3. Materials and Methods
3.1. General Methods
The IR spectra were recorded on a JASCO FTIR-4100 Type A spectrometer (JASCO corporation, Tokyo, Japan) using a NaCl cell. The 1H NMR and 13C NMR spectra were recorded using a JNM-EX 400 (400 MHz and 100 MHz) spectrometer (JEOL Ltd., Tokyo, Japan). Chemical shifts were reported in ppm relative to CHCl3 in CDCl3 for 1H NMR (δ = 7.26) and 13C NMR (δ = 77.0) and CHD2OH in CD3OD for 1H NMR (δ = 3.35) and 13C NMR (δ = 49.3). Splitting patterns for 1H NMR were designated as “s, d, t, q, m, dt, dd, and td”. These symbols indicate “singlet, doublet, triplet, quartet, multiplet, doublettriplet, doubletdoublet, and tripletdoublet”, respectively. All commercially obtained reagents were employed as received. Analytical TLC was carried out using pre-coated silica gel plates (Wako TLC Silicagel 70F254, FUJIFILM Wako Pure Chemical Coporation, Osaka, Japan). Wakogel 60N 63-212 μm was used for column chromatography.
3.2. Pht-Dpv-NH(8-quinoline) 5
Amide 4 (1.00 g, 2.67 mmol), AgOAc (891 mg, 5.36 mmol), Pd(OAc)2 (118 mg, 0.540 mmol) and iodobenzene (0.600 mL, 5.36 mmol) were added to a flask. The mixture was stirred for 6 h at 90 °C under Ar atmosphere. After being cooled to room temperature, the reaction was diluted with AcOEt and then filtered through a pad of celite wash with AcOEt and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane:EtOAc = 80:20 then 70:30) to afford 5 (840 mg, 70%) as a white solid: −36.1 (c 0.18, CHCl3); IR (neat) 3650, 2924, 1717, 1530, 1487, 1384, 1327, 1261, 1070, 792, 721 cm–1; 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, d, J = 6.8 Hz), 2.43 (1H, dd, J = 13.4, 10.5 Hz), 3.23 (1H, dd, J = 13.7, 3.4 Hz), 3.36–3.40 (1H, m), 4.88 (1H, d, J = 10.7 Hz), 7.16–7.31 (5H, m), 7.46 (1H, dd, J = 8.3, 4.4 Hz), 7.53 (2H, d, J = 3.4 Hz), 7.74 (2H, dd, J = 5.4, 2.9 Hz), 7.88 (2H, dd, J = 5.4, 2.9 Hz), 8.16 (1H, dd, J = 8.3, 2.0 Hz), 8.77 (1H, t, J = 4.4 Hz), 8.86 (1H, dd, J = 4.4, 2.0 Hz), 10.68 (1H, brs); 13C NMR (CDCl3, 100 MHz) δ 15.9, 34.6, 40.8, 61.7, 117.3, 121.9, 122.3, 124.0, 126.4, 127.5, 128.6, 129.7, 131.8, 134.1, 134.5, 136.5, 139.0, 139.8, 148.8, 166.7, 168.3; HRMS (ESI) m/z:[M + Na]+; Calcd for C28H23N3O3Na 472.1631; Found 472.1632.
3.3. Boc-Dpv-OH 7
Amide 5 (148 mg, 0.330 mmol) was dissolved in 3.0 mL of aqueous HCl (6.0 M) and refluxed for 24 h and then cooled to room temperature. The reaction mixture was concentrated in vacuo. To a solution of compound in THF (3.0 mL), saturated NaHCO3 (3.0 mL) was added Boc2O (151 µL, 0.660 mmol) at 0 °C. The mixture was stirred at room temperature for 24 h. The reaction was diluted with EtOAc, washed with HCl (0.10 M) and brine, dried over Na2SO4. The residue was purified using silica gel column chromatography (Hexane:EtOAc = 50:50) to afford 7 (45.7 mg, 47%) as a colorless oil:
3.4. Pht-Dpv(OTBS)-NH(8-quinoline) 6
Amide 4 (840 mg, 2.24 mmol), AgOAc (750 mg, 4.48 mmol), Pd(OAc)2 (0.100 g, 0.450 mmol), and p-iodophenyl tert-butyldimethylsilyl ether (3.00 g, 8.97 mmol) were added to a flask. The mixture was stirred at 90 °C for 6 h under Ar atmosphere. After being cooled to room temperature, the reaction was diluted with AcOEt and then filtered through a pad of celite wash with AcOEt and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane:EtOAc = 90:10 then 80:20) to afford 6 (590 mg, 1.02 mmol, 86%) as a white solid: −56.7 (c 0.45, CHCl3); IR (neat) 2929, 1772, 1718, 1530, 1509, 1382, 1260, 914, 826, 787, 719 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.17 (6H, s), 0.86 (3H, d, J = 7.3 Hz), 0.97 (9H, s), 2.38 (1H, dd, J = 13.7, 10.3 Hz), 3.17 (1H, dd, J = 13.7, 3.4 Hz), 3.33–3.35 (1H, m), 4.88 (1H, d, J = 10.7 Hz), 6.74 (2H, d, J = 7.6 Hz), 7.16 (2H, d, J = 7.6 Hz), 7.43 (1H, dd, J = 8.3, 4.4 Hz), 7.51 (2H, d, J = 3.4 Hz), 7.71 (2H, dd, J = 5.4, 2.9 Hz), 7.87 (2H, dd, J = 5.4, 2.9 Hz), 8.13 (1H, dd, J = 8.3, 1.5 Hz), 8.78 (1H, dd, J = 5.4, 3.4 Hz), 8.84 (1H, dd, J = 4.4, 1.5 Hz), 10.68 (1H, brs); 13C NMR (100 MHz, CDCl3) δ 4.5, 15.6, 18.1, 25.6, 34.4, 61.4, 117.0, 119.7, 121.6, 122.0, 123.4, 127.2, 127.8, 130.2, 131.5, 132.1, 134.1, 136.1, 138.7, 148.5, 153.9, 166.5, 168.0; HRMS (ESI) m/z: [M + Na]+; Calcd for C34H37N3O4SiNa 602.2447; Found 602.2446.
3.5. Boc-Dpv(OH)-OH 8
Amide 6 (290 mg, 0.500 mmol) was dissolved in 14 mL of aqueous HCl (6.0 M) was heated at 130 °C in a sealed tube for 24 h and then cooled to room temperature. The reaction mixture was concentrated in vacuo. To a solution of compound in THF (2.5 mL), saturated NaHCO3 (2.5 mL) was added Boc2O (0.200 mL, 1.00 mmol) at 0 °C. The mixture was stirred at room temperature for 24 h. The solution was diluted with EtOAc, washed with HCl (0.1 M) and brine, dried over Na2SO4. The residue was purified using silica gel column chromatography (Hexane:EtOAc = 50:50) to afford 8 (120 mg, 0.387 mmol, 77%) as a colorless oil: +3.5 (c 0.49, CH3OH); IR (neat) 3748, 2976, 1702, 1514, 1398, 1244, 1160, 1073, 774, 641 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, brs), 1.47 (9H, s), 2.33–2.43 (1H, m), 2.62–2.66 (1H, m), 4.34 (1H, brs), 5.10 (1H, d, J = 9.3 Hz), 6.73 (2H, d, J = 7.8 Hz), 6.98 (2H, d, J = 7.8 Hz); 13C NMR (100 MHz, CD3OD) δ 4.7, 28.4, 38.6, 39.8, 57.7, 80.3, 115.9, 129.6, 130.8, 131.8, 156.5, 171.0; HRMS (ESI) m/z: [M + Na]+; Calcd for C16H23NO5Na 332.1471; Found 332.1468.
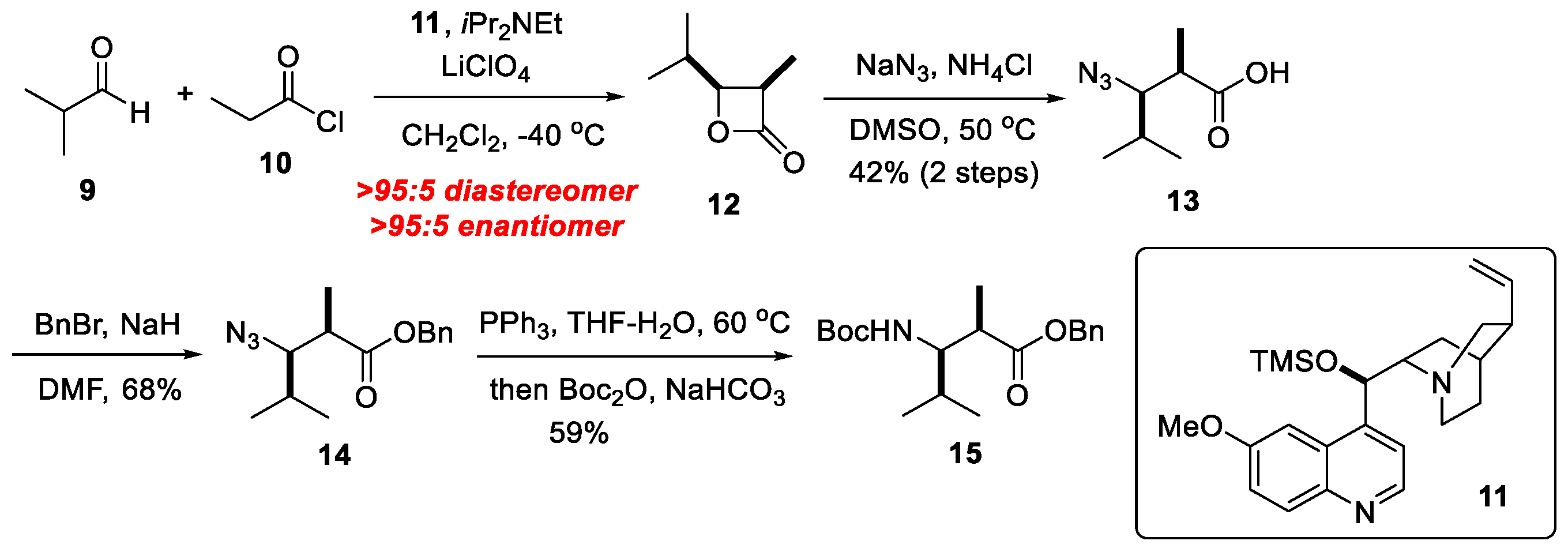
3.6. N2-Dml-OH 13
Lithium perchlorate (2.12 g, 20.0 mmol), was dissolved in 10 mL anhydrous Et2O. TMS-quinine 11 (400 mg, 1.00 mmol) and CH2Cl2 (20 mL) were added to this solution which was then cooled to −40 °C. DIEA (4.36 mL, 25.0 mmol) and isobutyraldehyde (0.920 mL, 10.0 mmol) were then added to the solution. Propionyl chloride (1.74 mL, 20.0 mmol) was dissolved in CH2Cl2 (5.0 mL). The solution of propionyl chloride was then added dropwise to the reaction over the course of 3 h. Upon completion of the addition, the reaction was allowed to stir at −40 °C for 16 h. After this time, Et2O was added to the solution. The resulting mixture was filtered through a pad of celite and washed with Et2O. The solution was concentrated at a light vacuum and diluted with CH2Cl2. The solution was washed with sat. NH4Cl and brine, dried over Na2SO4 and concentrated at a light vacuum to give crude lactone, which was used in the next step without further purification.
To a solution of the crude lactone in DMSO (30 mL) were added NaN3 (1.30 g, 20.0 mmol) and NH4Cl (535 mg, 10.0 mmol) at room temperature. The mixture was heated at 50 °C, diluted with aqueous HCl, extracted with EtOAc, washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified using silica gel column chromatography (Hexane:EtOAc = 20:80) to afford 14 (718 mg, 4.20 mmol, 42%) as a colorless oil: +8.1 (c 0.92, CHCl3); IR (neat) 2967, 2878, 2157, 2106, 1713, 1419, 893, 852, 663 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, d, J = 6.8 Hz), 1.08 (3H, d, J = 6.8 Hz), 1.23 (3H, d, J = 7.3 Hz), 1.95–2.05 (1H, m), 2.61–2.68 (1H, m), 3.42 (1H, dd, J = 8.8, 4.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.5, 15.7, 20.5, 29.5, 42.4, 70.6, 180.4; HRMS (ESI) m/z: [M + Na]+; Calcd for C7H13N3O2Na 194.0906; Found 194.0910.
3.7. N2-Dml-OBn 14
To a solution of 13 (20.0 mg, 0.120 mmol) in DMF (0.60 mL) were added BnBr (0.0200 mL, 0.180 mmol), NaH (4.00 mg, 0.160 mmol) at 0 °C under Atmosphere. The mixture was stirred at room temperature for overnight, quenched with saturated NaHCO3, extracted with EtOAc, washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified using silica gel column chromatography (Hexane:EtOAc = 20:80) to afford 14 (20.0 mg, 0.0800 mmol, 64%) as a colorless oil: +19.8 (c 0.68, CHCl3); IR (neat) 2966, 2103, 1734, 1456, 1366, 1341, 1261, 1171, 1147, 1027, 978, 907, 751, 697 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, d, J = 6.8 Hz), 1.05 (3H, d, J = 6.8 Hz), 1.17 (3H, d, J = 7.3 Hz), 1.93–1.99 (1H, m), 2.62–2.69 (1H, m), 3.45 (1H, dd, J = 9.3, 3.9 Hz), 5.17 (1H, s), 7.25–7.37 (5H, m); 13C NMR (100 MHz, CDCl3) δ14.5, 15.5, 20.6, 29.3, 42.7, 66.6, 70.8, 128.27, 128.28, 128.5, 135.7, 174.4; HRMS (ESI) m/z: [M + Na]+; Calcd for C14H19N3O2Na 284.1375; Found 284.1377.
3.8. Boc-Dml-OBn 15
To a solution of 14 (1.52 g, 5.82 mmol) in THF/water (10:1 v/v, 29 mL) were added Ph3P (4.60 g, 17.4 mmol) at 60 °C for 2 h. The reaction solution was cooled to room temperature, concentrated under reduced pressure, and the residue obtained was dissolved in THF/NaHCO3 (1:1 v/v, 17 mL). Cool the solution to 0 °C, add Boc2O (1.60 mL, 6.80 mmol), return to ambient temperature and stir overnight. Extracted with EtOAc, washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified using silica gel column chromatography (Hexane:EtOAc = 20:80) to afford 15 (1.09 g, 3.40 mmol, 59%) as a colorless oil: +18.1 (c 0.23, CHCl3); IR (neat) 3750, 2974, 2876, 1716, 1507, 1166, 772, 669 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, d, J = 6.8 Hz), 0.91 (3H, d, J = 6.8 Hz), 1.22 (3H, d, J = 6.8 Hz), 1.43 (9H, s), 1.61 (1H, sep, J = 6.8 Hz), 2.80-2.87 (1H, m), 3.38 (1H, ddd, J = 10.1, 7.3, 4.4 Hz), 5.10 (1H, d, J = 15.6 Hz), 5.11 (1H, d, J = 15.6 Hz), 5.24 (1H, d, J = 10.2 Hz), 7.32 -7.39 (5H, m); 13C NMR (100 MHz, CDCl3) δ 15.7, 19.2, 19.9, 28.4, 31.8, 40.5, 58.6, 66.3, 78.8, 128.1, 128.3, 128.6, 135.7, 156.4, 175.6; HRMS (ESI) m/z: [M + Na]+ Calcd for C19H29NO4Na 358.1989; Found 358.1992.
3.9. Boc-Dpv(OH)-Pro-OBn 16
To a solution of 8 (850 mg, 2.70 mmol) and L-proline benzylester (670 mg, 3.24 mmol) in CH3CN (13.5 mL) was added EDCI (620 mg, 3.24 mmol), HOAt (440 mg, 3.24 mmol) and NaHCO3 (230 mg, 2.70 mmol) under Ar atmosphere. After 24 h of stirring at room temperature, the mixture was concentrated in vacuo. The residue was purified using silica gel column chromatography (Hexane:EtOAc = 50:50) to afford the dipeptide 16 (1.02 g, 2.05 mmol, 76%) as colorless oil: −35.4 (c 1.20, CHCl3); IR (neat) 3326, 2927, 2931, 1744, 1715, 1637, 1514, 1260, 1168, 1016, 753 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.85 (3H, d, J = 6.8 Hz), 1.45 (9H, s), 1.82–1.89 (2H, m), 2.01–2.02 (1H, m), 2.13–2.17 (1H, m), 2.35 (1H, dd, J = 13.7, 7.3 Hz), 2.67 (1H, dd, J = 13.7, 6.8 Hz), 3.18–3.24 (1H, m), 3.30–3.32 (1H, m), 4.41 (1H, d, J = 6.3 Hz), 4.55 (1H, dd, J = 8.3, 4.4 Hz), 5.13 (2H, s), 5.30 (1H, d, J = 9.8 Hz), 5.91 (1H, brs), 6.74 (2H, d, J = 8.3 Hz), 7.05 (2H, d, J = 8.3 Hz), 7.28–7.33 (5H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ 14.2, 15.3, 24.9, 28.4, 28.9, 36.3, 38.4, 39.1, 46.5, 58.8, 65.9, 66.9, 115.1, 128.1, 128.3, 128.5, 130.5, 132.2, 135.6, 154.2, 171.9; HRMS (ESI) m/z: [M + Na]+; Calcd for C28H36N2O6Na 519.2465; Found 519.2466.
3.10. Boc-Dpv(OAc)-Pro-OBn 17
To a solution of 16 (520 mg, 1.05 mmol) in CH2Cl2 (5.25 mL) was added Ac2O (120 μL, 1.26 mmol) and DMAP (50.0 mg, 0.42 mmol) under Ar atmosphere. After 2 h of stirring at room temperature, the mixture was concentrated in vacuo. The residue was purified using silica gel column chromatography (Hexane:EtOAc = 50:50) to afford the dipeptide 17 (510 mg, 0.97 mmol, 92%) as colorless oil: −38.7 (c 1.20, CHCl3); IR (neat) 3735, 3308, 2970, 2963, 2931, 1744, 1715, 1651, 1510, 1433, 1168, 753, 711 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.84 (3H, d, J = 6.8 Hz), 1.34 (9H, s), 1.72–1.89 (3H, m), 1.97–2.07 (1H, m), 2.13–2.17 (1H, m), 2.25 (3H, s), 2.35 (1H, dd, J = 13.7, 7.3 Hz), 2.67 (1H, dd, J = 13.7, 6.8 Hz), 3.17–3.24 (1H, m), 3.29–3.32 (1H, m), 4.41 (1H, d, J = 6.3 Hz), 4.55 (1H, dd, J = 8.3, 4.4 Hz), 5.13 (2H, s), 5.30 (1H, d, J = 9.8 Hz), 5.91 (1H, brs), 6.74 (2H, d, J = 8.3 Hz), 7.05 (2H, d, J = 8.3 Hz), 7.28–7.33 (5H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ 14.3, 15.3, 24.0, 28.2, 28.9, 36.3, 37.8, 39.1, 46.5, 58.8, 65.9, 67.0, 115.1, 128.1, 128.2, 128.5, 130.5, 132.2, 135.6, 154.2, 172.3; HRMS (ESI) m/z: [M + Na]+; Calcd for C30H39N2O7Na 562.2463; Found 562.2467.
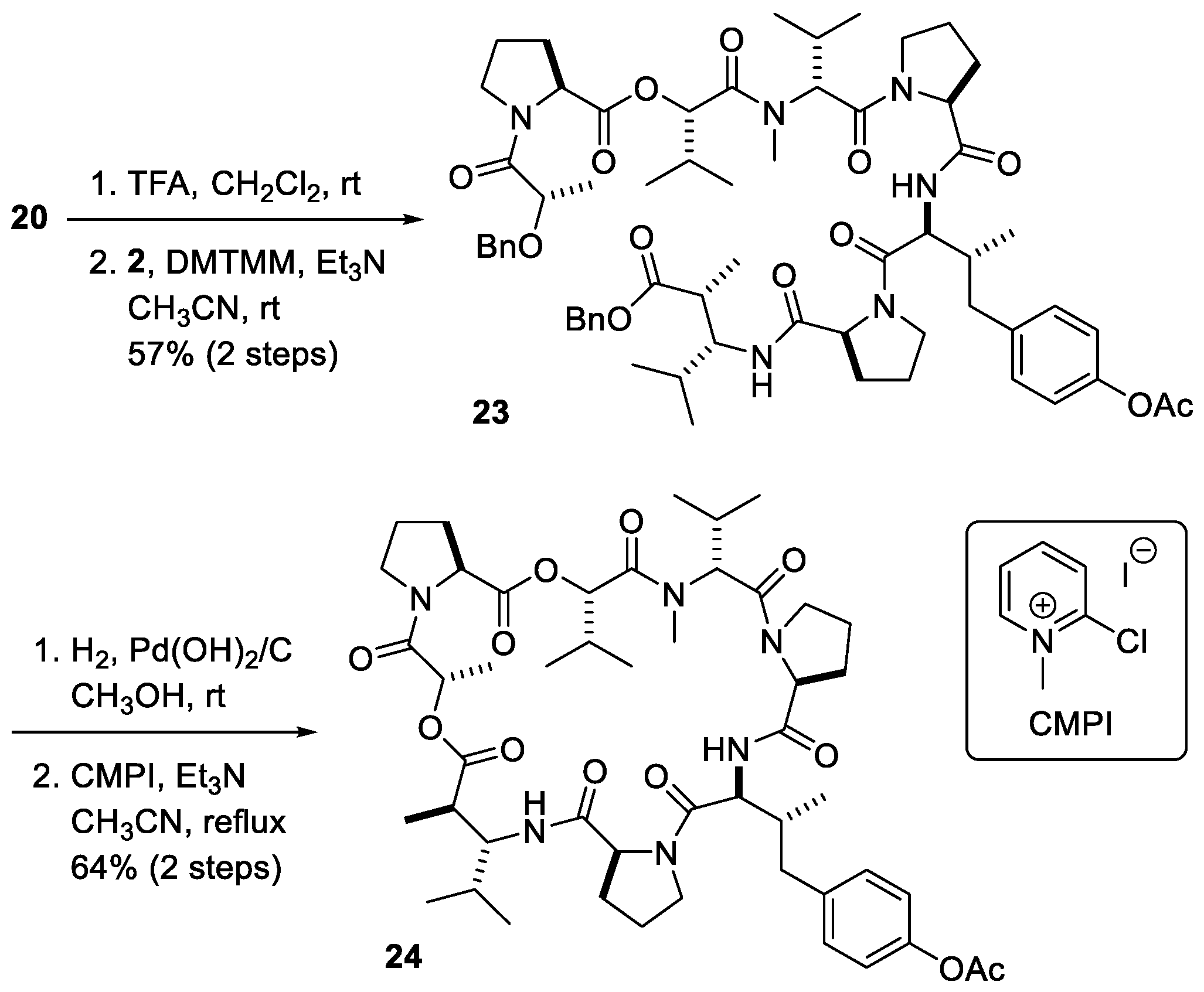
3.11. Boc-Dpv(OAc)-Pro-Dml-OBn 20
To 15 (1.09 g, 3.40 mmol) was added TFA/CH2Cl2 (1:4 v/v, 25 mL). After 1 h of stirring at room temperature, the solution was concentrated in vacuo to afford crude 19, which was used in the next step without further purification.
To a solution of 17 (550 mg, 1.05 mmol) in CH3OH (5.25 mL) was carefully added Pd(OH)2/C (110 mg, 20 wt%) under Ar atmosphere. The solution was purged with H2 gas and stirring was continued under H2 atmosphere at room temperature for 16 h. The solution was filtered through celite and concentrated in vacuo to afford crude 18, which was used in the next step without further purification.
To a solution of the crude 19 (610 mg, 2.60 mmol) and crude 18 (1.25 g, 2.60 mmol) in CH3CN (26 mL) were added DMTMM (740 mg, 2.60 mmol) and Et3N (2.17 mL, 15.6 mmol) under Ar atmosphere. After 24 h of stirring at room temperature, the mixture was concentrated in vacuo. The residue was purified using silica gel column chromatography (Hexane:EtOAc = 20:80) to afford 20 (1.01 g, 1.51 mmol, 58%) as a colorless oil: +8.8 (c 2.40, CHCl3); IR (neat) 3734, 3413, 3309, 2973, 2877, 1715, 1507, 1366, 1168, 754 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.85 (3H, d, J = 6.8 Hz), 0.86 (3H, d, J = 6.8 Hz), 0.88 (3H, d, J = 6.8 Hz), 1.17 (3H, d, J = 7.3 Hz), 1.46 (9H, s), 1.89 (2H, t, J = 5.4 Hz), 1.98–2.09 (2H, m), 2.17–2.23 (1H, m), 2.26 (3H, s), 2.48 (1H, dd, J = 13.7, 6.8 Hz), 2.74 (1H, dd, J = 13.2, 7.8 Hz), 2.86 (1H, dd, J = 6.8, 3.4 Hz), 3.15–3.18 (1H, m), 3.23–3.28 (1H, m), 3.68 (1H, td, J = 9.8, 3.4 Hz), 4.47–4.52 (2H, m), 5.01 (1H, d, J = 12.2 Hz), 5.06 (2H, d, J = 12.2 Hz), 5.37 (1H, d, J = 9.8 Hz), 6.80 (1H, d, J = 10.7 Hz), 6.98 (2H, d, J = 8.8 Hz), 7.29–7.37 (7H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ 0.3, 14.5, 16.2, 19.8, 20.1, 21.4, 25.2, 28.6, 28.7, 29.4, 32.2, 38.9, 40.0, 47.0, 53.8, 57.3, 61.0, 66.6, 79.8, 121.7, 128.3, 128.6, 128.9, 130.8, 135.9, 138.4, 149.3, 169.7, 171.8, 172.1, 176.3; HRMS (ESI) m/z: [M + Na]+; Calcd for C37H51N3O8Na 688.3566; Found 688.3568.
3.12. Boc-Dpv(OH)-Pro-Dml-OBn 21
To a solution of 20 (184 mg, 0.276 mmol) in CH3OH (0.14 mL) were added TEA (7.00 µL, 0.502 µmol) under Ar atmosphere. After 24 h of stirring at room temperature, the mixture was concentrated in vacuo. The crude product was purified using silica gel column chromatography (Hexane:Acetone = 75:25) to afford 21 (155 mg, 0.0205 mmol, 90%) as a colorless foam: +6.3 (c 1.18, CHCl3); IR (neat) 3403, 3311, 3009, 2975, 2935, 2878, 1715, 1652, 1615, 1594, 1516, 1455, 1391, 1367, 1235, 1172, 1101, 1057, 1003, 877, 826, 755, 698, 666 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.72–0.94 (9H, m), 1.20 (3H, d, J = 7.3 Hz), 1.35–1.58 (10H, m), 1.70–1.92 (2H, m), 1.96–2.10 (4H, m), 2.15–2.25 (1H, m), 2.40 (1H, dd, J = 13.7, 6.8 Hz), 2.59–2.69 (1H, m), 2.81–2.92 (1H, m), 3.21–3.35 (2H, m), 3.69 (1H, td, J = 9.76, 3.4 Hz), 4.45–4.55 (2H, m), 5.06 (2H, s), 5.38 (1H, d, J = 9.8 Hz), 6.74 (2H, d, J = 8.3 Hz), 6.90 (1H, d, J = 10.2 Hz), 7.06 (2H, d, J = 7.8 Hz), 7.18–7.41 (5H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ 13.9, 15.8, 19.6, 19.8, 24.9, 28.3, 29.1, 31.8, 38.0, 39.5, 39.7, 46.7, 53.7, 57.2, 60.6, 66.3, 79.8, 115.1, 128.0, 128.1, 128.3, 128.5, 128.6, 130.1, 130.3, 131.1, 135.5, 155.0, 156.2, 171.9, 172.1, 175.9; HRMS (ESI) m/z: [M + Na]+ Calcd for C35H49N3O7Na 646.3478; Found 646.3463
3.13. Boc-Dpv(OBn)-Pro-Dml-OBn 22
To a solution of 21 (12.1 mg, 0.0194 mmol) in CH3CN (0.16 mL) were added K2CO3 (8.90 mg, mmol), BnBr (8.00 µL, 0.0669 mmol) and KI (1.10 mg, 6.63 µmol) under Ar atmosphere at room temperature. The mixture was stirred for 24 h, quenched with sat. NaHCO3, extracted with EtOAc, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified using silica gel column chromatography (Hexane:Acetone = 80:20) to afford 22 (14.6 mg, 0.0205 mmol, 95%) as a colorless oil: +13.1 (c 1.22, CHCl3); IR (neat) 3416, 3315, 2972, 2932, 2876, 1714, 1652, 1507, 1456, 1417, 1392, 1367, 1241, 1172, 1099, 1026, 907, 808, 751, 697, 666 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.72–0.95 (9H, m), 1.17 (3H, d, J = 7.3 Hz), 1.35–1.52 (10H, m), 1.79–2.23 (5H, m), 2.44 (1H, dd, J = 13.7, 6.8 Hz), 2.61–2.75 (1H, m), 2.80–2.92 (1H, m), 3.20–3.39 (2H, m), 3.69 (1H, td, J = 10.1, 3.4 Hz), 4.41–4.59 (2H, m), 4.92–5.08 (4H, m), 5.35 (1H, d, J = 8.8 Hz), 6.72–6.95 (3H, m), 7.15–7.50 (12H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ 14.0, 15.9, 19.5, 19.8, 24.9, 29.2, 29.6, 31.7, 31.8, 38.4, 39.6, 39.7, 46.7, 53.4, 53.8, 53.8, 57.0, 60.6, 66.3, 69.9, 79.5, 114.6, 127.3, 127.8, 128.0, 128.3, 128.5, 128.6, 130.4, 132.7, 135.6, 137.1, 155.9, 157.1, 171.5, 172.0, 175.9; HRMS (ESI) m/z: [M + Na]+ Calcd for C42H55N3O7Na 736.3932; Found 736.3931.
3.14. BnO-Lac-Pro-O-Hiv-D-MeVal-Pro-Dpv(OAc)-Pro-Dml-OBn 23
To 20 (380 mg, 0.570 mmol) was added TFA/CH2Cl2 (1:4 v/v, 19 mL). After 1 h of stirring at room temperature, the solution was concentrated in vacuo to afford crude amine, which was used in the next step without further purification.
To a solution of the crude amine and 2 (340 mg, 0.570 mmol) in CH3CN (5.70 mL) were added Et3N (0.480 mL, 3.42 mmol) and DMTMM (160 mg, 0.570 mmol) under Ar atmosphere. After 24 h of stirring at room temperature, the solution was concentrated in vacuo. The residue was purified using silica gel column chromatography to afford 23 (368 mg, 0.325 mmol, 57% over 2 steps) as a colorless foam: −12.7 (c 1.20, CHCl3); IR (neat) 3733, 3410, 3309, 2973, 2965, 2876, 1730, 1663, 1510, 1430, 1183, 1100, 753 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.69–1.17 (20H, m), 1.24–1.27 (4H, m), 1.38–1.59 (4H, m), 1.70–2.16 (9H, m), 2.28 (3H, s), 2.23–2.94 (11H, m), 3.08 (3H, s), 3.10–3.77 (5H, m), 4.05–4.93 (11H, m), 6.78 (0.5H, d, J = 10.3 Hz), 6.95 (1.5H, d, J = 8.3 Hz), 7.23–7.35 (14H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ13.8, 14.2, 15.83, 15.85, 16.0, 16.6, 17.0, 17.2, 17.8, 18.0, 19.2, 19.4, 19.60, 19.67, 19.7, 20.0, 20.9, 21.0, 24.6, 24.9, 25.5, 26.3, 29.0, 29.8, 30.0, 31.7, 31.8, 38.6, 39.5, 46.6, 46.7, 56.8, 58.6, 59.1, 59.9, 60.5, 60.6, 66.15, 66.19, 70.8, 71.0, 74.8, 75.6, 121.1, 121.2, 127.56, 127.59, 127.77, 127.82, 127.84, 128.23, 128.25, 128.27, 128.4, 128.5, 130.3, 130.4, 135.4, 137.6, 137.9, 148.8, 167.5, 169.2, 169.7, 170.9, 171.0, 171.2, 171.3, 171.5, 171.6, 175.86, 175.88; HRMS (ESI) m/z: [M + Na]+; Calcd for C63H86N13O6Na 1157.3893; Found 1157.6145.
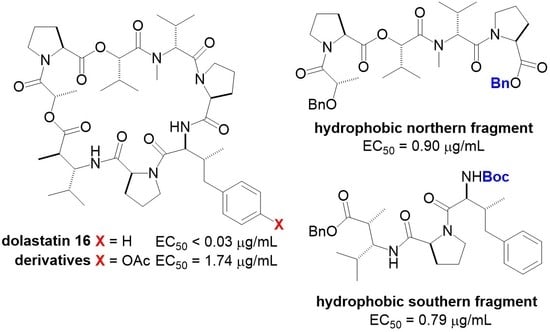
3.15. Dolastatin 16 Acetate 24
To a solution of 23 (110 mg, 0.100 mmol) in CH3OH (1.00 mL) was carefully added Pd(OH)2/C (22.0 mg, 20 wt%) under Ar atmosphere. The solution was purged with H2 gas and stirring was continued under H2 atmosphere at room temperature for 16 h. The solution was filtered through celite and concentrated in vacuo to afford crude carboxylic acid, which was used in the next step without further purification.
A solution of the crude carboxylic acid 10a (9.50 mg, 0.0100 mmol) in CH3CN (6.3 mL) was dropwised to a refluxing solution of 2-chloro-1-methylpyridinium iodide (13.0 mg, 0.0130 mmol) and Et3N (0.0150 mL, 0.110 mmol) in CH3CN (3.1 mL) over a 3 h period via addition funnel. The addition funnel was rinsed with a total of 0.6 mL of CH3CN. The mixture was refluxed for overnight. After being cooled to ambient temperature, the mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography on silica gel (Hexane:EtOAc = 20:80) to afford 24 as a colorless oil (4.90 mg, 0.005 mmol, 64% over 2 steps): +29.4 (c 0.38, CHCl3); IR (neat) 3394, 3324, 2965, 2876, 1750, 1732, 1650, 1507, 1458, 1426, 1386, 1298, 1194, 1090, 1019, 753, 666 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.82–0.93 (14H, m), 1.01–1.09 (9H, m), 1.43 (3H, d, J = 6.8 Hz), 1.45–1.60 (2H, m), 1.65–2.26 (9H, m), 2.28 (3H, s), 2.30–2.44 (6H, m), 2.45–2.55 (2H, m), 2.78–2.90 (2H, m), 3.08 (3H, s), 3.35–3.50 (2H, m), 3.60–3.70 (2H, m), 3.85–3.92 (1H, m), 4.44 (1H, d, J = 6.8 Hz), 4.54 (1H, d, J = 7.8 Hz), 4.60–4.64 (1H, m), 4.94 (1H, d, J = 8.8 Hz), 5.12–5.20 (2H, m), 5.41 (1H,d, J = 2.9 Hz), 6.72 (1H, d, J = 8.8 Hz), 6.74 (1H, d, J = 8.7 Hz), 7.00 (2H, d, J = 8.3 Hz), 7.41 (2H, d, J = 8.8 Hz), 7.68 (1H, d, J = 10.2 Hz); 13C NMR (100 MHz, CDCl3) δ 14.8, 15.2, 16.0, 17.1, 17.7, 19.6, 19.7, 20.3, 21.1, 21.7, 24.7, 24.8, 24.9, 25.4, 25.5, 28.2, 29.5, 30.6, 30.7, 32.2, 38.6, 41.0, 45.9, 46.4, 47.5, 50.2, 56.2, 57.8, 58.8, 59.4, 59.5, 61.2, 66.6, 120.1, 121.4, 130.6, 138.2, 149.0, 161.8, 169.0, 169.50, 169.54, 170.95, 170.99, 171.1, 172.3, 174.6; HRMS (ESI) m/z: [M + Na]+; Calcd for C49H72N6O12Na 959.5108; Found 959.5100.
3.16. BnO-Lac-Pro-O-Hiv-D-MeVal-Pro-OBn 25
To a solution of 2 (6.3 mg, 0.0107 mmol) in CH2Cl2 (0.30 mL) were added BnOH (1.30 μL, 0.0126 mmol), DMAP (catalytic) and EDCI (3.00 mg, 0.0156 mmol) under Ar atmosphere. The mixture was stirred for 27 h at room temperature and concentrated in vacuo. The crude product was purified using column chromatography (5% EtOAc in hexane) to afford 25 (4.70 mg, 0.00694 mmol, 64%) as a colorless oil:
+35.1 (c 0.47, CHCl3); IR (neat) 2963, 2927, 2874, 1742, 1646, 1454, 1426, 1370, 1295, 1255, 1186, 1092, 1014, 738, 699 cm−1; 1H NMR (CDCl3, 400 MHz, mixture of rotamers) δ 0.68–1.01 (12H, m), 1.33–1.42 (3H, m), 1.70–2.32 (10H, m), 2.82 (2H, s), 2.95 (1H, s), 3.30–3.72 (4H, m), 4.10–4.35 (2H, m), 4.40–4.75 (3H, m), 4.90–5.10 (4H, m), 7.19–7.31 (10H, m); 13C NMR (CDCl3, 100 MHz, mixture of rotamers) δ 14.1, 15.9, 16.1, 17.1, 17.2, 17.8, 19.7, 19.8, 20.1, 24.9, 25.1, 26.2, 26.3, 28.0, 28.8, 28.90, 28.99, 29.6, 29.81, 29.85, 46.52, 46.56, 58.5, 58.8, 59.1, 60.1, 66.7, 67.1, 70.9, 72.6, 75.2, 75.4, 77.2, 127.6, 127.72, 127.75, 128.1, 128.2, 128.31, 128.38, 128.52, 128.58, 135.5, 137.7, 167.8, 168.1, 169.1, 169.6, 171.3, 171.71, 171.78, 172.0, 172.3; HRMS (ESI) m/z: [M + Na]+ Calcd for C38H51N3O8Na 700.3574; Found 700.3578.
3.17. Boc-Pro-O-Hiv-D-MeVal-Pro-CH2OBn 29
To peptide 2745 (51.4 mg, 0.127 mmol) was added TFA/DCM (1:4 v/v, 2.2 mL). After 1 h of stirring at room temperature, the mixture was concentrated in vacuo. The residue TFA salt was added 0.5 M NaOH aq, extracted with DCM, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to afford crude amine, which was used in the next step without further purification.
To a solution of the crude amine in MeCN (0.65 mL) was added 4 N HCl in dioxane (31.0 µL, 0.127 mmol) under Ar atmosphere. After 30 min of stirring at room temperature, to the mixture were added 28 (51.5 mg, 0.127 mmol), PyBroP (71.1 mg, 0.152 mmol) and iPr2NEt (66.0 µL, 0.381 mmol) The mixture was stirred for 24 h, quenched with sat. NH4Cl, extracted with EtOAc, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified using silica gel column chromatography (Hexane:Acetone = 80:20) to afford 29 (56.1 mg, 0.0932 mmol, 73%) as a colorless oil: +18.0 (c 1.03, CHCl3); IR (neat), 2970, 2933, 2875, 1747, 1701, 1643, 1454, 1397, 1365, 1297, 1254, 1188, 1168, 1119, 1088, 1029, 1011, 917, 890, 772, 740, 699 cm−1; 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 0.80 (3H, t, J = 7.3 Hz), 0.85–1.13 (9H, m), 1.32–1.52 (9H, m), 1.69–2.41 (10H, m), 2.89 (1.9H, s), 2.97 (0.5H, s), 2.99 (0.6H, s), 3.25–3.59 (5.5H, m), 3.60–3.70 (0.5H, m), 4.23–4.38 (1.5H, m), 4.40–4.60 (2.5H, m), 4.94 (0.6H, d, J = 10.7 Hz), 4.95–5.05 (1H, m), 5.20 (0.4H, dd, J = 10.7, 8.3), 7.22–7.40 (5H, m); 13C NMR (100 MHz, CDCl3, mixture of rotamers) δ 16.3, 16.3, 16.4, 18.1, 18.2, 19.5, 19.6, 19.6, 19.7, 19.8, 20.0, 20.0, 21.7, 23.2, 24.0, 24.0, 26.5, 26.5, 27.4, 28.1, 28.3, 28.5, 28.5, 28.8, 29.0, 29.1, 29.6, 29.9, 29.9, 29.9, 30.4, 30.5, 45.7, 46.3, 46.3, 46.9, 55.5, 56.6, 56.6, 58.2, 58.5, 58.5, 59.5, 60.3, 60.3, 69.6, 71.3, 72.8, 73.0, 75.1, 75.2, 75.5, 79.6, 79.6, 79.7, 127.4, 127.4, 127.5, 127.6, 128.3, 128.4, 138.6, 153.9, 167.8, 168.0, 169.2, 127.8; HRMS (ESI) m/z: [M + Na]+ Calcd for C33H51O7N3Na 524.3614; Found 524.3619.
3.18. Antifouling Assay
Antifouling assay against larvae of the barnacle
Amphibalanus amphitrite was conducted according to the previous literature [
18,
19,
38]. The adult barnacles,
A. amphitrite, obtained from oyster farms in Lake Hamana and a pier of Shimizu bay, Shizuoka, were kept in an aquarium at 20 °C and were fed on
Artemia salina nauplii. Broods were released as I–II stage nauplii upon immersion in seawater after drying overnight. The nauplii (1.0~3.0 indiv./mL) thus obtained were cultured in 2.0 L filtered (0.2 μm) natural seawater (diluted by DW: salinity 28) containing penicillin G (20 μg/mL) and streptomycin sulfate (30 μg/mL) at 25 °C and were fed on the diatom
Chaetoceros gracillis at concentrations of 40 × 10
4 cells/mL. Larvae reached the cyprid stage in 5 days. The cyprids were collected, then stored at 4 °C until use (0-day-old).
The test compounds were dissolved in ethanol and aliquots of the solution (20 μL) were transferred to wells of a 24-well polystyrene culture plates and then air-dried for 3 h at room temperature and CuSO4 was used as positive compound. Four wells were used for each concentration (0.03, 0.1, 0.3, 1.0, 3.0, 10.0 μg/mL). To each well were added filtered (0.2 mm) natural seawater (2.0 mL, salinity 28) and six 2-day-old cyprids. The plates were kept in the dark at 25 °C for 48 h. The numbers of cyprids that attached, metamorphosed, died, or did not settle were counted under a microscope. Three or four trials were carried out for each concentration. Antifouling activity (EC50) indicates the concentration reducing the larval settlement to 50% of the control (non-treatment) by Probit analysis. Toxicity of compounds were expressed as LC50 value, which indicates the concentration showing 50% mortality estimated by Probit analysis. If mortality rate did not show over 50% at most hagh concentration (10.0 μg/mL), then LC50 value was indicated as over 10.0 μg/mL.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}