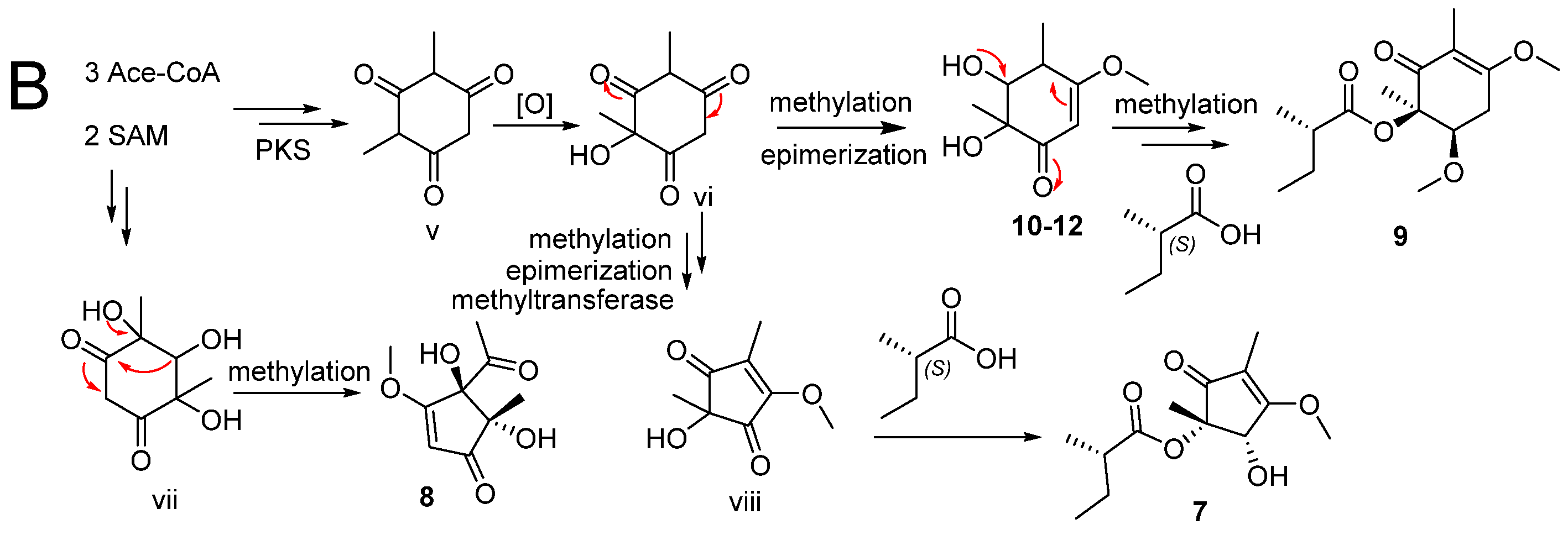
2.2. Proposed Biosynthesis Pathway
2.3. α-Glucosidase Inhibitory Activity
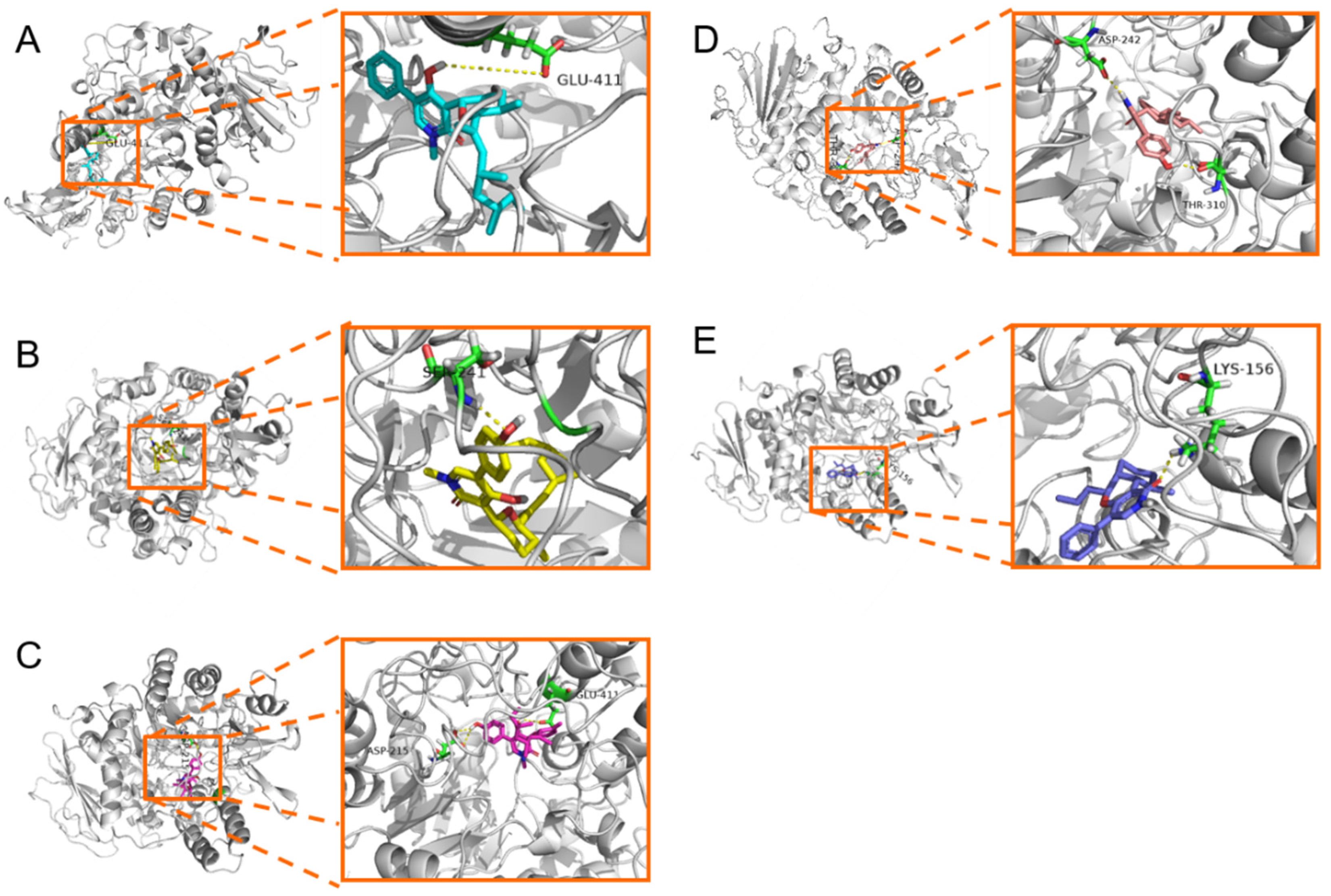
2.4. Molecular Docking Study
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation
3.4. Extraction and Purification
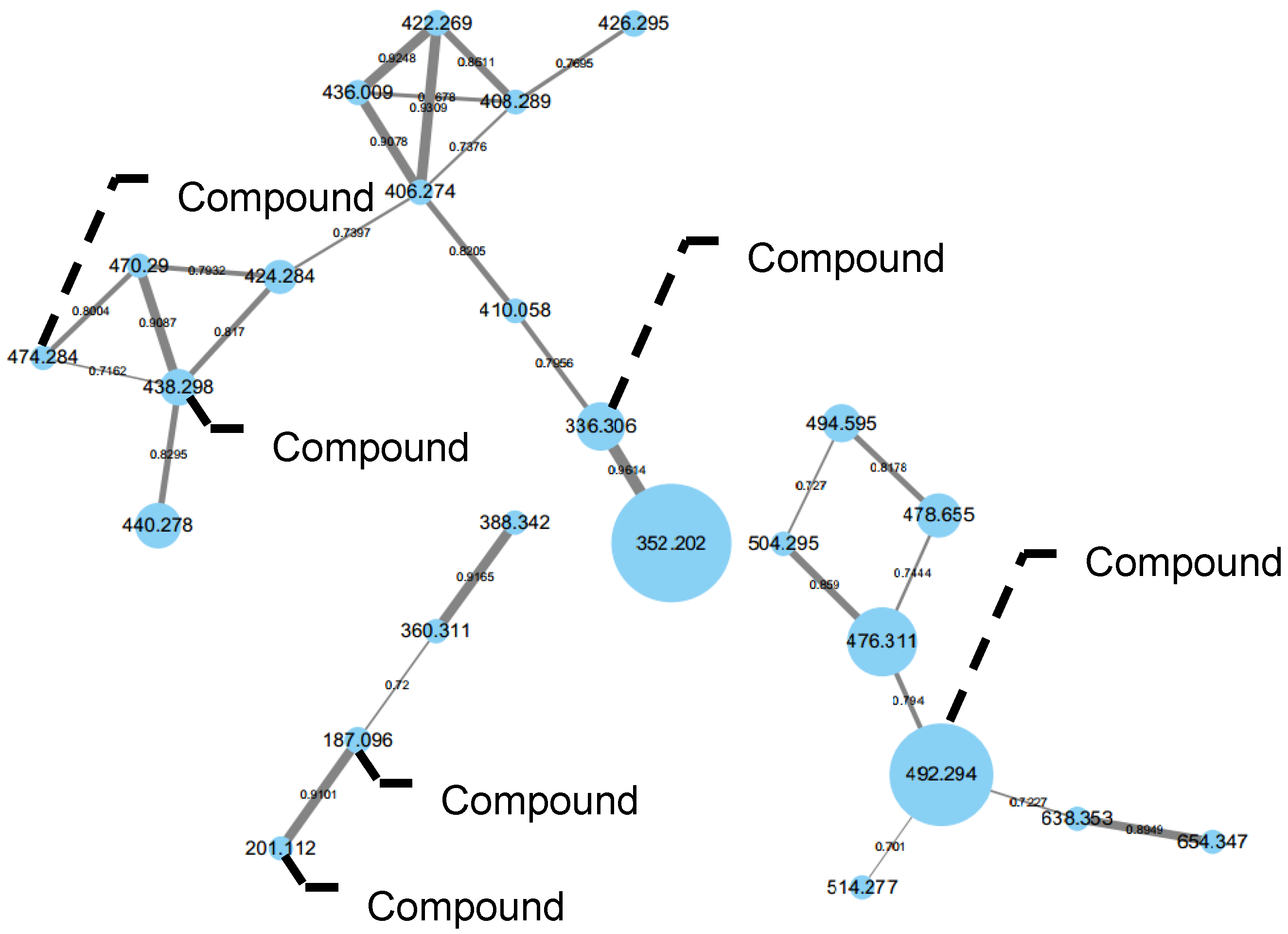
3.5. Molecular Networking
3.6. ECD and 13C NMR Calculations
3.7. Bioassay
3.8. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhameja, M.; Gupta, P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 2019, 176, 343–377. [Google Scholar] [CrossRef] [PubMed]
- Rajalakshmi, R.; Lalitha, P.; Sharma, S.C.; Rajiv, A.; Chithambharan, A.; Ponnusamy, A. In Silico studies: Physicochemical properties, drug score, toxicity predictions and molecular docking of organosulphur compounds against Diabetes mellitus. J. Mol. Recognit. 2021, e2925. [Google Scholar] [CrossRef]
- Carroll, A.R.; Coop, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Coop, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 16, 319–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Y.; Yang, Q.; Xia, G.P.; Huang, H.B.; Li, H.X.; Ma, L.; Lu, Y.J.; He, L.; Xia, X.K.; She, Z.G. Polyketides with α-glucosidase inhibitory activity from a mangrove endophytic fungus, Penicillium sp. HN29-3B1. J. Nat. Prod. 2015, 78, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chen, D.N.; Cai, R.L.; Cui, H.; Long, Y.H.; Lu, Y.J.; Li, C.Y.; She, Z.G. Cytotoxic and antibacterial preussomerins from the mangrove endophytic fungus Lasiodiplodia theobromae ZJ-HQ1. J. Nat. Prod. 2016, 79, 2397–2402. [Google Scholar] [CrossRef]
- Liang, Z.Y.; Shen, N.X.; Zheng, Y.Y.; Wu, J.T.; Miao, L.; Fu, X.M.; Chen, M.; Wang, C.Y. Two new unsaturated fatty acids from the mangrove rhizosphere soil-derived fungus Penicillium javanicum HK1-22. Bioorg. Chem. 2019, 93, 103331. [Google Scholar] [CrossRef]
- An, C.Y.; Li, X.M.; Li, C.S.; Wang, M.H.; Xu, G.M.; Wang, B.G. Aniquinazolines A-D, four new quinazolinone alkaloids from marine-derived endophytic fungus Aspergillus nidulans. Mar. Drugs 2013, 11, 2682–2694. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Z.M.; Huang, Y.; Liu, L.; He, J.G.; Wang, L.; Yuan, J.; She, Z.G. Ascomylactams A−C, cytotoxic 12- or 13-membered-ring macrocyclic alkaloids isolated from the mangrove endophytic fungus Didymella sp. CYSK-4, and structure eevisions of phomapyrrolidones A and C. J. Nat. Prod. 2019, 82, 1752–1758. [Google Scholar] [CrossRef]
- Peng, J.X.; Lin, T.; Wang, W.; Xin, Z.H.; Zhu, T.J.; Gu, Q.Q.; Li, D.H. Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013, 76, 1133–1140. [Google Scholar] [CrossRef]
- Zou, G.; Tan, Q.; Chen, Y.; Yang, W.C.; Zang, Z.M.; Jiang, H.M.; Chen, S.Y.; Wang, B.; She, Z.G. Furobenzotropolones A, B and 3-hydroxyepicoccone B with antioxidative activity from mangrove endophytic fungus Epicoccum nigrum MLY-3. Mar. Drugs 2021, 19, 395. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, W.C.; Zou, G.; Yan, Z.Y.; Qiu, P.; Long, Y.H.; She, Z.G. Metabolites with anti-inflammatory and alpha-glucosidase inhibitory activities from the mangrove endophytic fungus Phoma sp. SYSU-SK-7. Tetrahedron Lett. 2020, 61, 152578. [Google Scholar] [CrossRef]
- Cai, R.L.; Wu, Y.N.; Chen, S.H.; Cui, H.; Liu, Z.M.; Li, C.Y.; She, Z.G. Peniisocoumarins A–J: Isocoumarins from Penicillium commune QQF-3, an endophytic fungus of the mangrove plant Kandelia candel. J. Nat. Prod. 2018, 81, 1376–1383. [Google Scholar] [CrossRef]
- Wu, Y.N.; Chen, Y.; Huang, X.S.; Pan, Y.H.; Liu, Z.M.; Yan, T.; Cao, W.H.; She, Z.G. α-Glucosidase inhibitors: Diphenyl ethers and phenolic bisabolane sesquiterpenoids from the mangrove endophytic fungus Aspergillus flavus QQSG-3. Mar. Drugs 2018, 16, 307. [Google Scholar] [CrossRef] [Green Version]
- Allard, P.M.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.L. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef]
- Nie, Y.Y.; Yang, W.C.; Liu, Y.Y.; Yang, J.M.; Lei, X.L.; Gerwick, W.H.; Zhang, Y. Acetylcholinesterase inhibitors and antioxidants mining from marine fungi: Bioassays, bioactivity coupled LC–MS/MS analyses and molecular networking. Mar. Life Sci. Technol. 2020, 2, 386–397. [Google Scholar] [CrossRef]
- Gu, B.; Wu, Y.; Tang, J.; Jiao, W.; Li, L.; Sun, F.; Wang, S.; Yang, F.; Lin, H. Azaphilone and isocoumarin derivatives from the sponge-derived fungus Eupenicillium sp. 6A-9. Tetrahedron Lett. 2018, 59, 3345–3348. [Google Scholar] [CrossRef]
- Huo, C.; Lu, X.; Zheng, Z.; Li, Y.; Xu, Y.; Zheng, H.; Niu, Y. Azaphilones with protein tyrosine phosphatase inhibitory activity isolated from the fungus Aspergillus deflectus. Phytochemistry 2020, 170, 112224. [Google Scholar] [CrossRef]
- Kim, J.C.; Lee, Y.W.; Tamura, H.; Yoshizawa, T. Sambutoxin: A new mycotoxin isolated from Fusarium sambucinum. Tetrahedron Lett. 1995, 36, 1047–1050. [Google Scholar] [CrossRef]
- Williams, D.R.; Turske, R.A. Construction of 4-hydroxy-2-pyridinones. Total synthesis of (+)-sambutoxin. Org. Lett. 2000, 2, 3217–3220. [Google Scholar] [CrossRef] [PubMed]
- Chunyu, W.X.; Zhao, J.Y.; Ding, Z.G.; Han, X.L.; Wang, Y.X.; Ding, J.H.; Wang, F.; Li, M.G.; Wen, M.L. A new cyclohexenone from the tin mine tailingsderived fungus Aspergillus flavus YIM DT 10012. Nat. Prod. Res. 2019, 33, 113–116. [Google Scholar] [CrossRef]
- Matsumoto, M.; Minato, H. Structure of ilicicolin H, an antifungal antibiotic. Tetrahedron Lett. 1976, 42, 3827–3830. [Google Scholar] [CrossRef]
- Sinder, B.B.; Lu, Q. Total synthesis of (±)-leporin A. J. Org. Chem. 1996, 61, 2839–2844. [Google Scholar] [CrossRef]
- Zhang, C.; Jin, L.; Mondie, B.; Mitchell, S.S.; Castelhano, A.L.; Cai, W.; Bergenhem, N. Leporin B: A novel hexokinase II gene inducing agent from an unidentified fungus. Bioorg. Med. Chem. Lett. 2003, 13, 1433–1435. [Google Scholar] [CrossRef]
- Zhang, Z.; Jamieson, C.S.; Zhao, Y.; Li, D.; Ohashi, M.; Houk, K.N.; Tang, Y. Enzyme-catalyzed inverse-electron demand Diels-Alder reaction in the biosynthesis of antifungal ilicicolin H. J. Am. Chem. Soc. 2019, 141, 5659–5663. [Google Scholar] [CrossRef]
- Millot, M.; Dieu, A.; Tomosi, S. Dibenzofurans and derivatives from lichens and ascomycetes. Nat. Prod. Rep. 2016, 33, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yu, H.B.; Zhang, Y.; Leao, T.; Glukhov, E.; Pierce, M.L.; Zhang, C.; Kim, H.; Mao, H.H.; Fang, F.; et al. Pagoamide A, a cyclic depsipeptide isolated from a cultured marine chlorophyte, Derbesia sp., using MS/MS-based molecular networking. J. Nat. Prod. 2020, 83, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Liu, Y.N.; Li, J.; Huang, X.S.; Yan, T.; Cao, W.H.; Liu, H.J.; Long, Y.H.; She, Z.G. Diaporindenes A–D: Four unusual 2,3-dihydro-1H-indene analogues with anti-inflammatory activities from the mangrove endophytic fungus Diaporthe sp. SYSU. J. Org. Chem. 2018, 83, 11804–11813. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Ye, G.J.; Lan, T.; Huang, Z.X.; Cheng, X.N.; Cai, C.Y.; Ding, S.M.; Xie, M.L.; Wang, B. Design and synthesis of novel xanthone-triazole derivatives aspotential antidiabetic agents:α-glucosidase inhibition and glucoseuptake promotion. Eur. J. Med. Chem. 2019, 177, 362–373. [Google Scholar] [CrossRef]
- Chen, T.; Huang, Y.; Hong, J.; Wei, X.; Zeng, F.; Li, J.; Ye, G.; Yuan, J.; Long, Y. Preparation, COX-2 inhibition and anticancer activity of sclerotiorin derivatives. Mar. Drugs 2021, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on 30 August 2020).
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | 1 (CDCl3) | 2 (CDCl3) | 3 (CDCl3) | |||
|---|---|---|---|---|---|---|
| δC, Type | δH(J in Hz) | δC, Type | δH(J in Hz) | δC, Type | δH(J in Hz) | |
| 2 | 161.5, C | 161.7, C | 162.7, C | |||
| 3 | 110.4, C | 110.1, C | 110.4, C | |||
| 4 | 162.0, C | 162.5, C | 162.4, C | |||
| 5 | 115.2, C | 115.4, C | 115.9, C | |||
| 6 | 136.4, CH | 7.14, s | 136.2, CH | 7.11, s | 136.0, CH | 7.12, s |
| 7 | 78.0, CH | 5.02, d, (9.1) | 73.7, CH | 5.17, dd, (10.5, 3.1) | 77.8, CH | 5.02, d, (9.0) |
| 8 | 30.8, CH2 | 1.64–1.69, m, 2.09, d, (11.4) | 29.9, CH2 | 1.25–1.31, m, 2.25–2.35, m | 30.9, CH2 | 1.64–1.70, m, 2.09, d, (11.4) |
| 9 | 32.3, CH2 | 1.44, d, (10.1) 1.91, d, (13.1) | 121.9, CH | 5.72, d, (5.2) | 32.2, CH2 | 1.44, d, (10.1) 1.91, d, (13.1) |
| 10 | 32.5, CH | 1.64–1.69, m | 132.9, C | 32.1, CH | 1.64–1.70, m | |
| 11 | 92.7, CH | 3.53, s | 86.9, CH | 4.56, s | 92.7, CH | 3.53, s |
| 12 | 130.4, C | 130.2, C | 130.3, C | |||
| 13 | 138.1, CH | 5.18, d, (9.5) | 139.7, CH | 5.29, d, (9.6) | 138.1, CH | 5.18, d, (9.5) |
| 14 | 29.7, CH | 2.42–2.49, m | 30.0, CH | 2.40–2.50, m | 29.7, CH | 2.42–2.49, m |
| 15 | 44.8, CH2 | 1.01–1.07, m 1.16–1.21, m | 44.8, CH2 | 1.01–1.04, m, 1.25–1.31, m | 44.8, CH2 | 1.01–1.07, m 1.16–1.21, m |
| 16 | 32.1, CH | 1.29–1.35, m | 32.2, CH | 1.25–1.31, m | 32.4, CH | 1.29–1.34, m |
| 17 | 29.0, CH2 | 1.01–1.07, m 1.29–1.35, m | 29.0, CH2 | 1.01–1.04, m, 1.25–1.31, m | 29.0, CH2 | 1.01–1.07, m 1.29–1.34, m |
| 18 | 11.3, CH3 | 0.83, s | 11.0, CH3 | 1.53, d, (1.3) | 11.3, CH3 | 0.83, s |
| 19 | 19.7, CH3 | 0.82, d, (6.5) | 20.8, CH3 | 0.90, d, (6.6) | 19.7, CH3 | 0.82, d, (6.5) |
| 20 | 20.8, CH3 | 0.90, d, (6.6) | 19.7, CH3 | 0.82, d, (6.4) | 20.8, CH3 | 0.90, d, (6.6) |
| 21 | 11.7, CH3 | 1.61, s | 11.3, CH3 | 1.49, s | 11.7, CH3 | 1.61, s |
| 22 | 17.8, CH3 | 0.74, d, (6.5) | 19.1, CH3 | 0.81, s | 17.7, CH3 | 0.74, d, (6.5) |
| 23 | 37.2, CH3 | 3.50, s | 37.3, CH3 | 3.50, s | 37.4, CH3 | 3.50, s |
| 1′ | 134.2, C | 125.9, C | 125.2, C | |||
| 2′ | 129.3, CH | 7.35–7.44, m | 130.6, CH | 7.26, d, (9.0) | 130.5, CH | 7.26, d, (9.0) |
| 3′ | 128.4, CH | 7.35–7.44, m | 115.5, CH | 6.87, d, (8.1) | 115.6, CH | 6.92, d, (8.1) |
| 4′ | 127.5, CH | 7.29–7.34, m | 155.8, C | 156.5, C | ||
| 5′ | 128.4, CH | 7.35–7.44, m | 115.5, CH | 6.87, d, (8.1) | 115.6, CH | 6.92, d, (8.1) |
| 6′ | 129.3, CH | 7.35–7.44, m | 130.6, CH | 7.26, d, (9.0) | 130.5, CH | 7.26, d, (9.0) |
| 4′-OH | 9.99, s | 9.83, s | 9.83, s | |||
| Position | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|
| 3 | 4.98, s | 5.10, q, (6.3) | 3.51, d, (9.5) | 3.40, d, (7.2) | 3.80, d, (3.2) | |
| 4 | 2.55, dqd, (11.3, 6.8, 1.7) | 2.77–2.83, m | 3.02, qdd, (7.1, 3.2, 1.9) | |||
| 5 | 5.55, s | |||||
| 6 | 1.30, s | 1.42, s | 1.29, d, (6.4) | 5.30, d, (1.7) | 5.33, d, (1.2) | 5.34, d, (1.9) |
| 7 | 4.16, s | 3.98, s | 4.10, s | 1.28, d, (6.8) | 1.30, d, (7.2) | 1.27, d, (7.1) |
| 8 | 1.67, s | 1.56, t, (1.8) | 1.22, s | 1.34, s | 1.30, s | |
| 9 | 2.20, s | 3.75, s | 3.76, s | 3.75, s | ||
| 10 | 2.37, q, (7.0) | 2.19–2.28, m | ||||
| 11 | 1.44–1.50, m 1.58–1.65, m | 1.34–1.40, m 1.44–1.50, m | ||||
| 12 | 0.94, t, (7.5) | 0.80, t, (7.5) | ||||
| 13 | 1.12, d, (7.0) | 1.02, d, (7.0) | ||||
| 14 | 2.82, dq, (18.2, 1.8) 3.03, dq, (18.2, 1.8) | |||||
| 15 | 3.16, s |
| Position | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|
| 1 | 202.0 | 200.6 | 205.0 | 203.0 | 200.9 | 203.3 |
| 2 | 86.4 | 80.4 | 84.3 | 78.4 | 75.5 | 77.7 |
| 3 | 73.5 | 92.5 | 71.7 | 78.1 | 78.7 | 79.0 |
| 4 | 179.7 | 192.4 | 188.2 | 41.5 | 40.1 | 39.0 |
| 5 | 115.6 | 103.3 | 117.5 | 180.1 | 180.5 | 178.9 |
| 6 | 19.5 | 23.5 | 14.9 | 99.5 | 99.6 | 99.0 |
| 7 | 59.1 | 60.1 | 58.2 | 14.0 | 14.2 | 13.4 |
| 8 | 6.08 | 207.2 | 5.6 | 19.2 | 21.4 | 23.6 |
| 9 | 177.5 | 27.5 | 176.4 | 57.2 | 57.0 | 57.1 |
| 10 | 42.0 | 42.4 | ||||
| 11 | 27.8 | 27.8 | ||||
| 12 | 11.8 | 11.7 | ||||
| 13 | 16.8 | 16.6 | ||||
| 14 | 31.2 | |||||
| 15 | 51.8 |
| Compounds | IC50/μM | Compounds | IC50/μM |
|---|---|---|---|
| 1 | 12.6 ± 0.9 | 7 | >100 |
| 2 | 37.4 ± 1.4 | 8 | >100 |
| 3 | 16.9 ± 0.6 | 9 | >100 |
| 4 | 16.5 ± 0.7 | 10 | >100 |
| 5 | 57.3 ± 1.3 | 11 | >100 |
| 6 | >100 | 12 | >100 |
| 1-deoxynojirimycin | 80.8 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, W.; Tan, Q.; Yin, Y.; Chen, Y.; Zhang, Y.; Wu, J.; Gao, L.; Wang, B.; She, Z. Secondary Metabolites with α-Glucosidase Inhibitory Activity from Mangrove Endophytic Fungus Talaromyces sp. CY-3. Mar. Drugs 2021, 19, 492. https://doi.org/10.3390/md19090492
Yang W, Tan Q, Yin Y, Chen Y, Zhang Y, Wu J, Gao L, Wang B, She Z. Secondary Metabolites with α-Glucosidase Inhibitory Activity from Mangrove Endophytic Fungus Talaromyces sp. CY-3. Marine Drugs. 2021; 19(9):492. https://doi.org/10.3390/md19090492
Chicago/Turabian StyleYang, Wencong, Qi Tan, Yihao Yin, Yan Chen, Yi Zhang, Jianying Wu, Leyao Gao, Bo Wang, and Zhigang She. 2021. "Secondary Metabolites with α-Glucosidase Inhibitory Activity from Mangrove Endophytic Fungus Talaromyces sp. CY-3" Marine Drugs 19, no. 9: 492. https://doi.org/10.3390/md19090492