Phase II Randomized Study of Plitidepsin (Aplidin), Alone or in Association with L-carnitine, in Patients with Unresectable Advanced Renal Cell Carcinoma

Abstract
:1. Introduction
2. Results and Discussion
2.1. Patients and methods
Patient selection criteria
Study design and endpoints
Drug administration and concomitant medication
Diagnostic process
Statistics
2.2. Results
Study patients
Protocol adherence
Duration of treatment and reasons for treatment discontinuation
2.3. Progression free and response rate
2.4. Duration of response, progression-free interval and overall survival
2.5. Safety
2.6. Discussion
Acknowledgments
References and Notes
- Cancer Facts & Figures - 1996; American Cancer Society: Atlanta, GA, 1996.
- Linehan, WM; Shipley, W; Parkinson, D. Cancer of the kidney and ureter. In Cancer: Principles and Practice of Oncology, 5th Ed; De Vita, VTJ, Hellman, S, Rosenberg, SA, Eds.; Lippincott-Raven: Philadelphia, PA, 1997; pp. 1271–1300. [Google Scholar]
- Maher, ER; Iselius, L; Yates, JR; Littler, M; Benjamin, C; Harris, R; Sampson, J; Williams, A; Ferguson-Smith, MA; Morton, N. Von Hippel-Lindau disease: a genetic study. J. Med. Genet 1991, 28, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Pisters, LL; El-Naggar, AK; Luo, W; Malpica, A; Lin, SH. C-met proto-oncogene expression in benign and malignant human renal tissues. J. Urol 1997, 158, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, JR; Zhou, S; Merrill, MJ; Wagner, JR; Krumm, A; Papavassiliou, E; Oldfield, EH; Klausner, RD; Linehan, WM. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 10589–10594. [Google Scholar] [CrossRef] [PubMed]
- Sene, AP; Hunt, L; McMahon, RF; Carroll, RN. Renal carcinoma in patients undergoing nephrectomy: analysis of survival and prognostic factors. Br. J. Urol 1992, 70, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Motzer, RJ; Bander, NH; Nanus, DM. Renal-cell carcinoma. N. Engl. J. Med 1996, 335, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Negrier, S; Escudier, B; Lasset, C; Douillard, JY; Savary, J; Chevreau, C; Ravaud, A; Mercatello, A; Peny, J; Mousseau, M; Philip, T; Tursz, T. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. Groupe Francais d'Immunotherapie. N. Engl. J. Med 1998, 338, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, RM. Natural history and therapy of metastatic RCC: the role of interleukin-2. Cancer 1997, 80, 1198–1220. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, SA; Lotze, MT; Muul, LM; Chang, AE; Avis, FP; Leitman, S; Linehan, WM; Robertson, CN; Lee, RE; Rubin, JT; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Biscadi, M; Caporale, R; Balestri, F; Gavazzi, S; Jimeno, J; Grossi, A. VEGF inhibition and cytotoxic effect of aplidin in leukemia cell lines and cells from acute myeloid leukemia. Ann. Oncol 2005, 16, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Faircloth, G; Rinehart, K; Nunez, DC. Dehydrodidemnin B a new marine derived antitumor agent with activity against experimental tumor models. Ann. Oncol 1996, 7, 34. [Google Scholar]
- Casciari, JJ; Hollingshead, MG; Alley, MC; Mayo, JG; Malspeis, L; Miyauchi, S; Grever, MR; Weinstein, JN. Growth and chemotherapeutic response of cells in a hollow-fiber in vitro solid tumor model. J. Natl. Cancer Inst 1994, 86, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- Anthoney, A; Paz-Ares, L; Twelves, C. Phase I and pharmacokinetic (PK) study of Aplidin (APL) using a 24-hour, weekly schedule. Proc Am Soc Clin Oncol 2000, 734. [Google Scholar]
- Izquierdo, MA; Bowman, A; Garcia, M; Jodrell, D; Martinez, M; Pardo, B; Gómez, J; López-Martin, JA; Jimeno, J; Germá, JR; Smyth, JF. Phase I clinical and pharmacokinetic study of plitidepsin as a 1-hour weekly intravenous infusion in patients with advanced solid tumors. Clin. Cancer Res 2008, 14, 3105–3112. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S; Chièze, S; Delbaldo, C; Ady-Vago, N; Guzman, C; Lopez-Lazaro, L; Lozahic, S; Jimeno, J; Pico, F; Armand, JP; Martin, JA; Raymond, E. Phase I and pharmacokinetic study of aplidine aplidine, a new marine cyclodepsipeptide, in patients with advanced malignancies. J. Clin. Oncol 2005, 23, 7780–7782. [Google Scholar] [CrossRef] [PubMed]
- Maroun, JA; Belanger, K; Seymour, L; Ady-Vago, N; Guzman, C; Lopez-Lazaro, L; Lozahic, S; Jimeno, J; Pico, F; Armand, JP; Martin, JA; Raymond, E. Phase I study of Aplidine in a dailyx5 one hour infusion every 3 weeks in patients with solid tumors refractory to standard therapy.A National Cancer Institute of Canada Clinical Tirals Group Study : NCIC CTG IND 115. Ann. Oncol 2006, 17, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Cumming, WJ; Hardy, M; Hudgson, P; Walls, J. Carnitine-palmityl-transferase deficiency. J. Neurol. Sci 1976, 30, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J. Renal-cell carcinoma–molecular pathways and therapies. N. Engl. J. Med 2007, 356, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Speca, J; Yenser, S; Creel, P; George, D. Improving outcomes with novel therapies for patients with newly diagnosed RCC. Clin. Genitourin Cancer 2006, 5(Suppl 1), 24–30. [Google Scholar]
- Rock, EP; Goodman, V; Jiang, JX; Mahjoob, K; Verbois, SL; Mors, D; Dagher, R; Justice, R; Pazdur, R. Food and Drug Administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced RCC. Oncologist 2007, 12, 107–113. [Google Scholar] [PubMed]
- Escudier, B; Pluzanska, A; Koralewski, P; Ravaud, A; Bracarda, S; Szczylik, C; Chevreau, C; Filipek, M; Melichar, B; Bajetta, E; Gorbunova, V; Bay, JO; Bodrogi, I; Jagiello-Gruszfeld, A; Moore, N. AVOREN Trial investigators. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma : a randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [PubMed]
- Atkins, MB; Regan, M; McDermott, DF. Update on the role of interleukin 2 and other cytokines in the treatment of patients with stage IV renal carcinoma. Clin. Cancer Res 2004, 10, 6342–6346. [Google Scholar]
- McDermott, DF; Atkins, MB. Application of IL-2 and other cytokines in RCC. Expert Opin. Biol. Ther 2004, 4, 455–468. [Google Scholar] [PubMed]
- Atkins, MB; Hidalgo, M; Stadler, WM; Loga, TF; Dutcher, JP; Hudes, GR; Park, Y; Liou, SH; Marshall, B; Boni, JP; Dukart, G; Sherman, ML. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory RCC. J. Clin. Oncol 2004, 22, 909–918. [Google Scholar] [PubMed]
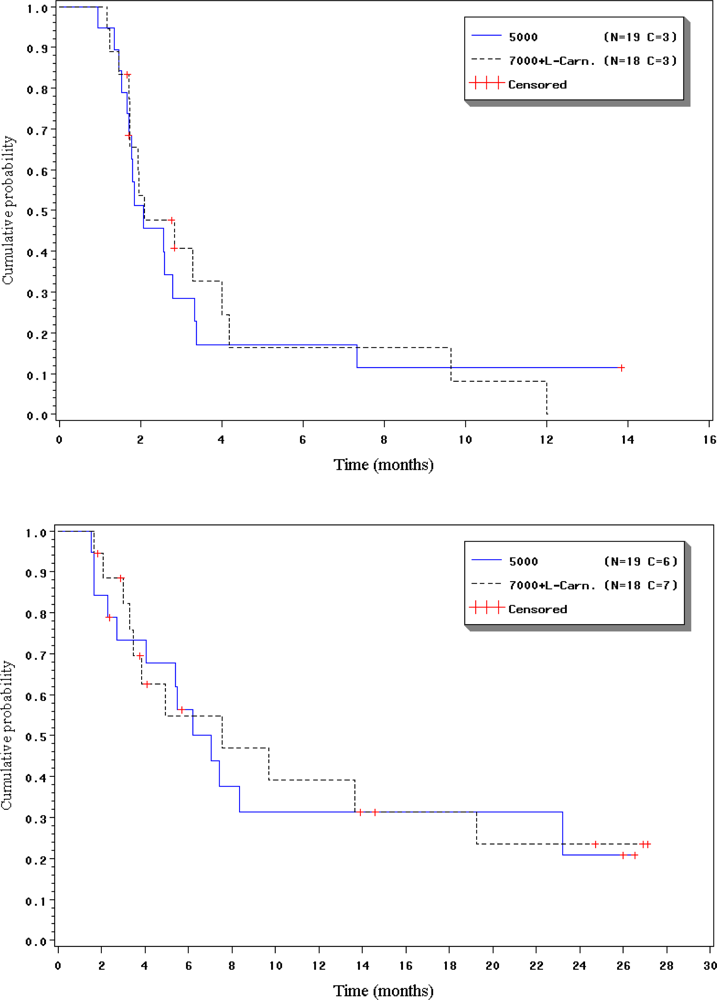

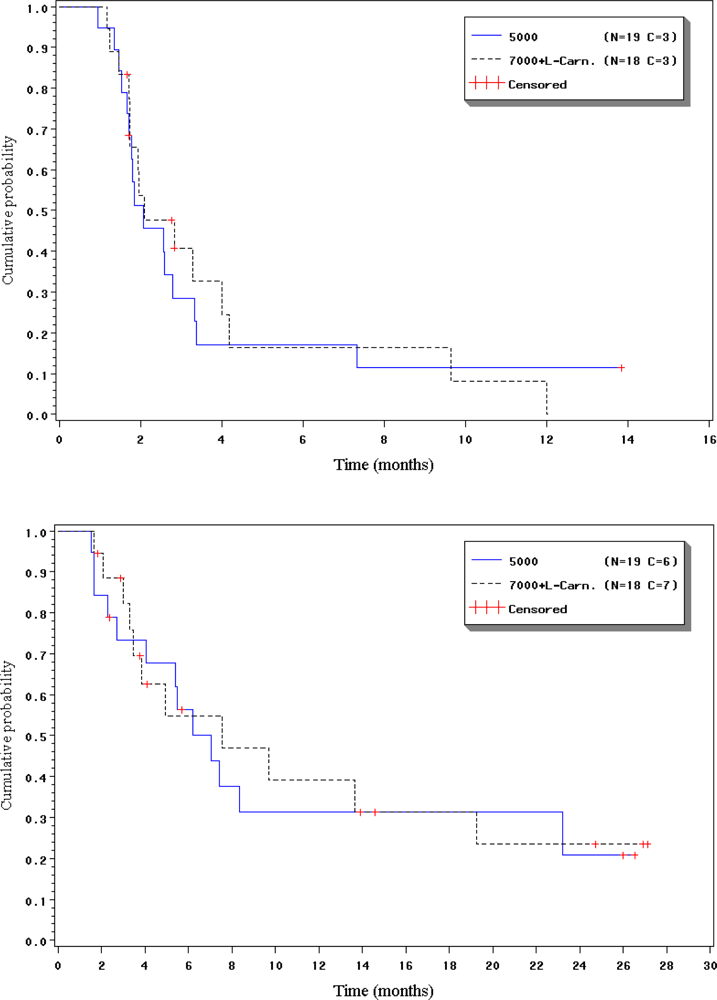{kind=link}
| Arm A | Arm B | Total | ||||
|---|---|---|---|---|---|---|
| n | % | n | % | % | ||
| No. of patients | 19 | 100.0 | 20 | 100.0 | 39 | 100.0 |
| Gender | ||||||
| Male | 13 | 68.4 | 16 | 80.0 | 29 | 74.4 |
| Female | 6 | 31.6 | 4 | 20.0 | 10 | 25.6 |
| Race | ||||||
| Caucasian | 19 | 100.0 | 18 | 90.0 | 37 | 94.9 |
| Hispanic | . | . | 2 | 10.0 | 2 | 5.1 |
| Age (years) | ||||||
| Median | 56.0 | . | 56.0 | . | 56.0 | . |
| Range | 28.0–75.0 | . | 42.0–74.0 | . | 28.0–75.0 | . |
| ECOG PS | ||||||
| 0 | 10 | 52.6 | 10 | 50.0 | 20 | 51.3 |
| 1 | 9 | 47.4 | 10 | 50.0 | 19 | 48.7 |
| Histology of primary tumor | ||||||
| Chromophobic | 1 | 5.3 | . | . | 1 | 2.6 |
| Clear cell carcinoma | 16 | 84.1 | 19 | 95.0 | 35 | 89.6 |
| Epidermoid and clear cell carcinoma | . | . | 1 | 5.0 | 1 | 2.6 |
| Mixed clear cell carcinoma | 1 | 5.3 | . | . | 1 | 2.6 |
| Papillary carcinoma | 1 | 5.3 | . | 1 | 2.6 | |
| No. of metastatic sites | 2 | . | 2 | . | 2 | . |
| Median (range) | (1–5) | . | (1–5) | . | (1–5) | . |
| Time from diagnosis to plitidepsin (months) | . | |||||
| Median | 13.8 | 19.1 | . | 13.8 | . | |
| Range | 1.0–109.5 | . | 1.0–109.9 | . | 1.0–109.9 | . |
| Surgery | ||||||
| Yes | 16 | 84.2 | 19 | 95.0 | 35 | 89.7 |
| No | 3 | 15.8 | 1 | 5.0 | 4 | 10.3 |
| Radiotherapy | ||||||
| Yes | 4 | 21.1 | 6 | 30.0 | 10 | 25.6 |
| No | 15 | 78.9 | 14 | 70.0 | 29 | 74.4 |
| Immunotherapy | ||||||
| Yes | 14 | 73.7 | 15 | 75.0 | 29 | 74.4 |
| No | 5 | 26.3 | 5 | 25.0 | 10 | 25.6 |
| Prior immunotherapy agents | 19 | 100.0 | 18 | 100.0 | 37 | 100.0 |
| n | 7 | 36.8 | 11 | 61.1 | 18 | 48.6 |
| Interferon | 7 | 36.9 | 4 | 22.2 | 11 | 29.8 |
| Interleukin-2 | 5 | 26.3 | 3 | 16.7 | 8 | 21.6 |
| Chemotherapy | ||||||
| Yes | 5 | 26.3 | 6 | 30.0 | 11 | 28.2 |
| No | 14 | 73.7 | 14 | 70.0 | 28 | 71.8 |
| ARM A | NCI-CTC grade | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| n | % | n | % | n | % | |
| Constipation | 2 | 10.5 | . | . | . | . |
| Diarrhea | 5 | 26.3 | . | . | . | . |
| Nausea | 10 | 52.6 | 4 | 21.1 | . | . |
| Vomiting | 9 | 47.4 | 2 | 10.5 | 1 | 5.3 |
| Asthenia | 4 | 21.1 | 6 | 31.6 | 3 | 15.8 |
| Edema | 3 | 15.8 | . | . | . | . |
| Injection site reaction | 3 | 15.8 | 2 | 10.5 | 1 | 5.3 |
| Weight decreased | 2 | 10.5 | . | . | . | . |
| Anorexia | 7 | 36.8 | 2 | 10.5 | . | . |
| Muscle cramps | 2 | 10.5 | 1 | 5.3 | . | . |
| Muscle weakness | 2 | 10.5 | 3 | 15.8 | . | . |
| Myalgia | 3 | 15.8 | 3 | 15.8 | 1 | 5.3 |
| Dizziness | 2 | 10.5 | . | . | . | . |
| Folliculitis | 2 | 10.5 | . | . | . | . |
| Rash | 1 | 5.3 | . | . | . | . |
| ARM B | NCI-CTC grade | |||||
| 1 | 2 | 3 | ||||
| n | % | n | % | n | % | |
| Constipation | 2 | 10.0 | . | . | . | . |
| Diarrhea | 4 | 20.0 | 3 | 15.0 | . | . |
| Nausea | 7 | 35.0 | 9 | 45.0 | . | . |
| Stomatitis | 2 | 10.0 | . | . | . | . |
| Vomiting | 10 | 50.0 | 6 | 30.0 | . | . |
| Asthenia | 2 | 10.0 | 10 | 50.0 | 1 | 5.0 |
| Injection site reaction | 3 | 15.0 | 2 | 10.0 | 1 | 5.0 |
| Anorexia | 6 | 30.0 | 3 | 15.0 | 1 | 5.0 |
| Myalgia | 5 | 25.0 | 5 | 25.0 | 1 | 5.0 |
| NCI-CTC grade | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| n | % | n | % | n | % | |
| Arm A | ||||||
| Hemoglobin | 7 | 36.8 | 11 | 57.9 | 1 | 5.3 |
| Lymphocytes | 2 | 10.5 | 3 | 15.8 | 3 | 15.8 |
| Neutrophils | 1 | 5.3 | . | . | . | . |
| Platelets | . | . | 1 | 5.3 | 1 | 5.3 |
| WBC | . | . | 1 | 5.3 | . | . |
| Arm B | ||||||
| Hemoglobin | 12 | 60.0 | 6 | 30.0 | 1 | 5.0 |
| Lymphocytes | . | . | 3 | 15.8 | 2 | 10.5 |
| Neutrophils | 1 | 5.3 | . | . | . | . |
| Platelets | 3 | 15.8 | . | . | 1 | 5.3 |
| WBC | 2 | 10.5 | . | . | . | . |
| NCI-CTC grade | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| n | % | n | % | n | % | n | % | |
| Arm A | ||||||||
| ALT | 5 | 26.3 | 3 | 15.8 | 4 | 21.1 | . | . |
| AP | 6 | 31.6 | 3 | 15.8 | . | . | . | . |
| AST | 9 | 47.4 | 2 | 10.5 | 2 | 10.5 | . | . |
| CPK | 3 | 15.8 | 2 | 10.5 | . | . | 1 | 5.3 |
| Creatinine | 10 | 52.6 | 2 | 10.5 | . | . | . | . |
| GGT | 4 | 21.1 | 4 | 21.1 | 7 | 36.8 | . | . |
| LDH | 10 | 55.6 | 2 | 11.1 | . | . | 1 | 5.6 |
| Total bilirubin | 3 | 15.8 | . | . | . | . | . | . |
| Hypoalbuminemia | 9 | 52.9 | 4 | 23.5 | . | . | . | . |
| Hypercalcemia | 7 | 36.8 | . | . | 1 | 5.3 | . | . |
| Hypocalcemia | 2 | 10.5 | 6 | 31.6 | . | . | . | . |
| Hyperglycemia | 11 | 57.9 | 4 | 21.1 | 2 | 10.5 | . | . |
| Hypoglycemia | 1 | 5.3 | 1 | 5.3 | . | . | . | . |
| Hyperkalemia | 2 | 10.5 | 3 | 15.8 | 1 | 5.3 | . | . |
| Hypokalemia | 5 | 26.3 | . | . | 2 | 10.5 | . | . |
| Hypernatremia | 2 | 10.5 | . | . | . | . | . | . |
| Hyponatremia | 6 | 31.6 | . | . | 1 | 5.3 | 1 | 5.3 |
| Arm B | ||||||||
| ALT | 6 | 30.0 | 3 | 15.0 | 6 | 30.0 | . | . |
| AP | 12 | 60.0 | 1 | 5.0 | . | . | . | . |
| AST | 5 | 25.0 | 6 | 30.0 | 1 | 5.0 | . | . |
| CPK | 5 | 25.0 | 3 | 15.0 | 1 | 5.0 | . | . |
| Creatinine | 8 | 40.0 | 4 | 20.0 | 2 | 10.0 | . | . |
| GGT | 5 | 25.0 | 6 | 30.0 | 4 | 20.0 | 2 | 10.0 |
| LDH | 10 | 50.0 | 2 | 10.0 | . | . | . | . |
| Total bilirubin | 1 | 5.0 | 1 | 5.0 | 1 | 5.0 | . | . |
| Hypoalbuminemia | 6 | 31.6 | 4 | 21.1 | . | . | . | . |
| Hypercalcemia | 7 | 35.0 | . | . | 1 | 5.0 | . | . |
| Hypocalcemia | 6 | 30.0 | 2 | 10.0 | 1 | 5.0 | 1 | 5.0 |
| Hyperglycemia | 8 | 40.0 | 5 | 25.0 | 3 | 15.0 | . | . |
| Hypoglycemia | 1 | 5.0 | 2 | 10.0 | . | . | . | . |
| Hyperkalemia | 4 | 20.0 | . | . | 3 | 15.0 | 1 | 5.0 |
| Hypokalemia | 5 | 25.0 | . | . | 1 | 5.0 | . | . |
| Hypernatremia | 2 | 10.0 | . | . | . | . | . | . |
| Hyponatremia | 6 | 30.0 | . | . | 1 | 5.0 | . | . |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schöffski, P.; Guillem, V.; Garcia, M.; Rivera, F.; Tabernero, J.; Cullell, M.; Lopez-Martin, J.A.; Pollard, P.; Dumez, H.; Garcia del Muro, X.; et al. Phase II Randomized Study of Plitidepsin (Aplidin), Alone or in Association with L-carnitine, in Patients with Unresectable Advanced Renal Cell Carcinoma. Mar. Drugs 2009, 7, 57-70. https://doi.org/10.3390/md7010057
Schöffski P, Guillem V, Garcia M, Rivera F, Tabernero J, Cullell M, Lopez-Martin JA, Pollard P, Dumez H, Garcia del Muro X, et al. Phase II Randomized Study of Plitidepsin (Aplidin), Alone or in Association with L-carnitine, in Patients with Unresectable Advanced Renal Cell Carcinoma. Marine Drugs. 2009; 7(1):57-70. https://doi.org/10.3390/md7010057
Chicago/Turabian StyleSchöffski, Patrick, Vincente Guillem, Margarita Garcia, Fernando Rivera, Josep Tabernero, Martin Cullell, Jose Antonio Lopez-Martin, Patricia Pollard, Herlinde Dumez, Xavier Garcia del Muro, and et al. 2009. "Phase II Randomized Study of Plitidepsin (Aplidin), Alone or in Association with L-carnitine, in Patients with Unresectable Advanced Renal Cell Carcinoma" Marine Drugs 7, no. 1: 57-70. https://doi.org/10.3390/md7010057
APA StyleSchöffski, P., Guillem, V., Garcia, M., Rivera, F., Tabernero, J., Cullell, M., Lopez-Martin, J. A., Pollard, P., Dumez, H., Garcia del Muro, X., & Paz-Ares, L. (2009). Phase II Randomized Study of Plitidepsin (Aplidin), Alone or in Association with L-carnitine, in Patients with Unresectable Advanced Renal Cell Carcinoma. Marine Drugs, 7(1), 57-70. https://doi.org/10.3390/md7010057