
Isolation of Nocuolin A and Synthesis of New Oxadiazine Derivatives. Design, Synthesis, Molecular Docking, Apoptotic Evaluation, and Cathepsin B Inhibition

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
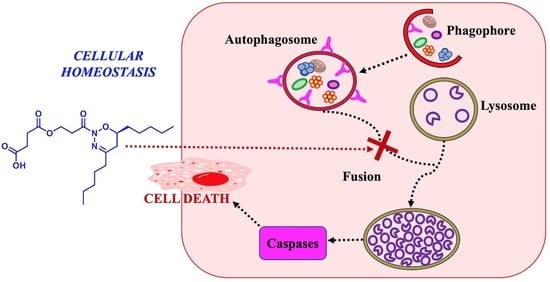
1. Introduction
2. Results and Discussion
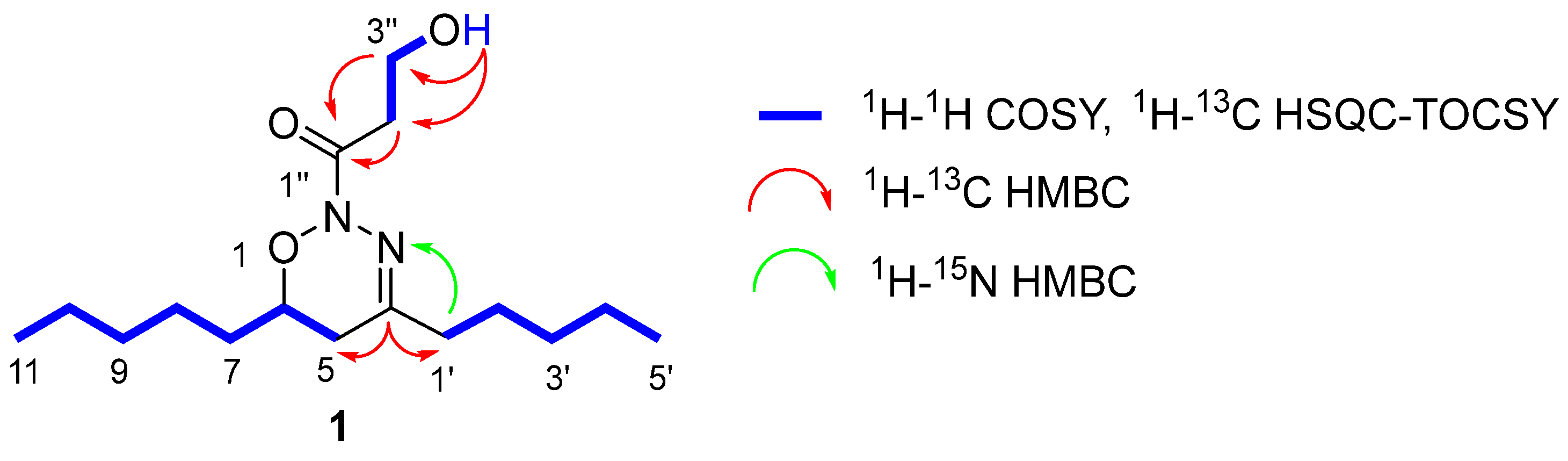
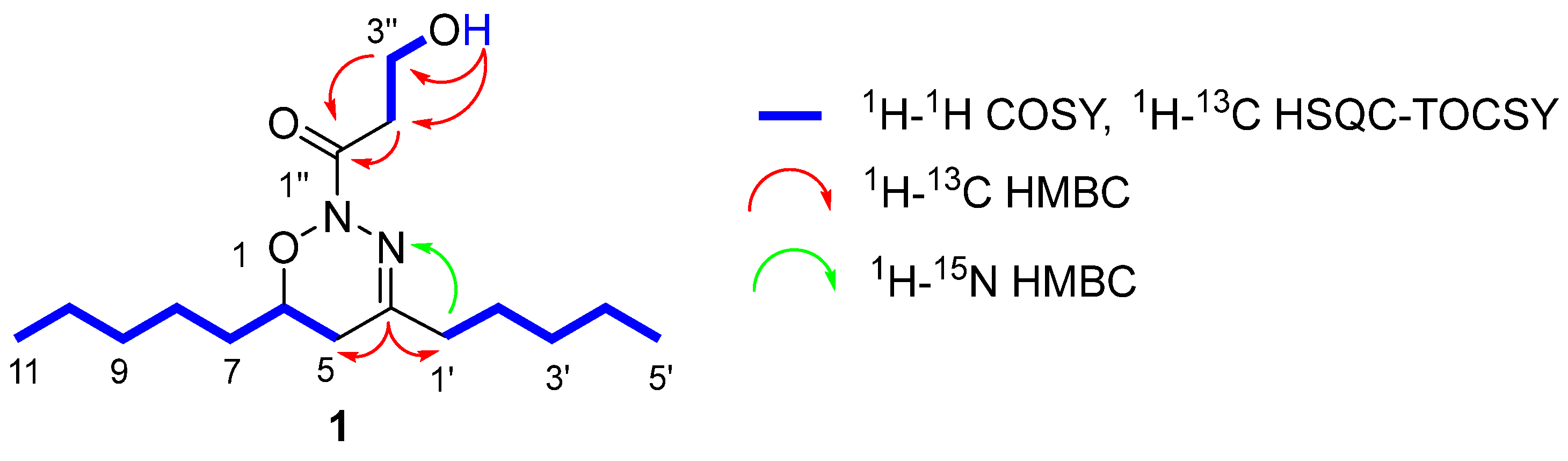
2.1. Structural Elucidation of the Natural Compound
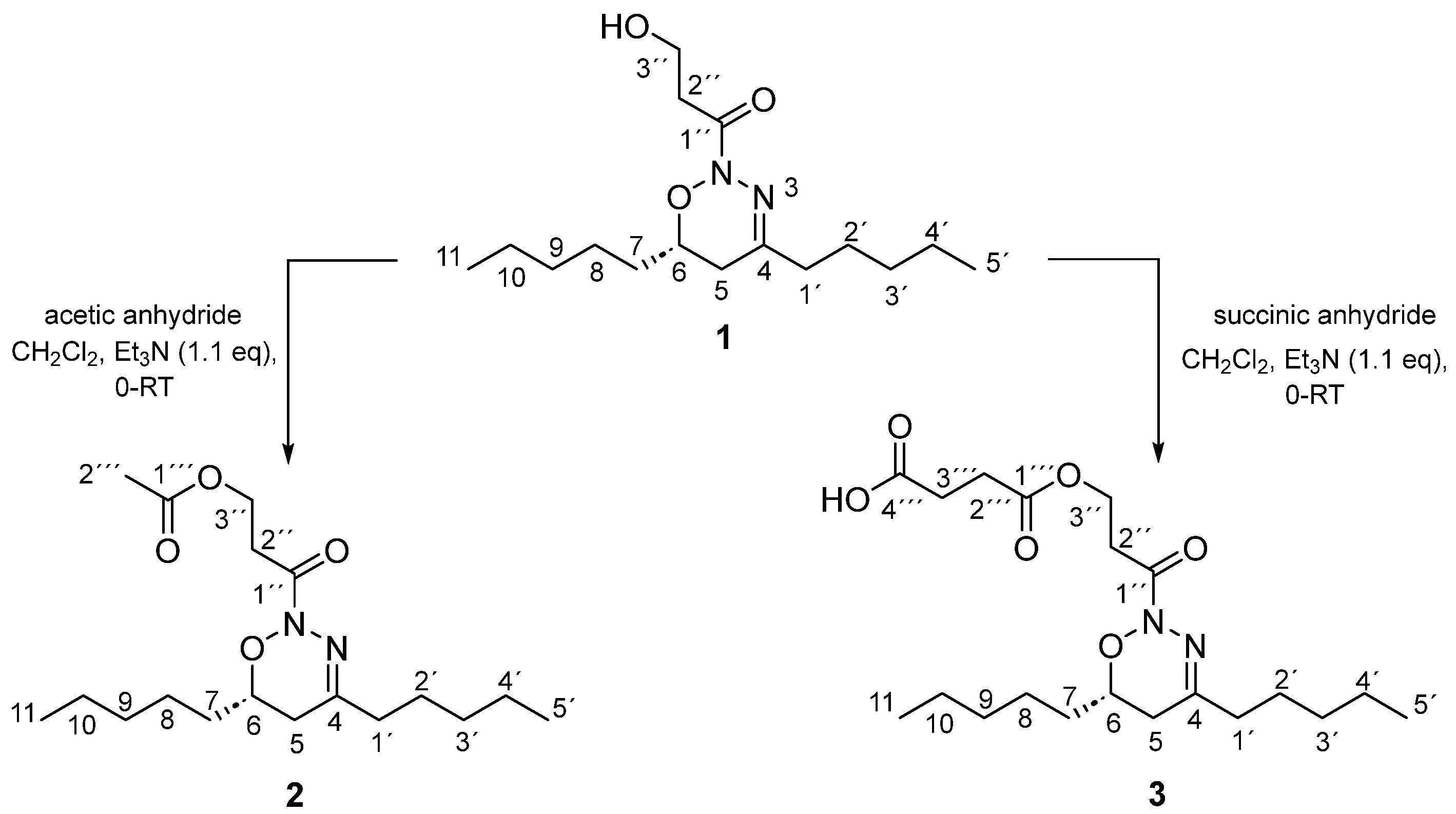
2.2. Synthesis of Analogues of the Natural Compound
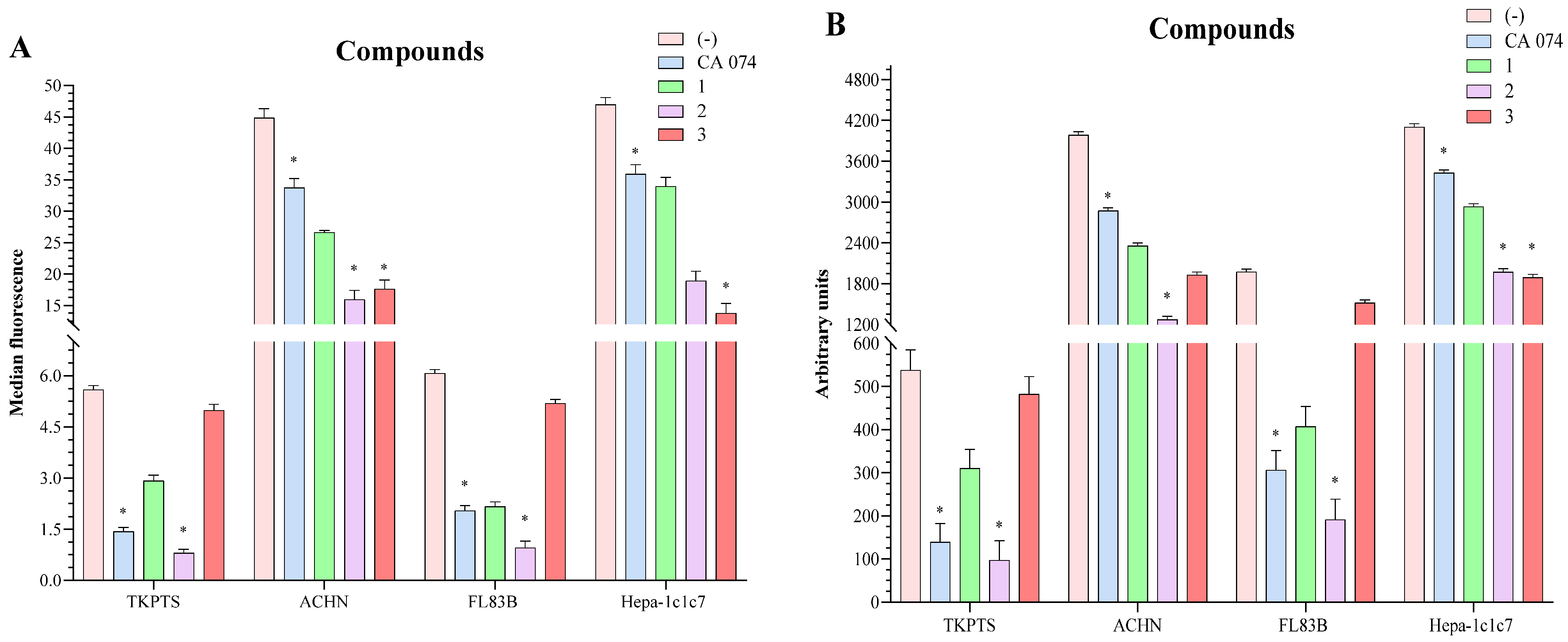
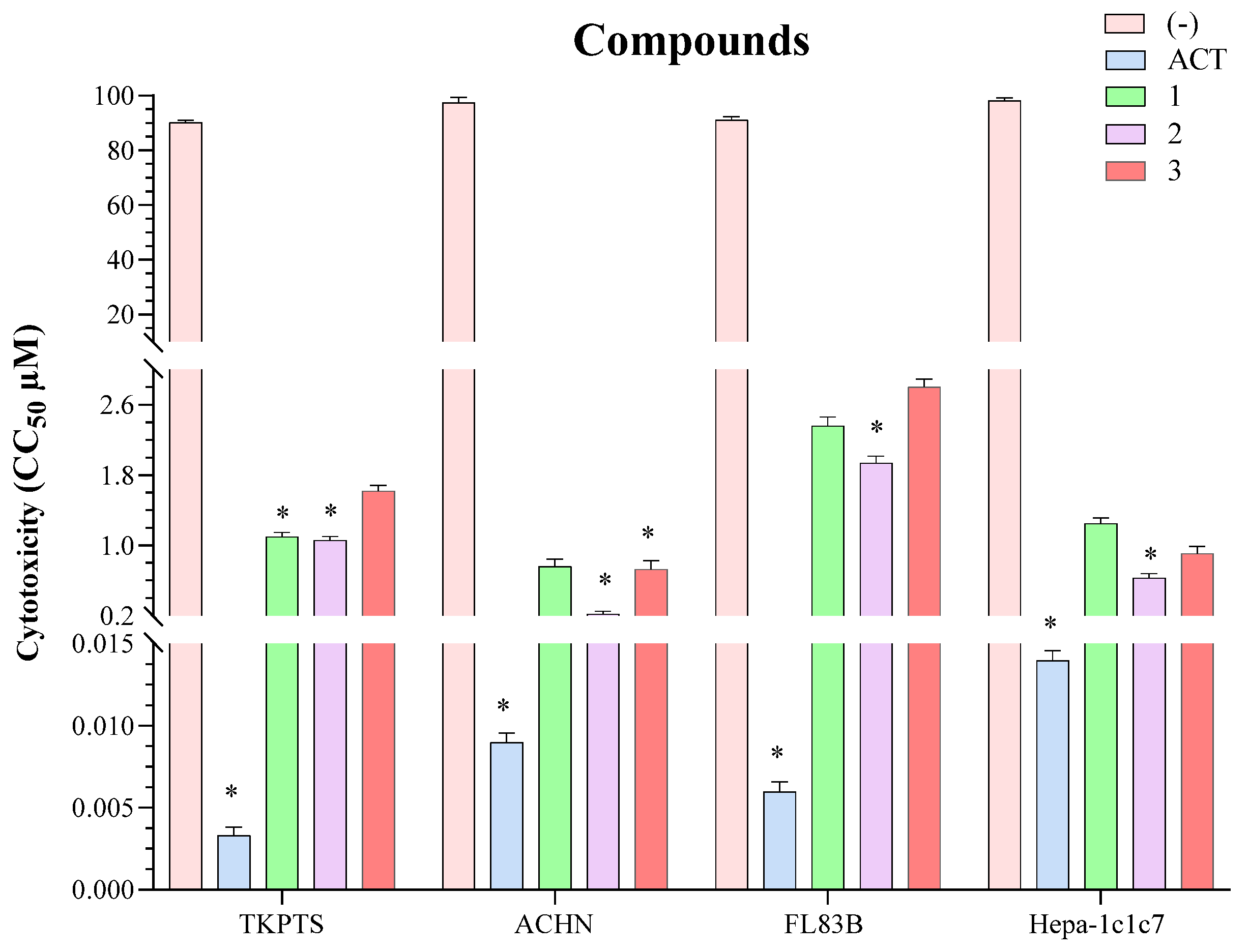
2.3. In Vitro Viability
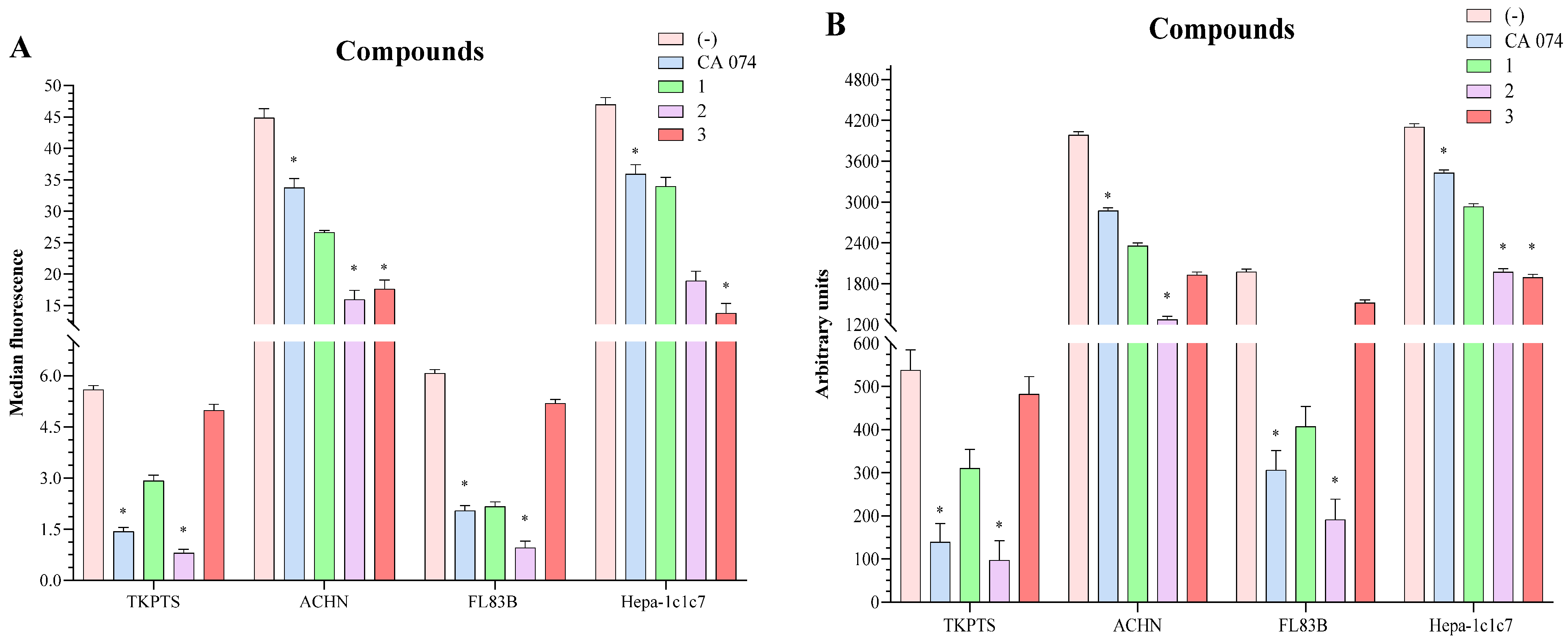
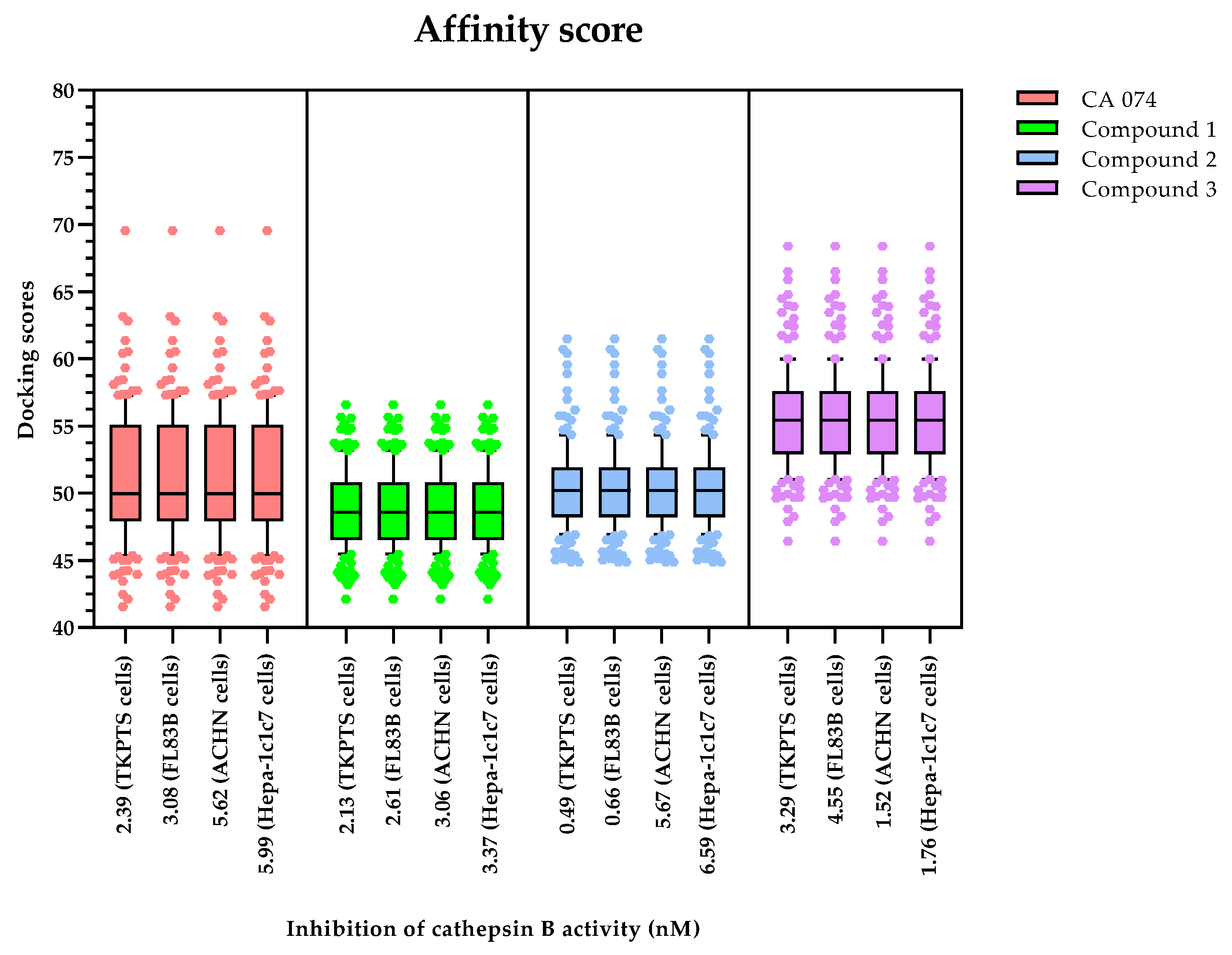
2.4. Inhibition of Cathepsin B Activity
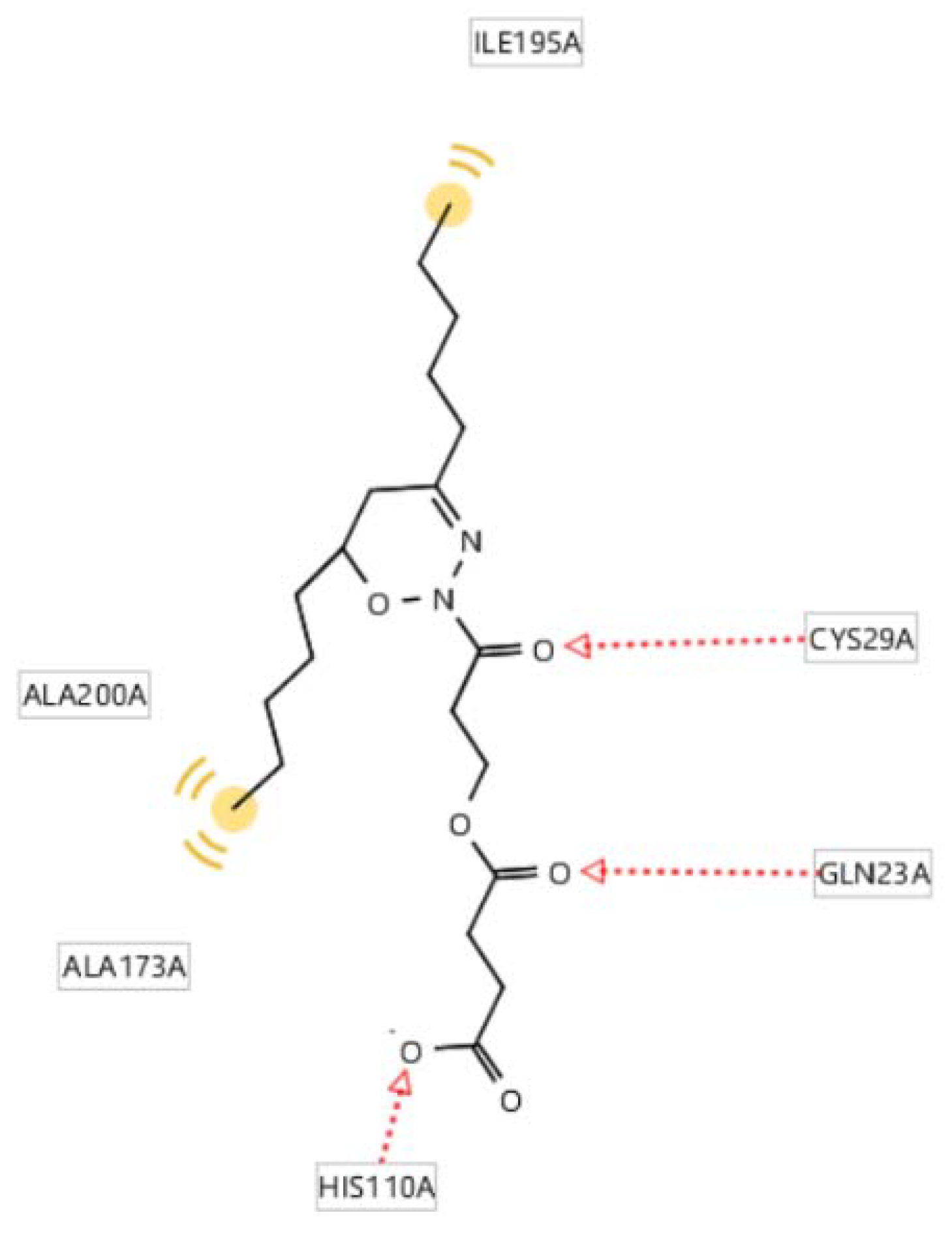
2.5. Molecular Docking of the Active Compounds
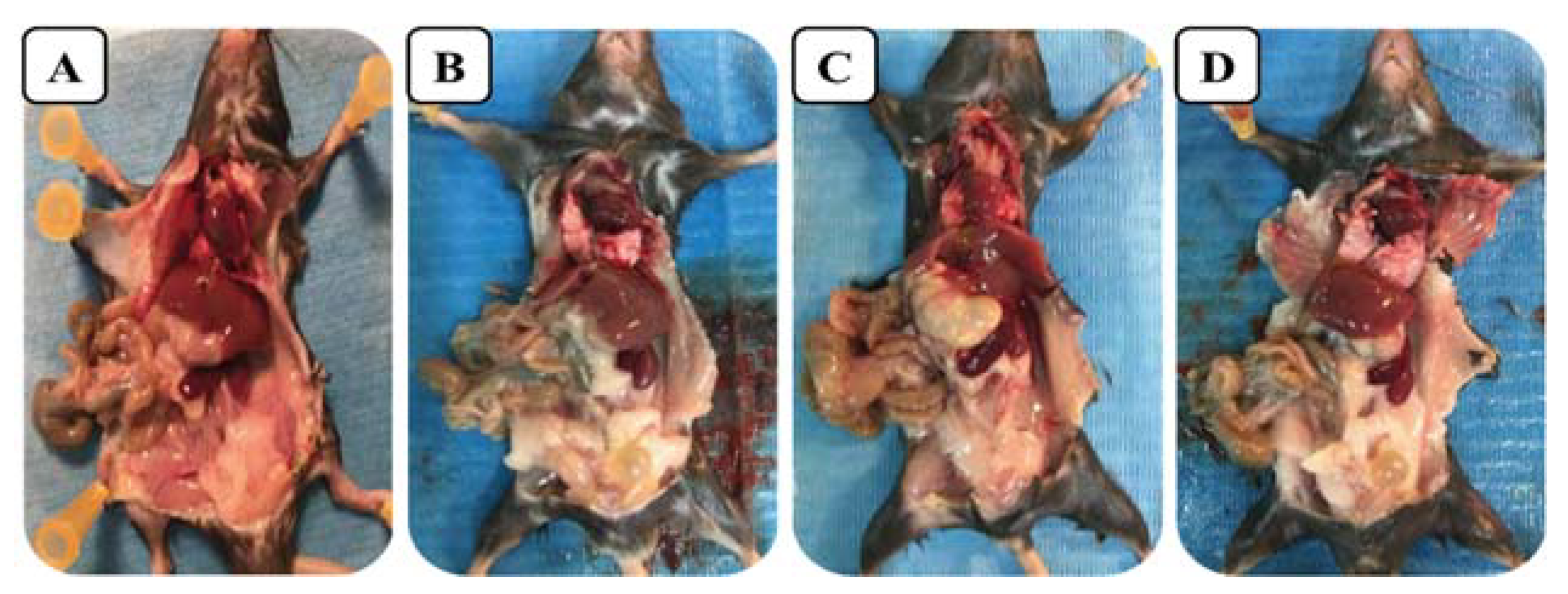
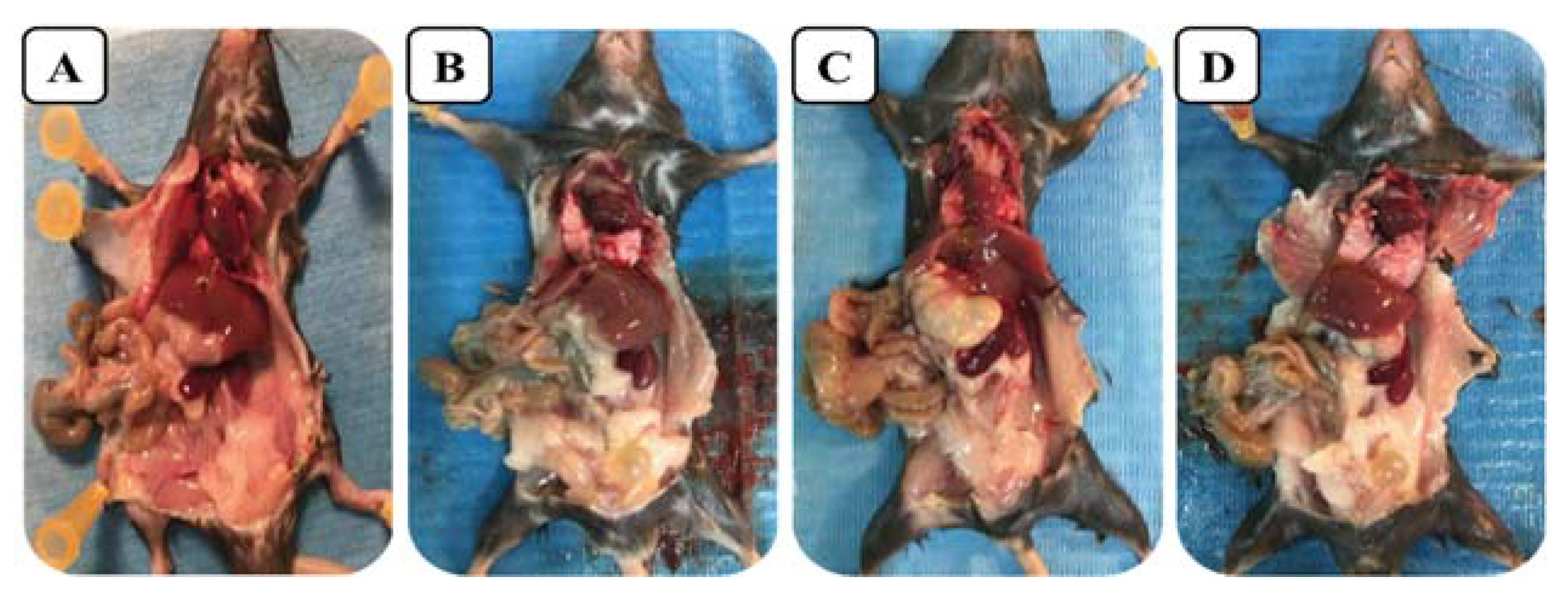
2.6. Acute Toxicity In Vivo
3. Material and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. General Procedure for the Synthesis of Oxadiazines Analogues
3.5. Spectroscopic Data
3.5.1. 1-[(6R)-5,6-Dihydro-4,6-dipentyl-2H-1,2,3-oxadiazin-2-yl]-3-hydroxypropan-1-one (1)
3.5.2. 3-[(6R)-5,6-Dihydro-4,6-dipentyl-2H-1,2,3-oxadiazin-2-yl]-3-oxopropyl Acetate (2)
3.5.3. 4-{3-[(6R)-5,6-Dihydro-4,6-dipentyl-2H-1,2,3-oxadiazin-2-yl]-3-oxopropoxy}-4-oxobutanoic Acid (3)
3.6. Cell Culture
3.7. Viability Assay
3.8. Assay for Activity of Cathepsin B
3.9. In Vivo Toxicity
3.10. Statistical Analysis
3.11. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Mohan, V.; Das, A.; Sagi, I. Emerging roles of ECM remodeling processes in cancer. Semin. Cancer Biol. 2020, 62, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Khaket, T.P.; Kwon, T.K.; Kang, S.C. Cathepsins: Potent regulators in carcinogenesis. Pharmacol. Ther. 2019, 198, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, H. Cathepsins. In xPharm The Comprehensive Pharmacology Reference, 1st ed.; Enna, S.J., Bylund, D.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 2567–2576. [Google Scholar] [CrossRef]
- Fonović, M.; Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Biophys. Acta 2014, 1840, 2560–2570. [Google Scholar] [CrossRef]
- Marí, M.; Fernandez-Checa, J.C. Damage Mediated by Dysfunction of Organelles and Cellular Systems: Lysosomes. Pathobiol. Human Dis. 2014, 97–107. [Google Scholar] [CrossRef]
- Lalmanach, G.; Saidi, A.; Bigot, P.; Chazeirat, T.; Lecaille, F.; Wartenberg, M. Regulation of the Proteolytic Activity of Cysteine Cathepsins by Oxidants. Int. J. Mol. Sci. 2020, 21, 1944. [Google Scholar] [CrossRef]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.J.; Peng, Z.K.; Lu, J.P.; Tang, F.Q. Cathepsins mediate tumor metastasis. World J. Biol. Chem. 2013, 4, 91–101. [Google Scholar] [CrossRef]
- Jokimaa, V.; Oksjoki, S.; Kujari, H.; Vuorio, E.; Anttila, L. Expression patterns of cathepsins B, H, K, L and S in the human endometrium. Mol. Hum. Reprod. 2001, 7, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Mijanović, O.; Branković, A.; Panin, A.N.; Savchuk, S.; Timashev, P.; Ulasov, I.; Lesniak, M.S. Cathepsin B: A sellsword of cancer progression. Cancer Lett. 2019, 449, 207–214. [Google Scholar] [CrossRef]
- Akinyemi, A.O.; Pereira, G.B.S.; Rocha, F.V. Role of Cathepsin B in Cancer Progression: A Potential Target for Coordination Compounds. Mini Rev. Med. Chem. 2021, 21, 1612–1624. [Google Scholar] [CrossRef]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef]
- Jung, M.; Lee, J.; Seo, H.Y.; Lim, J.S.; Kim, E.K. Cathepsin inhibition-induced lysosomal dysfunction enhances pancreatic beta-cell apoptosis in high glucose. PLoS ONE 2015, 10, e0116972. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Fang, J.; Ao, G.Z. Cathepsin B and L inhibitors: A patent review (2010–present). Expert Opin. Ther. Pat. 2017, 27, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Mitrović, A.; Mirković, B. The current stage of cathepsin B inhibitors as potential anticancer agents. Future Med. Chem. 2014, 6, 1355–1371. [Google Scholar] [CrossRef] [PubMed]
- Frlan, R.; Gobec, S. Inhibitors of cathepsin B. Curr. Med. Chem. 2006, 13, 2309–2327. [Google Scholar] [CrossRef]
- Tomoo, K. Development of cathepsin inhibitors and structure-based design of cathepsin B-specific inhibitor. Curr. Top. Med. Chem. 2010, 10, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T.; Phyo, M.Y. Marine Cyanobacteria: A Source of Lead Compounds and their Clinically Relevant Molecular Targets. Molecules 2020, 25, 2197. [Google Scholar] [CrossRef]
- Shahid, A.; Khurshid, M.; Aslam, B.; Muzammil, S.; Mehwish, H.M.; Rajoka, M.S.R.; Hayat, H.F.; Sarfraz, M.H.; Razzaq, M.K.; Nisar, M.A.; et al. Cyanobacteria derived compounds: Emerging drugs for cancer management. J. Basic Microbiol. 2022, 62, 1125–1142. [Google Scholar] [CrossRef]
- Hamida, R.S.; Albasher, G.; Bin-Meferij, M.M. Apoptotic Responses Mediated by Nostoc-Synthesized Silver Nanoparticles against Ehrlich Ascites Carcinoma Tumor-Bearing Mice. J. Nanomater. 2023, 5, 1–13. [Google Scholar] [CrossRef]
- Guo, M.; Ding, G.B.; Guo, S.; Li, Z.; Zhao, L.; Li, K.; Guo, X. Isolation and antitumor efficacy evaluation of a polysaccharide from Nostoc commune Vauch. Food Funct. 2015, 6, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Hernández Cabanillas, A.; Maderuelo Corral, S.; Ortega Domenech, M.; Rosero Valencia, D.; Rumbero Sánchez, Á.; Tena Pérez, V.; Apaza Ticona, L. Uses of Oxadiazine Compounds. EP3744328A1, 2 December 2020. [Google Scholar]
- Voráčová, K.; Hájek, J.; Mareš, J.; Urajová, P.; Kuzma, M.; Cheel, J.; Villunger, A.; Kapuscik, A.; Bally, M.; Novák, P.; et al. The cyanobacterial metabolite Nocuolin a is a natural oxadiazine that triggers apoptosis in human cancer cells. PLoS ONE 2017, 12, e0172850. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, S.A.C.; Preto, M.; Moreira, G.; Martins, T.P.; Abt, K.; Melo, A.; Vasconcelos, V.M.; Leão, P.N. Discovery of Cyanobacterial Natural Products Containing Fatty Acid Residues. Angew. Chem. Int. Ed. Engl. 2021, 60, 10064–10072. [Google Scholar] [CrossRef] [PubMed]
- Hernández Cabanillas, A.; Maderuelo Corral, S.; Ortega Domenech, M.; Rosero Valencia, D.; Rumbero Sánchez, Á.; Tena Pérez, V.; Apaza Ticona, L. Uses of Doxadiazine Compounds. EP3744329A1/WO2020/240037A1, 2 December 2020. [Google Scholar]
- Cavallo-Medved, D.; Moin, K.; Sloane, B. Cathepsin B: Basis Sequence: Mouse. AFCS Nat. Mol. Pages 2011, 2011, A000508. [Google Scholar]
- Mirković, B.; Renko, M.; Turk, S.; Sosič, I.; Jevnikar, Z.; Obermajer, N.; Turk, D.; Gobec, S.; Kos, J. Novel mechanism of cathepsin B inhibition by antibiotic nitroxoline and related compounds. ChemMedChem 2011, 6, 1351–1356. [Google Scholar] [CrossRef]
- Matarrese, P.; Ascione, B.; Ciarlo, L.; Vona, R.; Leonetti, C.; Scarsella, M.; Mileo, A.M.; Catricalà, C.; Paggi, M.G.; Malorni, W. Cathepsin B inhibition interferes with metastatic potential of human melanoma: An in vitro and in vivo study. Mol. Cancer 2010, 9, 1–14. [Google Scholar] [CrossRef]
- Mort, J.S. Cathepsin B. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 1784–1791. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development, and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: The structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar] [CrossRef]
- Yoon, M.C.; Christy, M.P.; Phan, V.V.; Gerwick, W.H.; Hook, G.; O’Donoghue, A.J.; Hook, V. Molecular Features of CA-074 pH-Dependent Inhibition of Cathepsin B. Biochem. J. 2022, 61, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Krupa, J.C.; Hasnain, S.; Nägler, D.K.; Ménard, R.; Mort, J.S. S2’ substrate specificity and the role of His110 and His111 in the exopeptidase activity of human cathepsin B. Biochem. J. 2002, 361, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Cotrin, S.S.; Puzer, L.; de Souza Judice, W.A.; Juliano, L.; Carmona, A.K.; Juliano, M.A. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: Assays with human cathepsin B. Anal. Biochem. 2004, 335, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.Y. Voltage-gated Sodium Channel Activity Promotes Cysteine Cathepsin-dependent Invasiveness and Colony Growth of Human Cancer Cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T.; Kohda, Y.; Tsuchiya, K.; Ueno, T.; Yamashita, J.; Yoshioka, T.; Kominami, E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: A novel strategy for neuroprotection based on ’calpain-cathepsin hypothesis’. Eur. J. Neurosci. 1998, 10, 1723–1733. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tena Pérez, V.; Apaza Ticona, L.; H. Cabanillas, A.; Maderuelo Corral, S.; Rosero Valencia, D.F.; Martel Quintana, A.; Ortega Domenech, M.; Rumbero Sánchez, Á. Isolation of Nocuolin A and Synthesis of New Oxadiazine Derivatives. Design, Synthesis, Molecular Docking, Apoptotic Evaluation, and Cathepsin B Inhibition. Mar. Drugs 2023, 21, 284. https://doi.org/10.3390/md21050284
Tena Pérez V, Apaza Ticona L, H. Cabanillas A, Maderuelo Corral S, Rosero Valencia DF, Martel Quintana A, Ortega Domenech M, Rumbero Sánchez Á. Isolation of Nocuolin A and Synthesis of New Oxadiazine Derivatives. Design, Synthesis, Molecular Docking, Apoptotic Evaluation, and Cathepsin B Inhibition. Marine Drugs. 2023; 21(5):284. https://doi.org/10.3390/md21050284
Chicago/Turabian StyleTena Pérez, Víctor, Luis Apaza Ticona, Alfredo H. Cabanillas, Santiago Maderuelo Corral, Diego Fernando Rosero Valencia, Antera Martel Quintana, Montserrat Ortega Domenech, and Ángel Rumbero Sánchez. 2023. "Isolation of Nocuolin A and Synthesis of New Oxadiazine Derivatives. Design, Synthesis, Molecular Docking, Apoptotic Evaluation, and Cathepsin B Inhibition" Marine Drugs 21, no. 5: 284. https://doi.org/10.3390/md21050284