Regulation of the Spontaneous Augmentation of NaV1.9 in Mouse Dorsal Root Ganglion Neurons: Effect of PKA and PKC Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
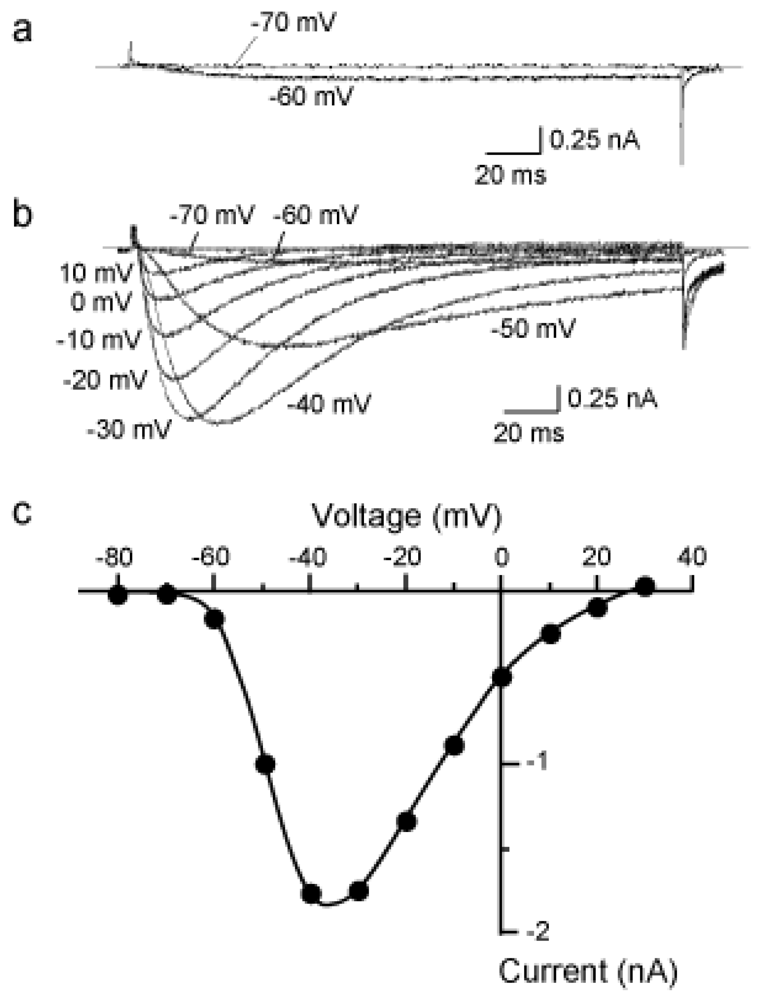
2.1. Characterization of the NaV1.9 current
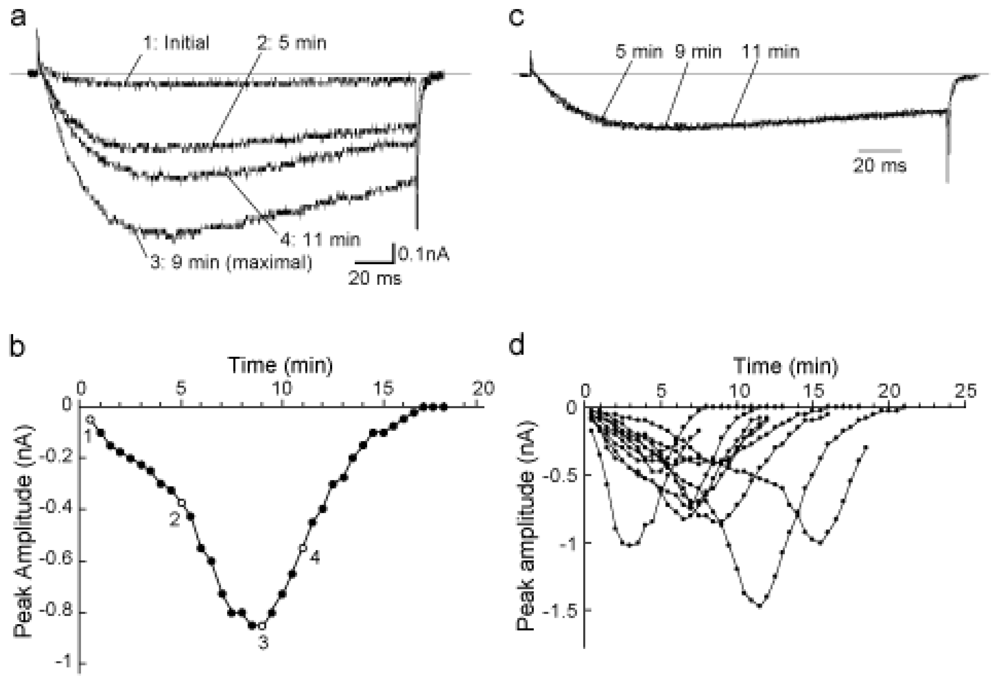
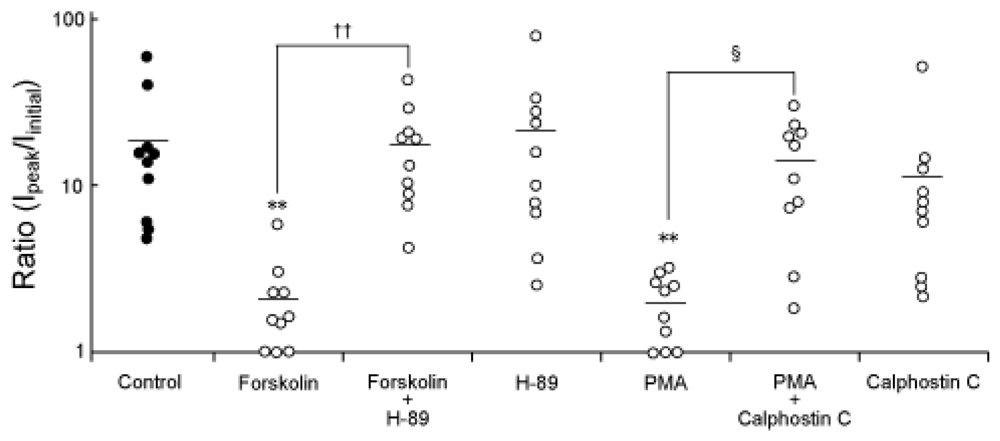
2.2. Modification of the spontaneous augmentation of NaV1.9 by PKA and PKC
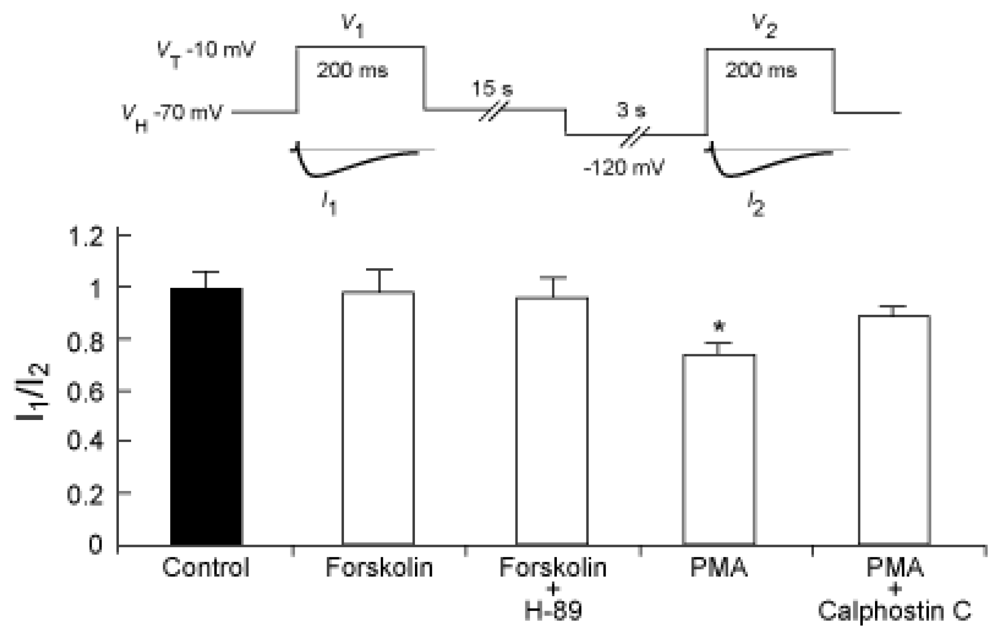
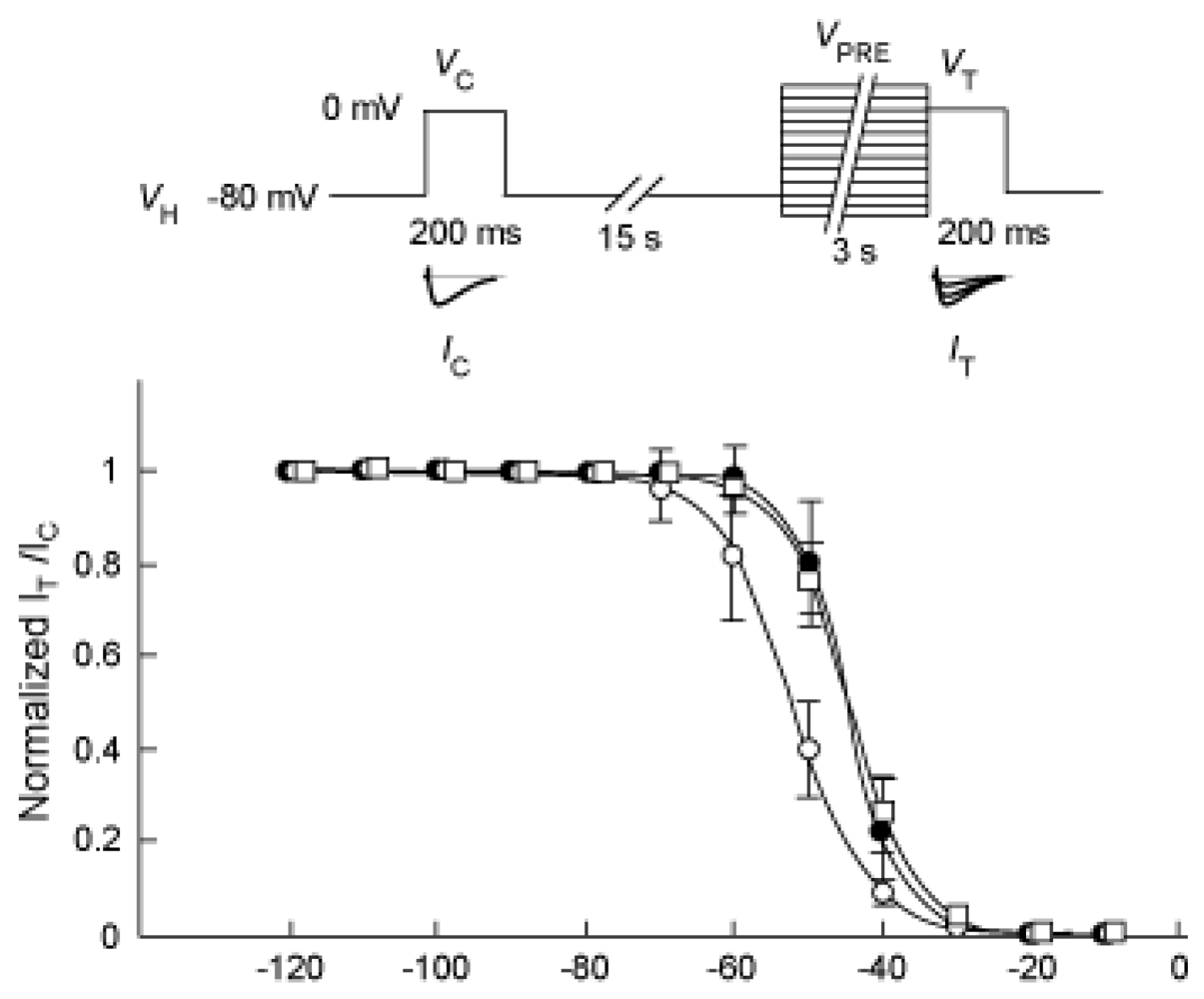
2.3. Possible involvement of PKA and PKC pathways in the voltage dependence of inactivation for the NaV1.9 current
2.4. The effect of PMA on the h∞ curve for the NaV1.9 current is reproducible under nystatin-perforated patch clamp recording
2.5. Discussion
3. Experimental Section
3.1. Isolation of single DRG neurons and cell culture
3.2. Electrophysiology
4. Conclusions
References
- Chahine, M; Ziane, R; Vijayaragaban, K; Okamura, Y. Regulation of NaV channels in sensory neurons. Trends Pharmacol Sci 2005, 26, 496–502. [Google Scholar]
- Cummins, TR; Rush, AM. Voltage-gated sodium channel blockers for the treatment of neuropathic pain. Expert Rev Neurother 2007, 7, 1597–612. [Google Scholar]
- Elliot, AA; Elliot, JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol 1993, 463, 39–56. [Google Scholar]
- Ogata, N; Tatebayashi, H. Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J Physiol 1993, 466, 9–37. [Google Scholar]
- Akopian, AN; Souslova, V; England, S; Okuse, K; Ogata, N; Ure, J; Smith, A; Kerr, BJ; McMahon, SB; Boyce, H; Hill, R; Stanfa, LC; Dickerson, AH; Wood, JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 1999, 2, 541–548. [Google Scholar]
- Renganathan, M; Cummins, TR; Waxman, SG. Contribution of NaV1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol 2001, 86, 629–640. [Google Scholar]
- Cummins, TR; Dib-Hajj, SD; Black, JA; Akopian, AN; Wood, JN; Waxman, SG. A novel persistent tetrodotoxin-resistant sodium current in sns-null and wild-type small primary sensory neurons. J Neurosci 1999, 19, 1–6. [Google Scholar]
- Dib-Hajj, SD; Tyrell, L; Escayg, A; Wood, PM; Meisler, MH; Waxman, SG. Coding sequence, genomic organization, and conserved chromosomal localization of the mouse gene Scn11a encoding the sodium channel NaN. Genomics 1999, 59, 309–318. [Google Scholar]
- Herzog, RI; Cummins, TR; Waxman, SG. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in stimulated spinal sensory neurons. J Neurophysiol 2001, 86, 1351–1364. [Google Scholar]
- Rugiero, F; Mistry, M; Sage, D; Black, JA; Waxman, SG; Crest, M; Clere, N; Delmas, P; Gola, M. Selective expression of a persistent tetrodotoxin-resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J Neurosci 2003, 23, 2715–2725. [Google Scholar]
- Maruyama, H; Yamamoto, M; Matsutomi, T; Zheng, T; Nakata, Y; Wood, JN; Ogata, N. Electrophysiological characterization of the tetrodotoxin-resistant Na+ channel, NaV1.9, in mouse dorsal root ganglion neurons. Pflugers Arch 2004, 449, 76–87. [Google Scholar]
- Coste, B; Osorio, N; Padilla, O; Crest, M; Delmas, P. Gating and modulation of presumptive NaV1.9 channels in enteric and spinal sensory neurons. Mol Cell Neurosci 2004, 26, 123–134. [Google Scholar]
- Matsutomi, T; Nakamoto, C; Zheng, T; Kakimura, J; Ogata, N. Multiple types of Na+ currents mediate action potential electrogenesis in small neurons of mouse dorsal root ganglia. Pflugers Arch 2006, 453, 83–96. [Google Scholar]
- Lu, T; Lee, HC; Kabat, JA; Shibata, EF. Modulation of rat cardiac sodium channel by the stimulatory G protein α subunit. J Physiol 1999, 518, 371–384. [Google Scholar]
- Aley, KO; Messing, RO; Mochly-Rosen, D; Levine, JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci 2000, 20, 4680–4685. [Google Scholar]
- Deschênes, I; Neyroud, N; DiSilvestre, D; Marbán, E; Yue, DT; Tomaselli, GF. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ Res 2002, 90, E49–57. [Google Scholar]
- Zheng, T; Kakimura, J; Matsutomi, T; Nakamoto, C; Ogata, N. Prostaglandin E2 has no effect on two components of tetrodotoxin-resistant Na+ current in mouse dorsal Root ganglion. J Pharmacol Sci 2007, 103, 93–102. [Google Scholar]
- Coggeshall, RE; Tate, S; Carlton, SM. Differential expression of tetrodotoxin resistant sodium channels NaV1.8 and NaV1.9 in normal and inflamed rats. Neurosci Lett 2004, 355, 45–48. [Google Scholar]
- Murphy, BJ; Rossie, S; De Jongh, KS; Catterall, WA. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel α subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. J Biol Chem 1993, 268, 27355–27362. [Google Scholar]
- Smith, RD; Goldin, AL. Potentiation of rat brain sodium channel currents by PKA in Xenopus oocytes involves the I–II linker. Am J Physiol Cell Physiol 2000, 278, C638–645. [Google Scholar]
- Cantrell, AR; Tibbs, VC; Yu, FH; Murphy, BJ; Sharp, EM; Qu, Y; Catterall, WA; Scheuer, T. Molecular mechanism of convergent regulation of brain Na+ channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol Cell Neurosci 2002, 21, 63–80. [Google Scholar]
- West, JW; Numann, R; Murphy, BJ; Scheuer, T; Catterall, WA. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science 1991, 254, 866–868. [Google Scholar]
- Qu, Y; Rogers, JC; Tanada, TN; Catterall, WA; Scheuer, T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J Gen Physiol 1996, 108, 375–379. [Google Scholar]
- Li, M; West, JW; Lay, Y; Scheuer, T; Catterall, WA. Functional modulation of brain sodium channels by cAMP-dependent phosphorylation. Neuron 1992, 8, 1151–1159. [Google Scholar]
- Numann, R; Hauschka, SD; Catterall, WA; Scheuer, T. Modulation of skeletal muscle sodium channels in a satellite cell line by protein kinase C. J Neurosci 1994, 14, 4226–4236. [Google Scholar]
- Numann, R; Catterall, WA; Scheuer, T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science 1991, 254, 115–118. [Google Scholar]
- Kausalia, V; Mohamed, B; Mohamed, C. Modulation of NaV1.7 and NaV1.8 Peripheral Nerve Sodium Channels by Protein Kinase A and Protein Kinase C. J Neurophysiol 2004, 91, 1556–1569. [Google Scholar]
- Baker, MD; Chandra, SY; Ding, Y; Waxman, SG; Wood, JN. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurons. J Physiol 2003, 548, 373–382. [Google Scholar]
- Baker, MD. Protein kinase C mediates up-regulation of tetrodotoxin-resistant, persistent Na+ current in rat and mouse sensory neurons. J Physiol 2005, 567, 851–867. [Google Scholar]
- Chen, H; Lei, J; He, X; Qu, F; Wang, Y; Wen, W; You, H; Arendt-Nielsen, L. Peripheral involvement of PKA and PKC in subcutaneous bee venom-induced persistent nociception, mechanical hyperalgesia, and inflammation in rats. Pain 2008, 135, 31–36. [Google Scholar]
- Cunha, FQ; Teixera, MM; Ferreira, SH. Pharmacological modulation of secondary mediator systems - cyclic AMP and cyclic GMP - on inflammatory hyperalgesia. Br J Pharmacol 1999, 127, 671–678. [Google Scholar]
- Cunha, JM; Rae, GA; Ferreira, SH; Cunha, FQ. Endothelins induce ETB receptor-mediated mechanical hypernociception in rat hindpaw: roles of cAMP and protein kinase C. Eur J Pharmacol 2004, 501, 87–94. [Google Scholar]
- Khasar, SG; Lin, YH; Martin, A; Dadgar, J; McMahon, T; Wang, D; Hundle, B; Aley, KO; Isenberg, W; McCarter, G; Green, PG; Hodge, CW; Levine, JD; Messing, RO. A novel nociceptor signaling pathway revealed in protein kinase Cɛ mutant mice. Neuron 1999, 24, 253–260. [Google Scholar]
- Martin, WJ; Liu, H; Wang, H; Malmberg, AB; Basbaum, AI. Inflammation-induced up-regulation of protein kinase Cγ immunoreactivity in rat spinal cord correlates with enhanced nociceptive processing. Neuroscience 1999, 88, 1267–1274. [Google Scholar]
- Aley, KO; Martin, A; McMahon, T; Mok, J; Levine, JD; Messing, RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci 2001, 21, 6933–6939. [Google Scholar]
- Igwe, OJ; Chronwall, BM. Hyperalgesia induced by peripheral inflammation is mediated by protein kinase C βII isozyme in the rat spinal cord. Neuroscience 2001, 104, 875–890. [Google Scholar]
- Priest, BT; Murphy, BA; Lindia, JA; Diaz, C; Abbadie, C; Ritter, AM; Liberator, P; Iyer, LM; Kash, SF; Kohler, MG; Kaczorowski, GJ; Maclntyre, DE; Martin, WJ. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci USA 2005, 102, 9382–9387. [Google Scholar]
- Hamill, OP; Marty, A; Neher, E; Sakmann, B; Sigworth, FJ. Improved patch-clamp techniques for high-resolution current recordings from cells and cell-free membrane patches. Pflugers Arch 1981, 391, 85–100. [Google Scholar]
- Horn, R; Marty, A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol 1988, 92, 145–159. [Google Scholar]





© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kakimura, J.-i.; Zheng, T.; Uryu, N.; Ogata, N. Regulation of the Spontaneous Augmentation of NaV1.9 in Mouse Dorsal Root Ganglion Neurons: Effect of PKA and PKC Pathways. Mar. Drugs 2010, 8, 728-740. https://doi.org/10.3390/md8030728
Kakimura J-i, Zheng T, Uryu N, Ogata N. Regulation of the Spontaneous Augmentation of NaV1.9 in Mouse Dorsal Root Ganglion Neurons: Effect of PKA and PKC Pathways. Marine Drugs. 2010; 8(3):728-740. https://doi.org/10.3390/md8030728
Chicago/Turabian StyleKakimura, Jun-ichi, Taixing Zheng, Noriko Uryu, and Nobukuni Ogata. 2010. "Regulation of the Spontaneous Augmentation of NaV1.9 in Mouse Dorsal Root Ganglion Neurons: Effect of PKA and PKC Pathways" Marine Drugs 8, no. 3: 728-740. https://doi.org/10.3390/md8030728