Mouse Models of Mutations and Variations in Autism Spectrum Disorder-Associated Genes: Mice Expressing Caps2/Cadps2 Copy Number and Alternative Splicing Variants

Abstract
:
1. Introduction
2. Risk Factors for ASD
2.1. Heredity Factors

{kind=link}
{kind=link}
{kind=link}
| Gene symbol (Cytoband) | Molecular function, property | Gene symbol (Cytoband) | Molecular function, property |
|---|---|---|---|
| DISC1 (1q42.1) | LIS1/NUDE1/14-3-3ε, PDE4B, GSK3/β−catenin pathway | PTPN11 (12q24) | Noonan syndrome, protein Tyr phospholyase |
| NRXN1 (2p16.3) | Cell adhesion, synapse assembly, interact with NLGL1 | CHD8 (14q11.2) | DNA helicase, transcription repressor, β−catenin binding |
| SLC25A12 (2q24) | Mitochondrial Asp/Glu transport | CYFIP1 (15q11) | Interact with FMR1 and WAVE |
| TBR1 (2q24) | T-box brain protein 1, transcriptional regulation | UBE3A (15q11.2) | Ubiquitin ligase, Angelman syndrome |
| OXTR (3p25) | Oxytocin receptor (Gq-PLC-coupled) | GABRB3 (15q11.2-q12) | GABA(A) receptor beta-3 subunit |
| CNTN4 (3p26-p25) | Immunoglobulin superfamily cell adhesion, axon connection | PRKCB (16p11.2) | Ca2+-activated protein kinase C-β |
| SLC9A9 (3q24) | Sodium/proton exchanger, late recycling endosome | CACNA1H (16p13.3) | Voltage dependent Ca2+ channel alpha 1H subunit |
| C3orf58 (3q24) | Uncharacterized, activity-dependent expression | TSC2 (16p13.3) | Tuberous sclerosis protein, regulate Rheb GTPase & mTOR |
| TBL1XR1 (3q26.32) | Transducin (beta)-like 1 X-linked receptor 1, | A2BP1/FOX1 (16p13.3) | RNA binding, RNA transport, splicing regulator |
| JAKMIP1 (4p16.1) | Interact with Janus kinase and microtubule | CREBBP (16p13.3) | Transcriptional coactivator, Rubinstein-Taybi syndrome |
| CDH10 (5p14.1) | Cell adhesion (type II classical cadherin) | RAI1 (17p11.2) | Transcription factor, Smith-Magenis syndrome |
| CDH9 (5p14.1) | Cell adhesion (type II classical cadherin) | NF1 (17q11.2) | Neurofibromatosis type I, negative regulator of Ras |
| SEMA5A (5p15.2) | Axon guidance molecule, neural development | SLC6A4 (17q11.1-q12) | Serotonin plasma membrane transporter |
| NIPBL (5p13.2) | Sister chromatid cohesion, Cornelia de Lange syndrome | ITGB3 (17q21.32) | Cell adhesion & cell surface signaling, integrin β-chain |
| NSD1 (5q35.3) | Transcription coregulator, Sotos syndrome, Weaver syndrome | PLAUR (19q13) | Receptor for urokinase plasminogen activator |
| GRIK2 (6q16.3-q21) | Glutamate receptor, ionotropic, kainate 2, GluR6 | DMPK (19q13.3) | Ser/Thr kinase, interact with Rho, Myotonic dystrophy type 1, |
| AHI1 (6q23.3) | Cerebellar & cortical development, Joubert syndrome | DYRK1A (21q22.13) | Dual-specificity protein kinase, Down syndrome critical region |
| AUTS2 (7q11.22) | Autism susceptibility candidate 2 | TBX1 (22q11.21) | T-box transcription factor, embryonic tissue development |
| RELN (7q22) | Extracellular matrix protein, neuronal migration & position | ADSL (22q13.1) | Adenylosuccinate lyase, purine synthesis, succinylpurinemic autism |
| SERPINE1 (7q22.1) | Serpin peptidase inhibitor, class E, member 1; serine protease inhibitor | SHANK3 (22q13.3) | Postsynaptic scaffold protein, synapse function & formation |
| FOXP2 (7q31) | Forkhead box P2, transcription factor | PTCHD1 (Xp22.11) | Receptor for Shh, neural tube formation & brain development |
| MET (7q31) | Proto-oncogene, HGF receptor protein Tyr kinase, | NLGN4X (Xp22.32-p22.31) | Cell adhesion, synapse assembly, interact with NRXN1 |
| CADPS2 (7q31.3) | Regulate release of neuropeptides & monamines | CDKL5 (Xp22) | Ser/Thr kinase, X-linked infantile spasm syndrome/Rett syndrome |
| EN2 (7q36) | Homeobox transcription factor, pattern formation of the CNS | ARX (Xp21) | Transcriptional regulation, CNS development, X-linked mental retardation, epilepsy |
| CNTNAP2 (7q36.1) | Cell adhesion, interact with K+ channel in myelinated axons | IL1RAPL1 (Xp22.1-p21.3) | Interleukin 1 accessary protein like, memory & leaning, X-linked mental retardation |
| CHD7 (8q12.2) | CHARGE syndrome, DNA helicase, chromatin remodeling | DMD (Xp21.2) | Dystrophin, Duchenne & Becker muscular dystrophy, cytoskeletal protein |
| FABP5 (8q21.13) | Fatty acid uptake, transport & metabolism | FGD1 (Xp11.21) | Cdc42 GEF, faciogenital dysplasia, X-linked mental retardation |
| VPS13B (8q22.2) | Cohen syndrome, vesicle transport & protein sorting | NLGN3 (Xq13.1) | Cell adhesion, synapse assembly, interact with NRXN1 |
| TSC1 (9q34) | Tuberous sclerosis protein, regulate Rheb GTPase & mTOR | ATRX (Xq21.1) | Chromatin remodeling, alpha-thelassemia/mental retardation syndrome X-linked, |
| PTEN (10q23.3) | PIP3dephospholyation, negative regulator of PI3K pathway | FMR1 (Xq27.3) | mRNA trafficking, fragile X mental retardation |
| DHCR7 (11q13.2-q13.5) | Biosynthesis of cholesterol | AFF2 (Xq28) | Putative transcriptional activator, fragile X E syndrome |
| SHANK2 (11q13.3-q13.4) | Postsynaptic scaffold protein, synapse function & formation | SLC6A8 (Xq28) | Creatine transporter, X-linked creatine deficiency syndrome |
| GRIN2B (12p12) | Glutamate receptor, ionotropic, N-methyl D-aspartate 2B | MECP2 (Xq28) | Methyl CpG binding protein 2, transcriptional repression, Rett syndrome, development |
| CACNA1C (12p13.3) | Voltage-dependent Ca2+ channel α 1C subunit (L type) | RPL10 (Xq28) | Ribosomal protein L10, a component of 60S subunit, translation |
| AVPR1A (12q14-q15) | Vasopressin receptor (Gq/11-PLC-coupled) |

2.2. Non-Heredity Factors
2.2.1. Environmental Factors
2.2.2. Epigenetic Factors
3. ASD-Related Animal Models
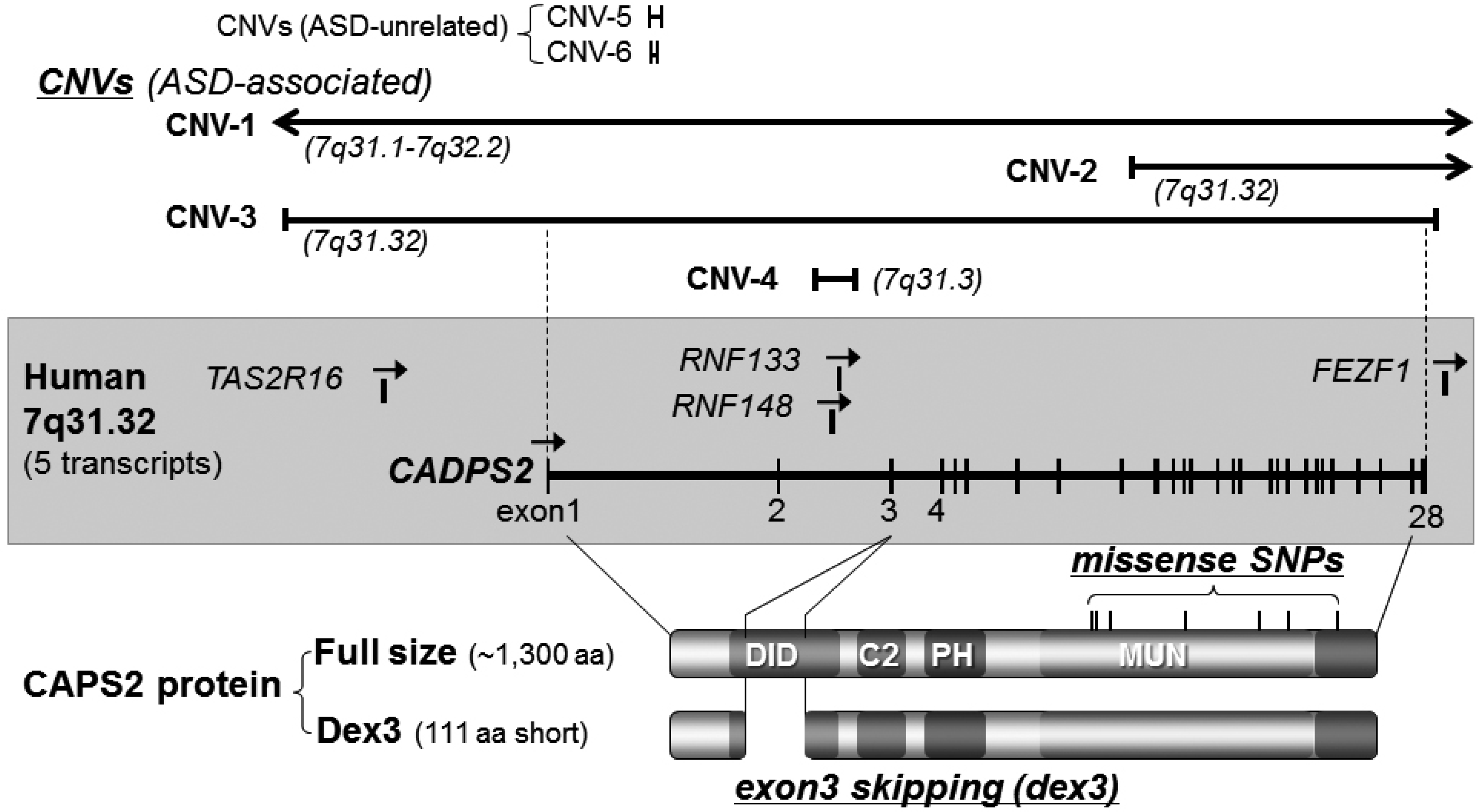
3.1. Caps2/Cadps2 Variant Mice

| Chromosome loci | Diagnosis (1) | Gender(2)/family (3) | CNV type (4) | CNV size/no. of affected genes | References |
|---|---|---|---|---|---|
| 7q31.1-q31.31 | ASD | M/SPX | loss | 25 genes | [40] |
| 7q31.1-q31.32 | autism, DVD | M/SPX | loss | 11 Mb/>50 genes | [37] |
| 7q31.1-q32.2 | ASD, DVD | F/SPX | loss | 15.49 Mb/77 genes | [40] |
| 7q31.2-q32 | ASD, DVD | M/SPX | loss | 26 Mb/ | [37] |
| 7q31.2-q32.2 | autism, DVD, Williams syndrome | F/SPX | loss | 15 Mb/>50 genes | [37,38] |
| 7q31.3 | ASD | -/MPX | loss | 1.52 Mb/2 genes | [18] |
| 7q31.3 | ASD | M/SPX | loss | 0.044 Mb/intron 2 of CADPS2 | [18] |
| 7q31.31-q31.33 | ASD | M/- | loss | 5.4 Mb | [41] |
| 7q31.32 | ASD | -/MPX | loss | 0.75 Mb/4 genes | [9] |
| 7q31.32 | ASD | -/MPX | loss | 0.94 Mb/4 genes | [39] |
| 7q31.32 | ASD | -/- | loss | 1.52 Mb/7 genes | [42] |
| 7q31.32-q34 | autism | M/- | gain | [1] | |
| 7q31.3 | ASD | -/- | gain | 0.43 Mb/exon of CADPS2 | [44] |
| Variant Type | Allele Change | Residue Change | Reference |
|---|---|---|---|
| Missense | T2405C | I761T | [43] |
| Missense | G2419C | V766L | [43] |
| Missense | C2504A | T794N | [43] |
| Missense | A2896G | T925A | [43] |
| Missense | G3286A | D1055N | [43] |
| Missense | G3286A | D1055N | [43] |
| Missense | G3457A | D1112N | [43] |
| Missense | C3722T | T1200M | [43] |
| Missense | G983A | Ala297Thr | [43] |
| Silent | C2461T | N/A ** | [36] |
| Silent | A2539C | N/A | [36] |
| Insertion | N/A | N/A | [36] |
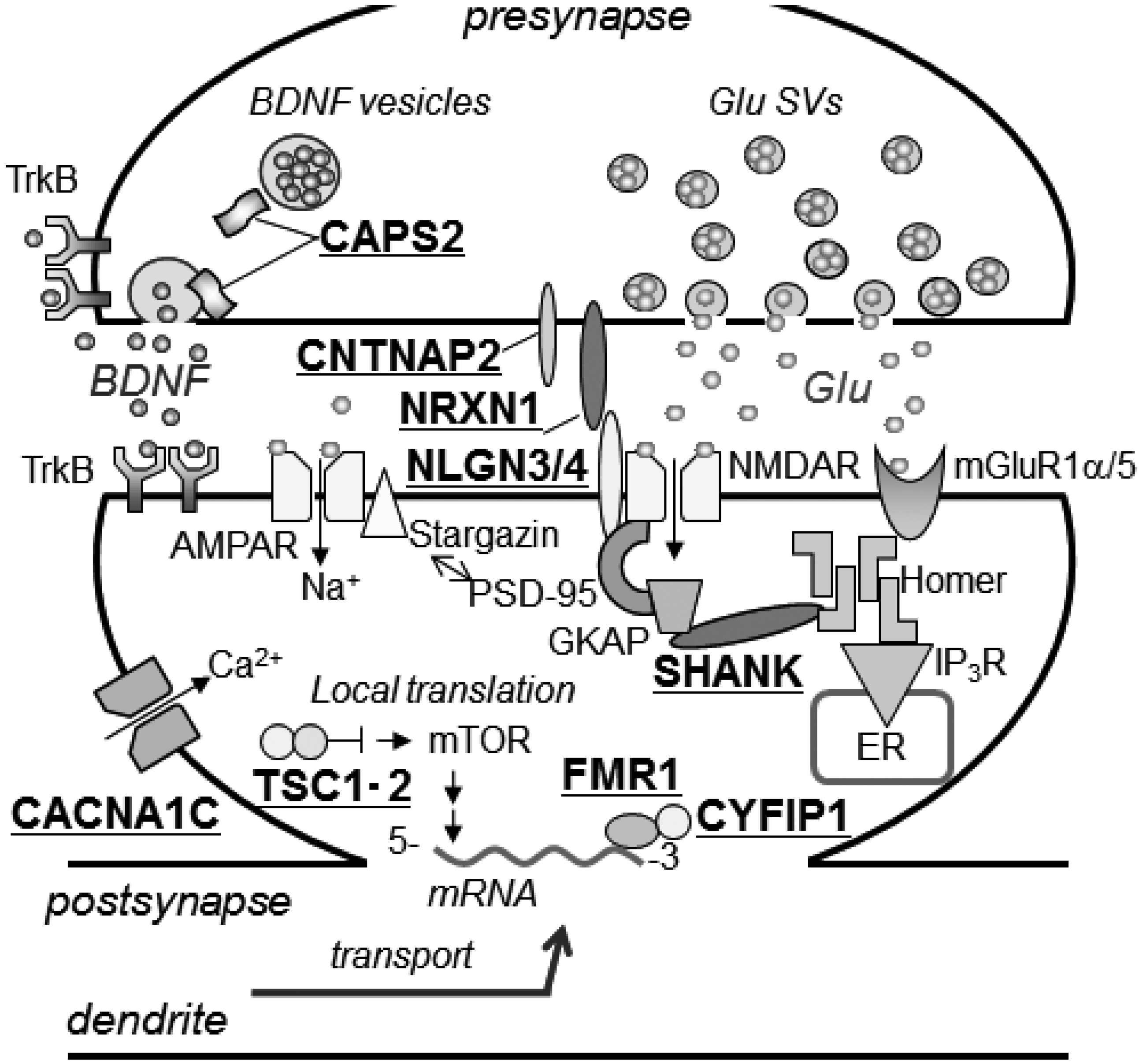
3.2. Other ASD-Associated Gene Modified Mice
3.2.1. Mediating Adhesion between Pre- and Post-Synaptic Membranes
3.2.2. Tethering of Signal Transduction-Associated Molecules in the Postsynaptic Density
3.2.3. Human Chromosome 15q11-13 Repeats
3.2.4. Secretory Pathways of Neuropeptide Hormones and Oxytocin
3.2.5. Mouse Models for Disorders Involving a Single Gene Accompanied by Autism Symptoms
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Folstein, S.E.; Rosen-Sheidley, B. Genetics of autism: Complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2001, 2, 943–955. [Google Scholar] [CrossRef]
- DiCicco-Bloom, E.; Lord, C.; Zwaigenbaum, L.; Courchesne, E.; Dager, S.R.; Schmitz, C.; Schultz, R.T.; Crawley, J.; Young, L.J. The developmental neurobiology of autism spectrum disorder. J. Neurosci. 2006, 26, 6897–6906. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef]
- Bill, B.R.; Geschwind, D.H. Genetic advances in autism: Heterogeneity and convergence on shared pathways. Curr. Opin. Genet. Dev. 2009, 19, 271–278. [Google Scholar] [CrossRef]
- Schanen, N.C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 2006, 15, R138–R150. [Google Scholar] [CrossRef]
- Grabrucker, A.M. Environmental factors in autism. Front. Psych. 2013, 3, 118. [Google Scholar]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma'ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 483, 242–246. [Google Scholar]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H.; et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef]
- Weiss, L.A.; Arking, D.E.; Daly, M.J.; Chakravarti, A.A. Genome-wide linkage and association scan reveals novel loci for autism. Nature 2009, 461, 802–808. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- The Autism Genome Project Consortium; Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [Green Version]
- Cook, E.H.; Scherer, S.W. Copy-number variations associated with neuropsychiatric conditions. Nature 2008, 455, 919–923. [Google Scholar]
- Bucan, M.; Abrahams, B.S.; Wang, K.; Glessner, J.T.; Herman, E.I.; Sonnenblick, L.I.; Retuerto, A.A.; Imielinski, M.; Hadley, D.; Bradfield, J.P.; et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009, 5, e1000536. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef]
- Persico, A.M.; Bourgeron, T. Searching for ways out of the autism maze: Genetic, epigenetic and environmental clues. Trends Neurosci. 2006, 29, 349–358. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef]
- Moy, S.S.; Nadler, J.J. Advances in behavioral genetics: Mouse models of autism. Mol. Psych. 2008, 13, 4–26. [Google Scholar] [CrossRef]
- Yang, M.; Weber, M.D.; Crawley, J.N. Light phase testing of social behaviors: Not a problem. Front. Neurosci. 2008, 2, 186–191. [Google Scholar] [CrossRef]
- Hahn, M.E.; Lavooy, M.J. A review of the methods of studies on infant ultrasound production and maternal retrieval in small rodents. Behav. Genet. 2005, 35, 31–52. [Google Scholar] [CrossRef]
- Lewis, M.H.; Tanimura, Y.; Lee, L.W.; Bodfish, J.W. Animal models of restricted repetitive behavior in autism. Behav. Brain Res. 2007, 176, 66–74. [Google Scholar] [CrossRef]
- Crawley, J.N. Behavioral phenotyping strategies for mutant mice. Neuron 2008, 57, 809–818. [Google Scholar] [CrossRef]
- Shinoda, Y.; Sadakata, T.; Furuichi, T. Animal models of Autism Spectrum Disorder (ASD): A synaptic-level approach to autistic-like behavior in mice. Exp. Anim. 2013, 62, 71–78. [Google Scholar]
- Berwin, B.; Floor, E.; Martin, T.F. CAPS (mammalian UNC-31) protein localizes to membranes involved in dense-core vesicle exocytosis. Neuron 1998, 21, 137–145. [Google Scholar] [CrossRef]
- Sadakata, T.; Shinoda, Y.; Sekine, Y.; Saruta, C.; Itakura, M.; Takahashi, M.; Furuichi, T. Interaction of calcium-dependent activator protein for secretion 1 (CAPS1) with the class II ADP-ribosylation factor small GTPases is required for dense-core vesicle trafficking in the trans-Golgi network. J. Biol. Chem. 2010, 285, 38710–38719. [Google Scholar]
- Sadakata, T.; Sekine, Y.; Oka, M.; Itakura, M.; Takahashi, M.; Furuichi, T. Calcium-dependent activator protein for secretion 2 interacts with the class II ARF small GTPases and regulates dense-core vesicle trafficking. FEBS J. 2011, 279, 384–394. [Google Scholar]
- Sadakata, T.; Kakegawa, W.; Shinoda, Y.; Hosono, M.; Katoh-Semba, R.; Sekine, Y.; Sato, Y.; Tanaka, M.; Iwasato, T.; Itohara, S.; Furuyama, K.; Kawaguchi, Y.; Ishizaki, Y.; Yuzaki, M.; Furuichi, T. CAPS1 deficiency perturbs dense-core vesicle trafficking and Golgi structure and reduces presynaptic release probability in the mouse brain. J. Neurosci. 2013, 33, 17326–17334. [Google Scholar] [CrossRef]
- Sadakata, T.; Mizoguchi, A.; Sato, Y.; Katoh-Semba, R.; Fukuda, M.; Mikoshiba, K.; Furuichi, T. The secretory granule-associated protein CAPS2 regulates neurotrophin release and cell survival. J. Neurosci. 2004, 24, 43–52. [Google Scholar] [CrossRef]
- Sadakata, T.; Furuichi, T. Developmentally regulated Ca2+-dependent activator protein for secretion 2 (CAPS2) is involved in BDNF secretion and is associated with autism susceptibility. Cerebellum 2008, 8, 312–322. [Google Scholar]
- Shinoda, Y.; Sadakata, T.; Nakano, K.; Katoh-Semba, R.; Kinameri, E.; Furuya, A.; Yanagawa, Y.; Hirase, H.; Furuichi, T. Calcium-dependent activator protein for secretion 2 (CAPS2) promotes BDNF secretion and is critical for the development of GABAergic interneuron network. Proc. Natl. Acad. Sci. USA 2011, 108, 373–378. [Google Scholar] [CrossRef]
- Cisternas, F.A.; Vincent, J.B.; Scherer, S.W.; Ray, P.N. Cloning and characterization of human CADPS and CADPS2, new members of the Ca2+-dependent activator for secretion protein family. Genomics 2003, 81, 279–291. [Google Scholar] [CrossRef]
- Feuk, L.; Kalervo, A.; Lipsanen-Nyman, M.; Skaug, J.; Nakabayashi, K.; Finucane, B.; Hartung, D.; Innes, M.; Kerem, B.; Nowaczyk, M.J.; et al. Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Amer. J. Hum. Genet. 2006, 79, 965–972. [Google Scholar] [CrossRef]
- Zeesman, S.; Nowaczyk, M.J.; Teshima, I.; Roberts, W.; Cardy, J.O.; Brian, J.; Senman, L.; Feuk, L.; Osborne, L.R.; Scherer, S.W. Speech and language impairment and oromotor dysparaxia due to deletion of 7q31 that involves FOXP2. Amer. J. Med. Genet. A 2006, 140, 509–514. [Google Scholar]
- Christian, S.L.; Brune, C.W.; Sudi, J.; Kumar, R.A.; Liu, S.; Karamohamed, S.; Badner, J.A.; Matsui, S.; Conroy, J.; McQuaid, D.; et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiat. 2008, 63, 1111–1117. [Google Scholar] [CrossRef]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Amer. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Okamoto, N.; Hatsukawa, Y.; Shimojima, K.; Yamamoto, T. Submicroscopic deletion in 7q31 encompassing CADPS2 and TSPAN12 in a child with autism spectrum disorder and PHPV. Amer. J. Med. Genet. Part A 2011, 155, 1568–1573. [Google Scholar] [CrossRef]
- Gai, X.; Xie, H.M.; Perin, J.C.; Takahashi, N.; Murphy, K.; Wenocur, A.S.; D'arcy, M.; O'Hara, R.J.; Goldmuntz, E.; Grice, D.E.; et al. Pare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry 2012, 17, 402–411. [Google Scholar] [CrossRef]
- Sadakata, T.; Washida, M.; Iwayama, Y.; Shoji, S.; Sato, Y.; Ohkura, T.; Katoh-Semba, R.; Nakajima, M.; Sekine, Y.; Tanaka, M.; et al. Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Invest. 2007, 117, 931–943. [Google Scholar] [CrossRef]
- Girirajan, S.; Dennis, M.Y.; Baker, C.; Malig, M.; Coe, B.P.; Campbell, C.D.; Mark, K.; Vu, T.H.; Alkan, C.; Cheng, Z.; et al. Refinementt and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 2013, 92, 221–237. [Google Scholar] [CrossRef]
- Sadakata, T.; Kakegawa, W.; Mizoguchi, A.; Washida, M.; Katoh-Semba, R.; Shutoh, F.; Okamoto, T.; Nakashima, H.; Kimura, K.; Tanaka, M.; et al. Impaired cerebellar development and function in mice lacking CAPS2, a protein involved in neurotrophin release. J. Neurosci. 2007, 27, 2472–2482. [Google Scholar] [CrossRef]
- Hamatake, M.; Miyazaki, N.; Sudo, K.; Matsuda, M.; Sadakata, T.; Furuya, A.; Ichisaka, S.; Hata, Y.; Nakagawa, C.; Nagata, K.; et al. Phase advance of the light-dark cycle perturbs diurnal rhythms of brain-derived neurotrophic factor and neurotrophin-3 protein levels, which reduces synaptophysin-positive presynaptic terminals in the cortex of juvenile rats. J. Biol. Chem. 2011, 286, 21478–21487. [Google Scholar]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halperm, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Deisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef]
- AutDB. Available online: http://autism.mindspec.org/autdb/CNVHome.do (accessed on 15 November 2013).
- Autism Chromosome Rearrangement Database. Available online: http://projects.tcag.ca/autism/ (accessed on 15 November 2013).
- Autism Database. Available online: http://autism.mindspec.org/autdb/ (accessed on 15 November 2013).
- Sadakata, T.; Shinoda, Y.; Oka, M.; Sekine, Y.; Furuichi, T. Autistic-like behavioral phenotypes in a mouse model with copy number variation of the CAPS2/CADPS2 gene. FEBS Lett. 2013, 587, 54–59. [Google Scholar] [CrossRef]
- Sadakata, T.; Shinoda, Y.; Oka, M.; Sekine, Y.; Sato, Y.; Saruta, C.; Miwa, H.; Tanaka, M.; Itohara, S.; Furuichi, T. Reduced axonal localization of a Caps2 splice variant impairs axonal release of BDNF and causes autistic-like behavior in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21104–21109. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammerm, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2009, 318, 71–76. [Google Scholar]
- Chadman, K.K.; Gong, S.; Scattoni, M.L.; Boltuck, S.E.; Gandhy, S.U.; Heintz, N.; Crawley, J.N. Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 2008, 1, 147–158. [Google Scholar]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Ferrante, A.; Ricceri, L. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. USA 2008, 105, 1710–1715. [Google Scholar]
- Etherton, M.R.; Blaiss, C.A.; Powell, C.M.; Südhof, T.C. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. USA 2009, 106, 17998–18003. [Google Scholar]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Südhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef] [Green Version]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Berkel, S.; Marshall, C.R.; Weiss, B.; Howe, J.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef]
- Sato, D.; Lionel, A.C.; Leblond, C.S.; Prasad, A.; Pinto, D.; Walker, S.; O'Connor, I.; Russell, C.; Drmic, I.E.; Hamdan, F.F.; et al. SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 2012, 90, 879–887. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef]
- Hung, A.Y.; Futai, K.; Sala, C.; Valtschanoff, J.G.; Ryu, J.; Woodworth, M.A.; Kidd, F.L.; Sung, C.C.; Miyakawa, T.; Bear, M.F.; et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 2008, 28, 1697–1708. [Google Scholar] [CrossRef]
- Nakatani, J.; Tamada, K.; Hatanaka, F.; Ise, S.; Ohta, H.; Inoue, K.; Tomonaga, S.; Watanabe, Y.; Chung, Y.J.; Banerjee, R.; et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–13 duplication seen in autism. Cell 2009, 137, 1235–1246. [Google Scholar]
- Tamada, K.; Tomonaga, S.; Hatanaka, F.; Nakai, N.; Takao, K.; Miyakawa, T.; Nakatani, J.; Takumi, T. Decreased exploratory activity in a mouse model of 15q duplication syndrome; implications for disturbance of serotonin signaling. PLoS One 2010, 5, e15126. [Google Scholar] [CrossRef]
- Higashida, H.; Yokoyama, S.; Munesue, T.; Kikuchi, M.; Minabe, Y.; Lopatina, O. CD38 gene knockout juvenile mice: A model of oxytocin signal defects in autism. Biol. Pharm. Bull. 2011, 34, 1369–1372. [Google Scholar] [CrossRef]
- Donaldson, Z.R.; Young, L.J. Oxytocin, vasopressin, and the neurogenetics of sociality. Science 2008, 322, 900–904. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A.; Domes, G.; Kirsch, P.; Heinrichs, M. Oxytocin and vasopressin in the human brain: Social neuropeptides for translational medicine. Nat. Rev. Neurosci. 2011, 12, 524–538. [Google Scholar] [CrossRef]
- Jin, D.; Liu, H.X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behavior by regulating oxytocin secretion. Nature 2007, 466, 41–45. [Google Scholar]
- Munesue, T.; Yokoyama, S.; Nakamura, K.; Anitha, A.; Yamada, K.; Hayashi, K.; Asaka, T.; Liu, H.X.; Jin, D.; Koizumi, K.; et al. Two genetic variants of CD38 in subjects with autism spectrum disorder and control. Neurosci. Res. 2010, 67, 181–191. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sadakata, T.; Shinoda, Y.; Sato, A.; Iguchi, H.; Ishii, C.; Matsuo, M.; Yamaga, R.; Furuichi, T. Mouse Models of Mutations and Variations in Autism Spectrum Disorder-Associated Genes: Mice Expressing Caps2/Cadps2 Copy Number and Alternative Splicing Variants. Int. J. Environ. Res. Public Health 2013, 10, 6335-6353. https://doi.org/10.3390/ijerph10126335
Sadakata T, Shinoda Y, Sato A, Iguchi H, Ishii C, Matsuo M, Yamaga R, Furuichi T. Mouse Models of Mutations and Variations in Autism Spectrum Disorder-Associated Genes: Mice Expressing Caps2/Cadps2 Copy Number and Alternative Splicing Variants. International Journal of Environmental Research and Public Health. 2013; 10(12):6335-6353. https://doi.org/10.3390/ijerph10126335
Chicago/Turabian StyleSadakata, Tetsushi, Yo Shinoda, Akira Sato, Hirotoshi Iguchi, Chiaki Ishii, Makoto Matsuo, Ryosuke Yamaga, and Teiichi Furuichi. 2013. "Mouse Models of Mutations and Variations in Autism Spectrum Disorder-Associated Genes: Mice Expressing Caps2/Cadps2 Copy Number and Alternative Splicing Variants" International Journal of Environmental Research and Public Health 10, no. 12: 6335-6353. https://doi.org/10.3390/ijerph10126335