Trypsin Binding with Copper Ions Scavenges Superoxide: Molecular Dynamics-Based Mechanism Investigation

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bacteria
2.2. Polychlorinated BiphenylsTreatment
2.3. Trypsin Treatment
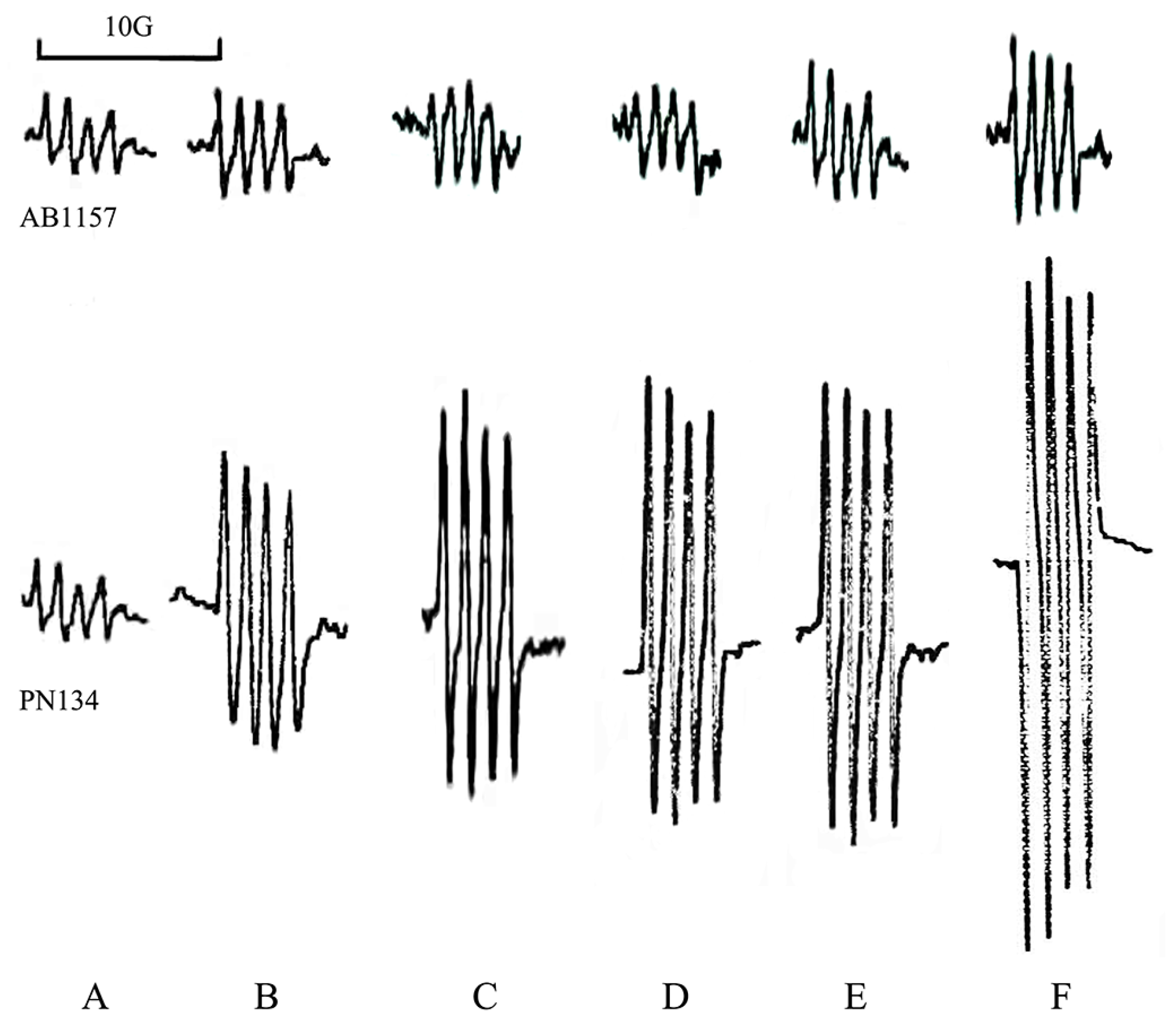
2.4. Quantitative Assay of Superoxide Anion
2.5. External Factors Treatments
2.6. Molecular Dynamics Simulation
2.7. Statistics
3. Results
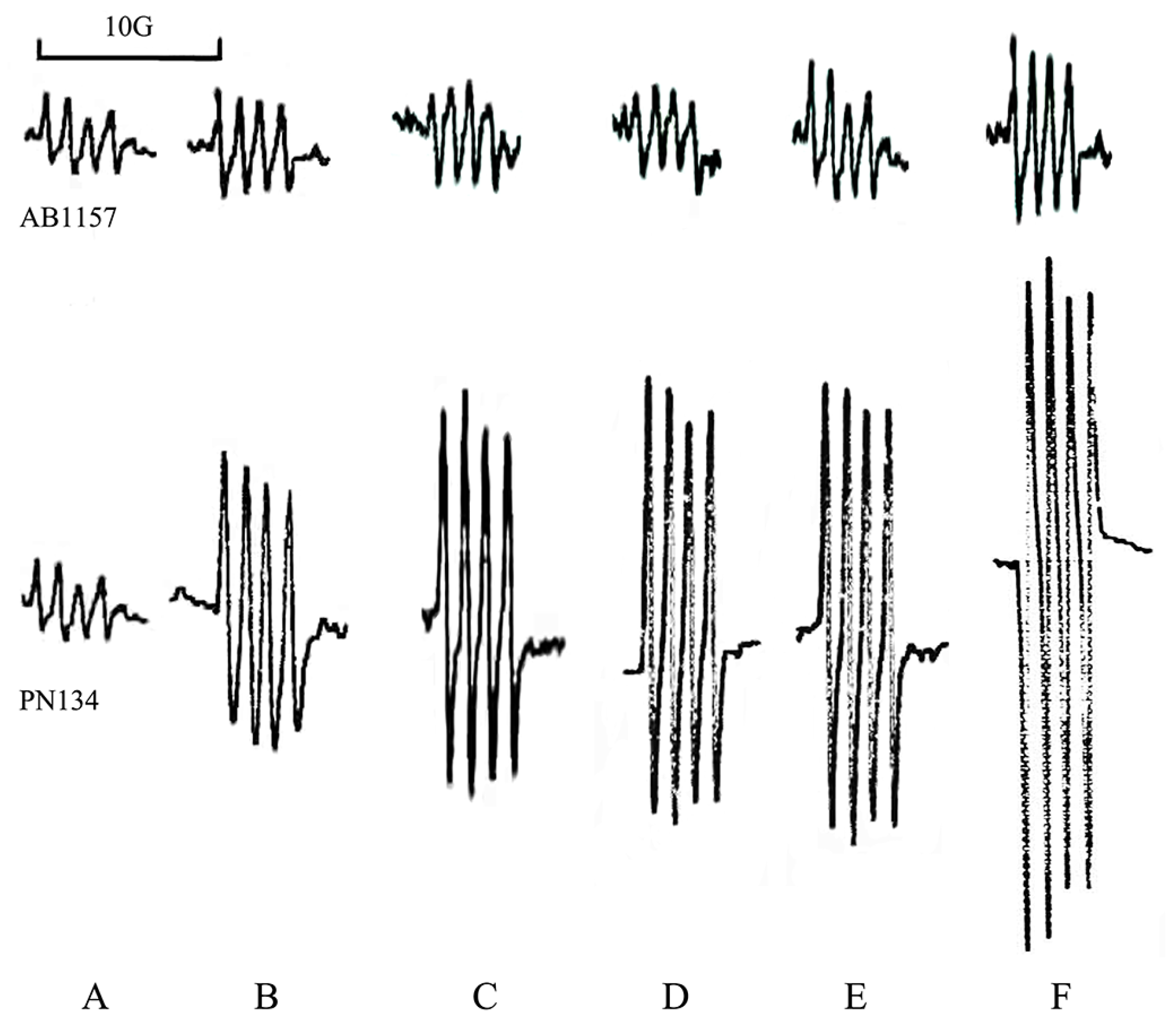
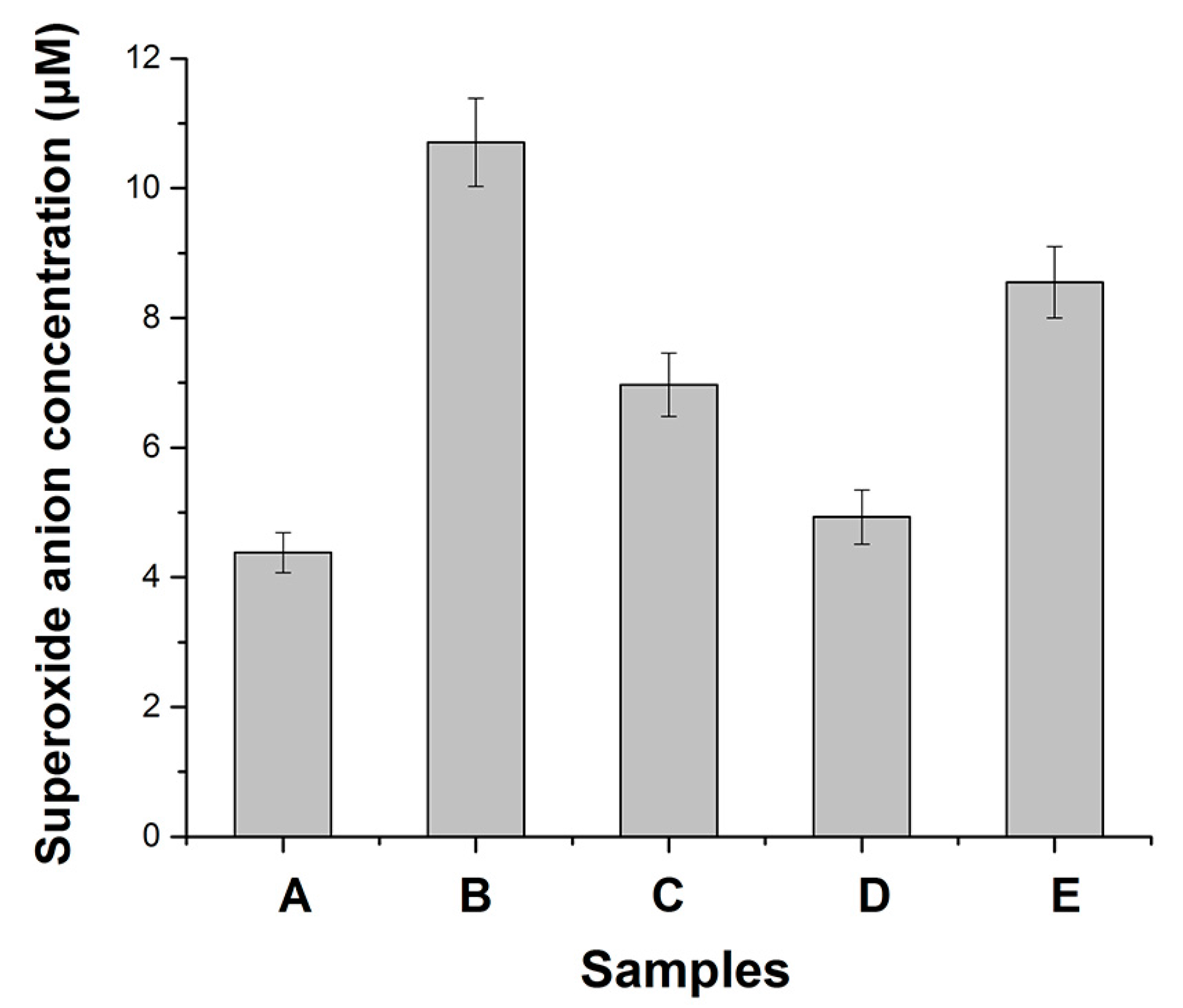
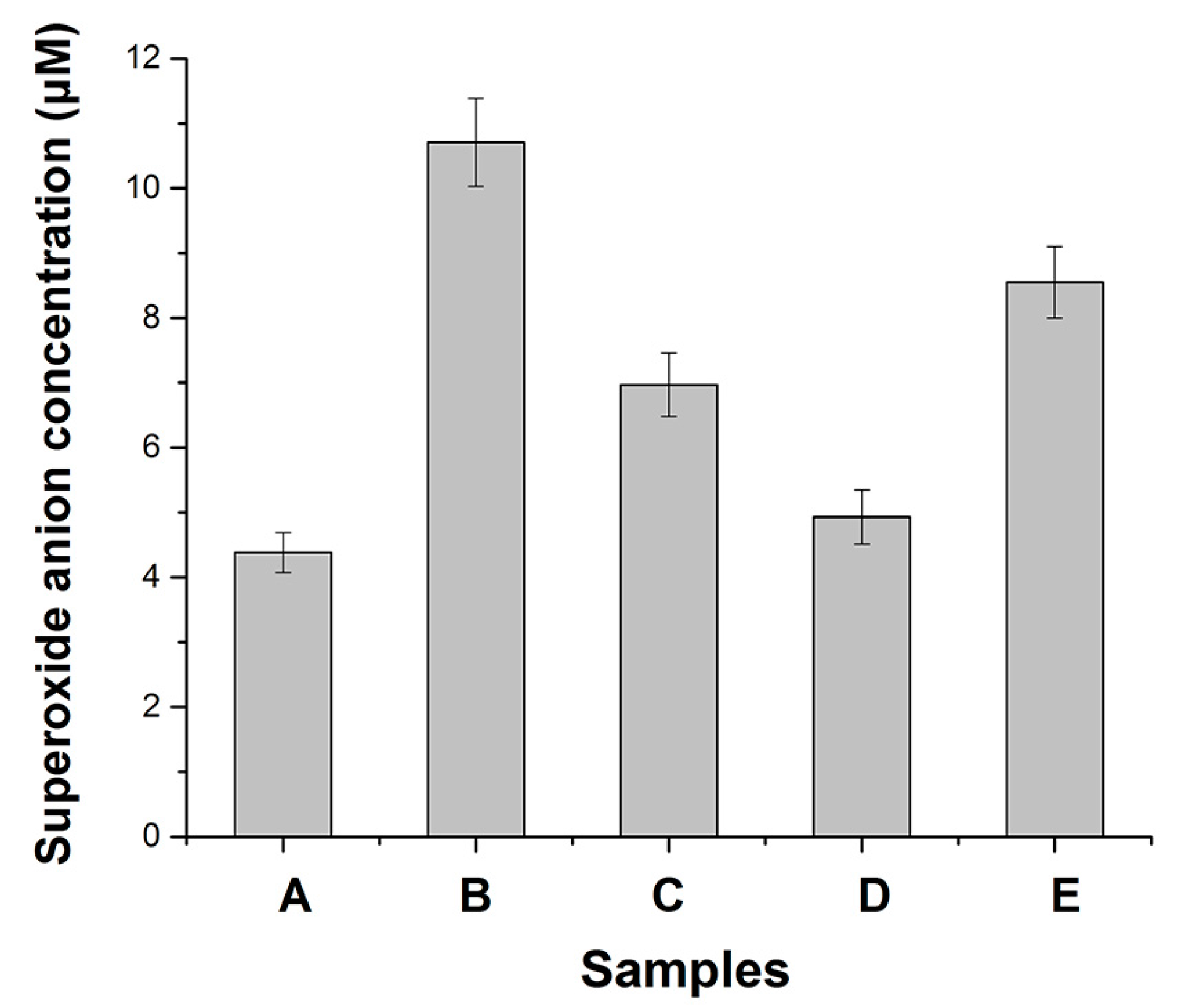
3.1. Effects of Metals on Trypsin Activity
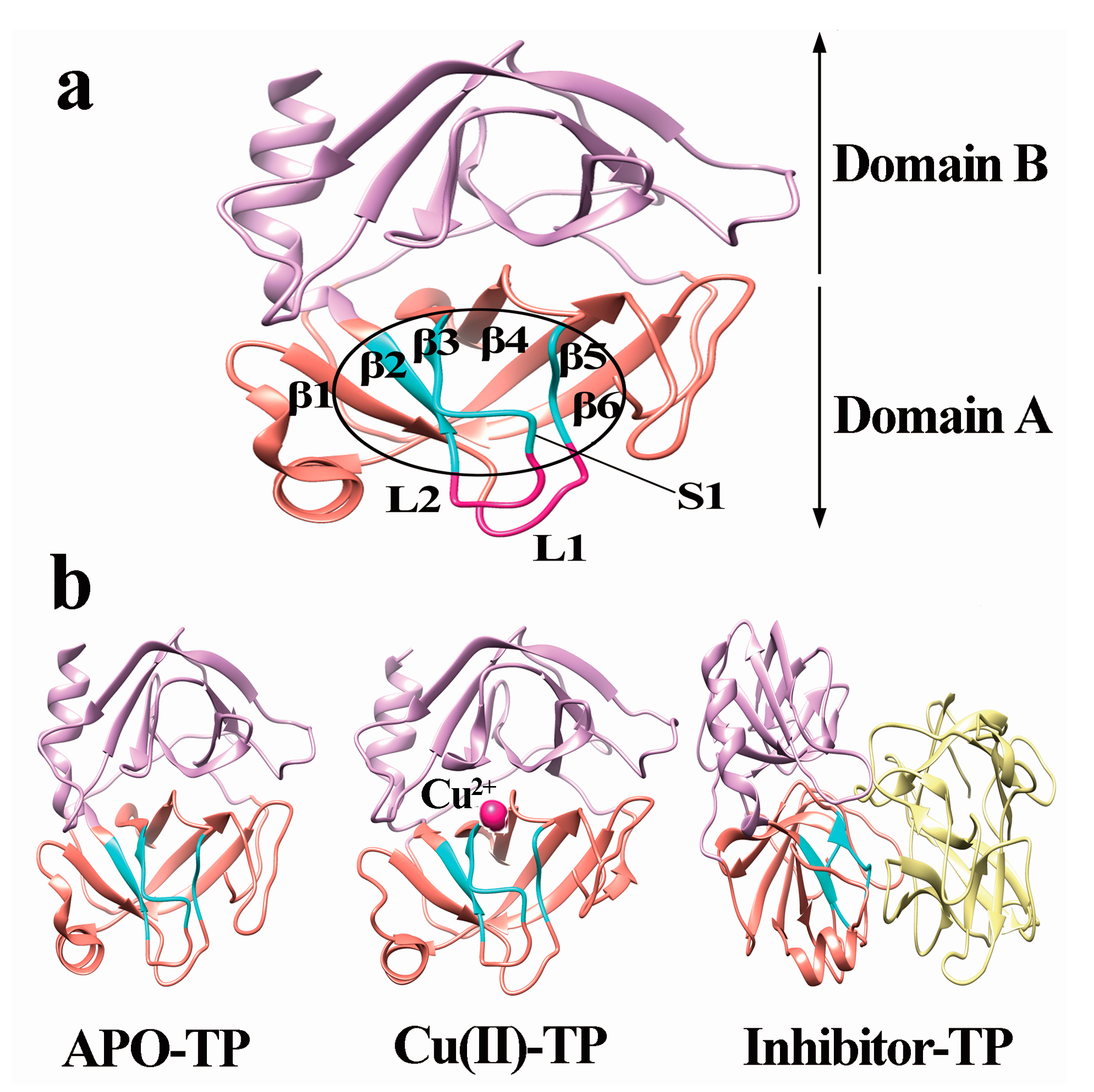
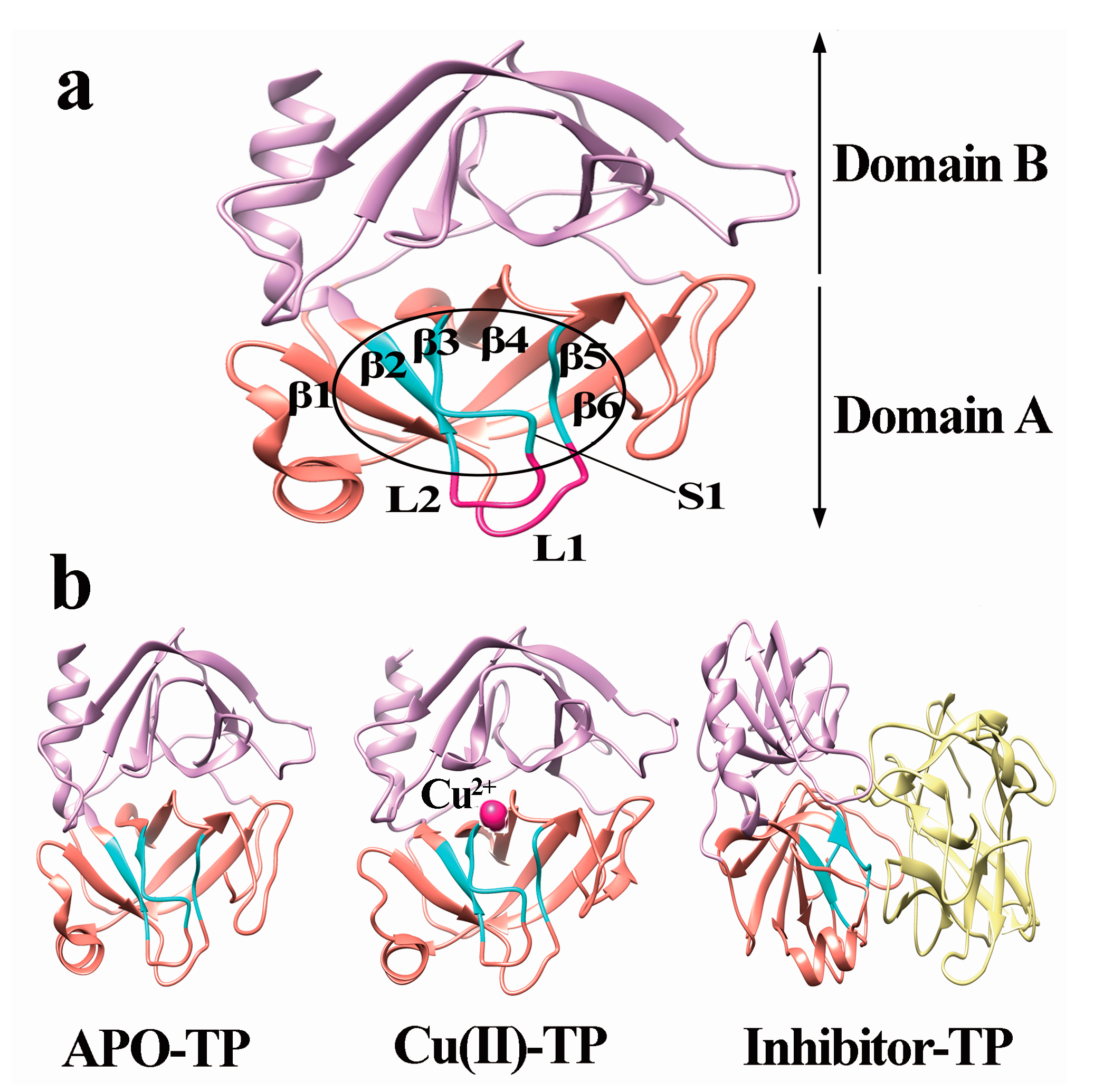
3.2. Model Construction
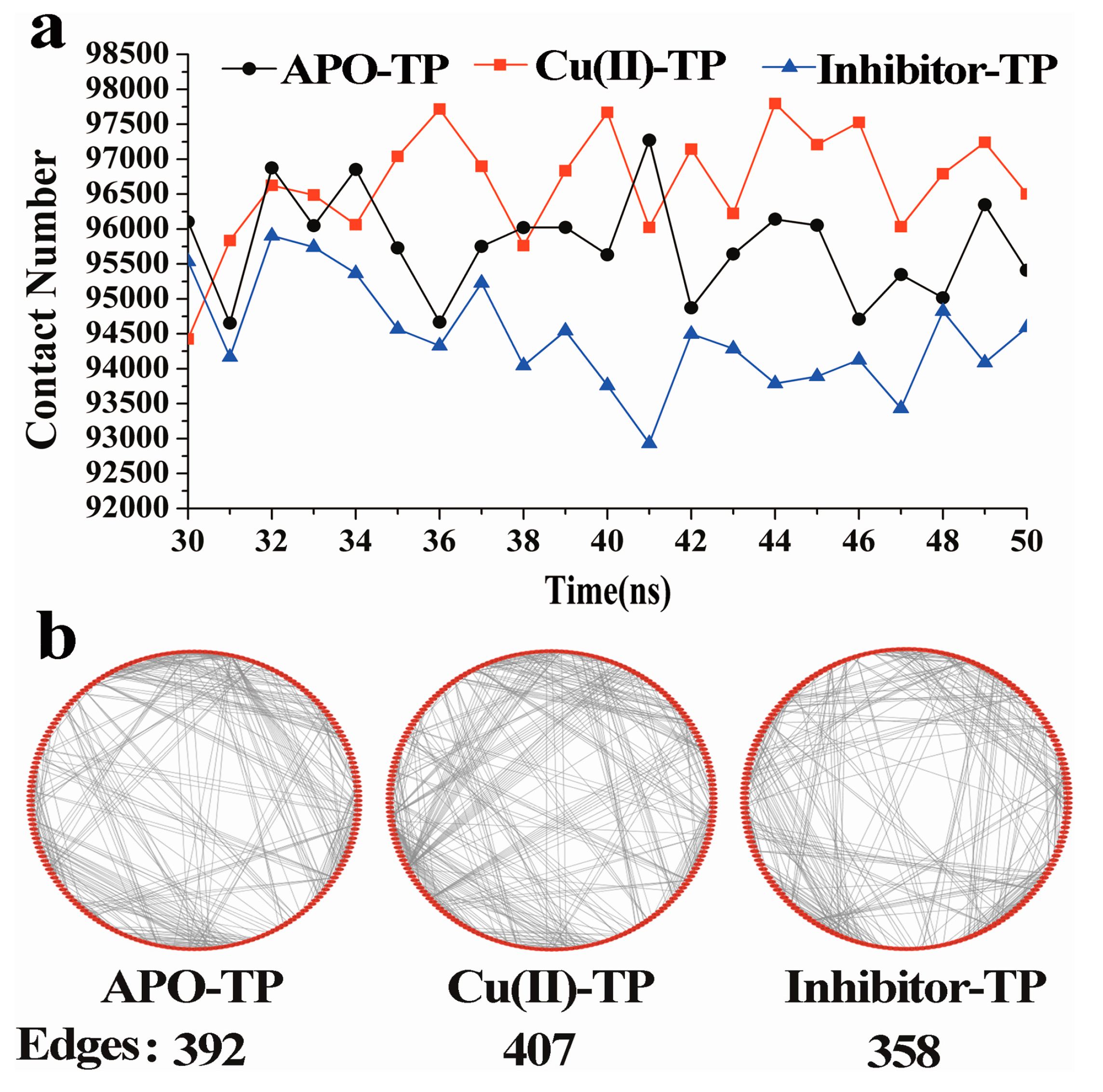
3.3. Residue–Residue Network Analysis
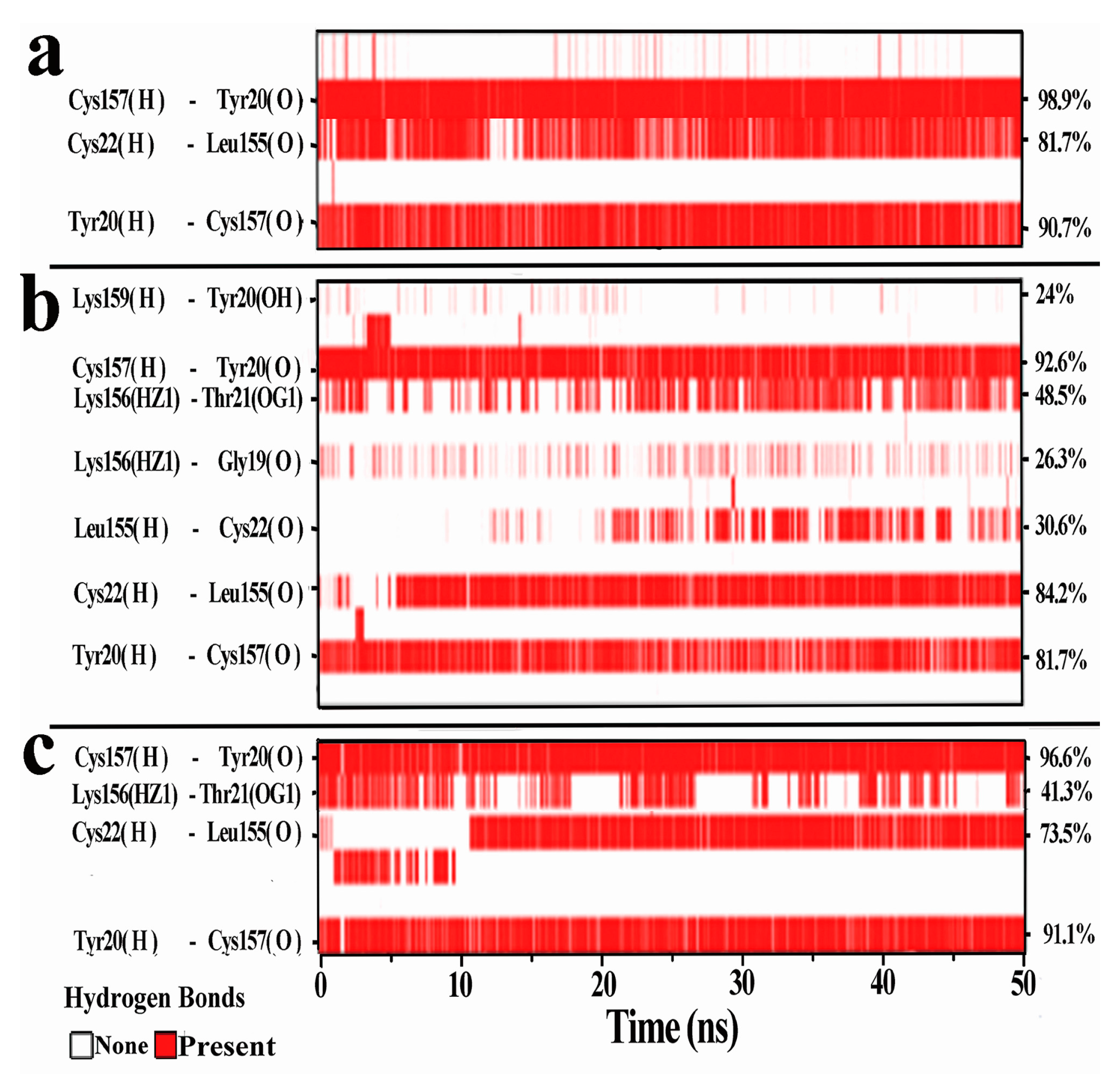
3.4. Hydrogen Bonds Analysis
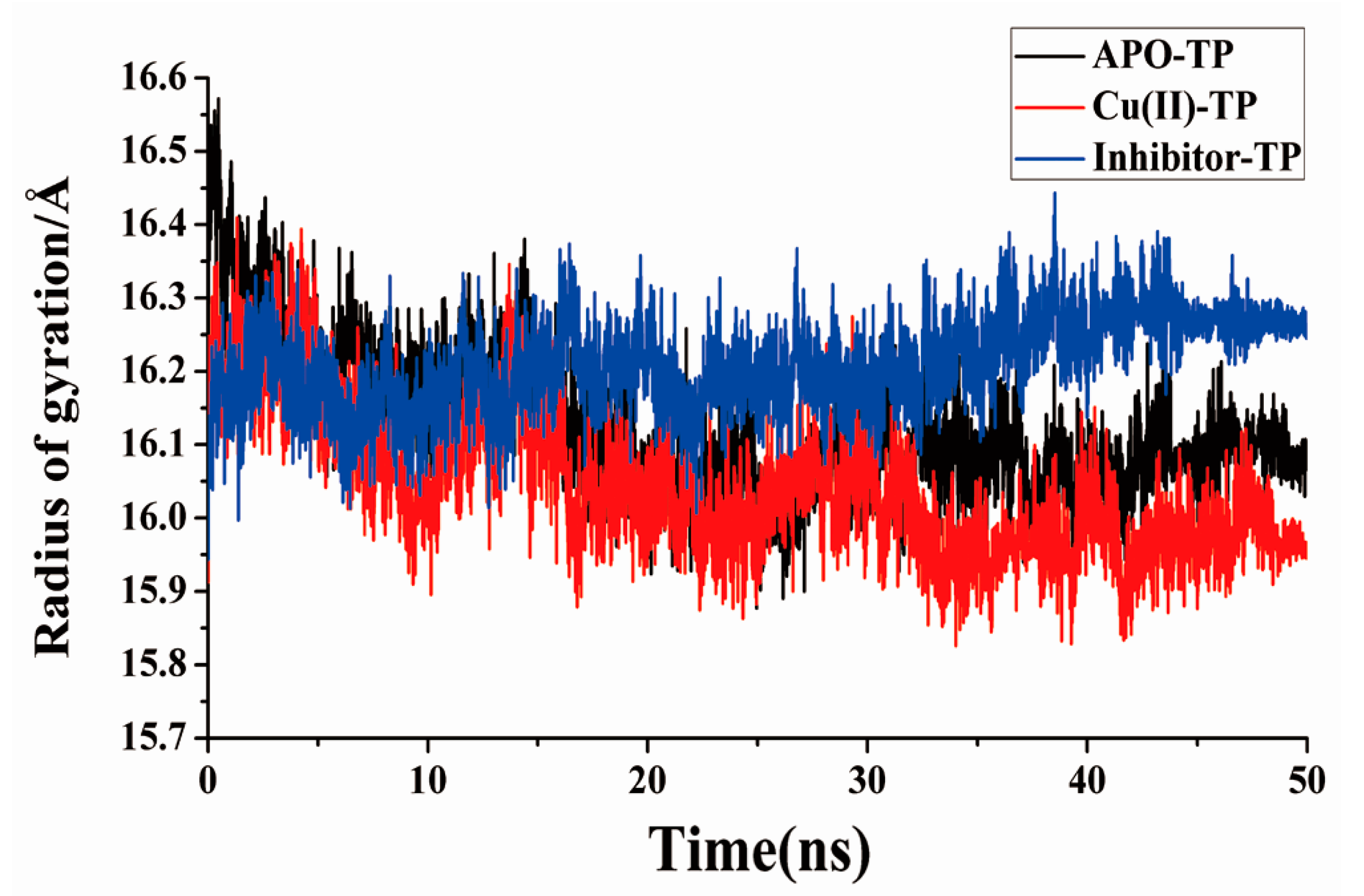
3.5. Radius of Gyration and SASA Analysis
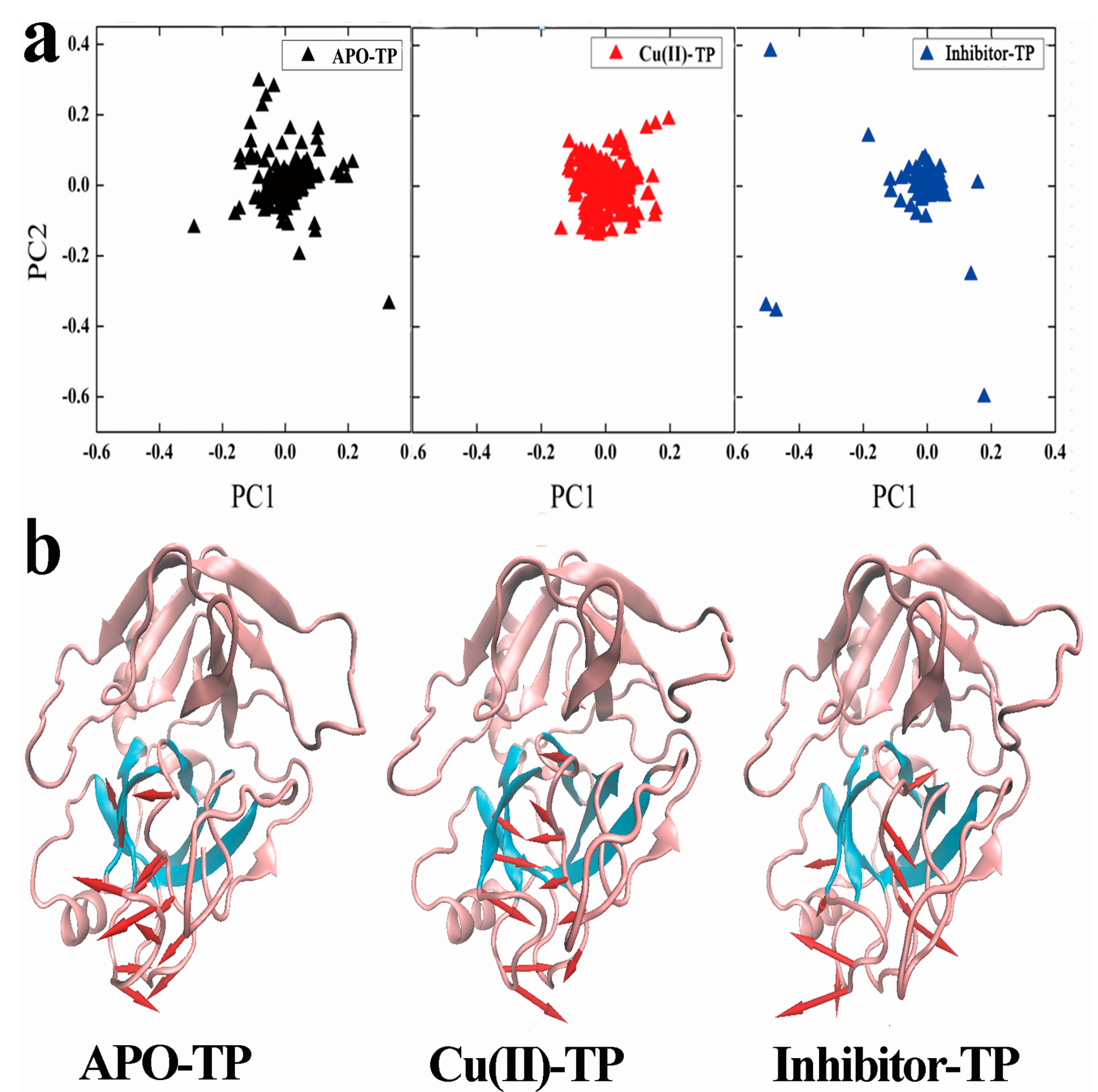
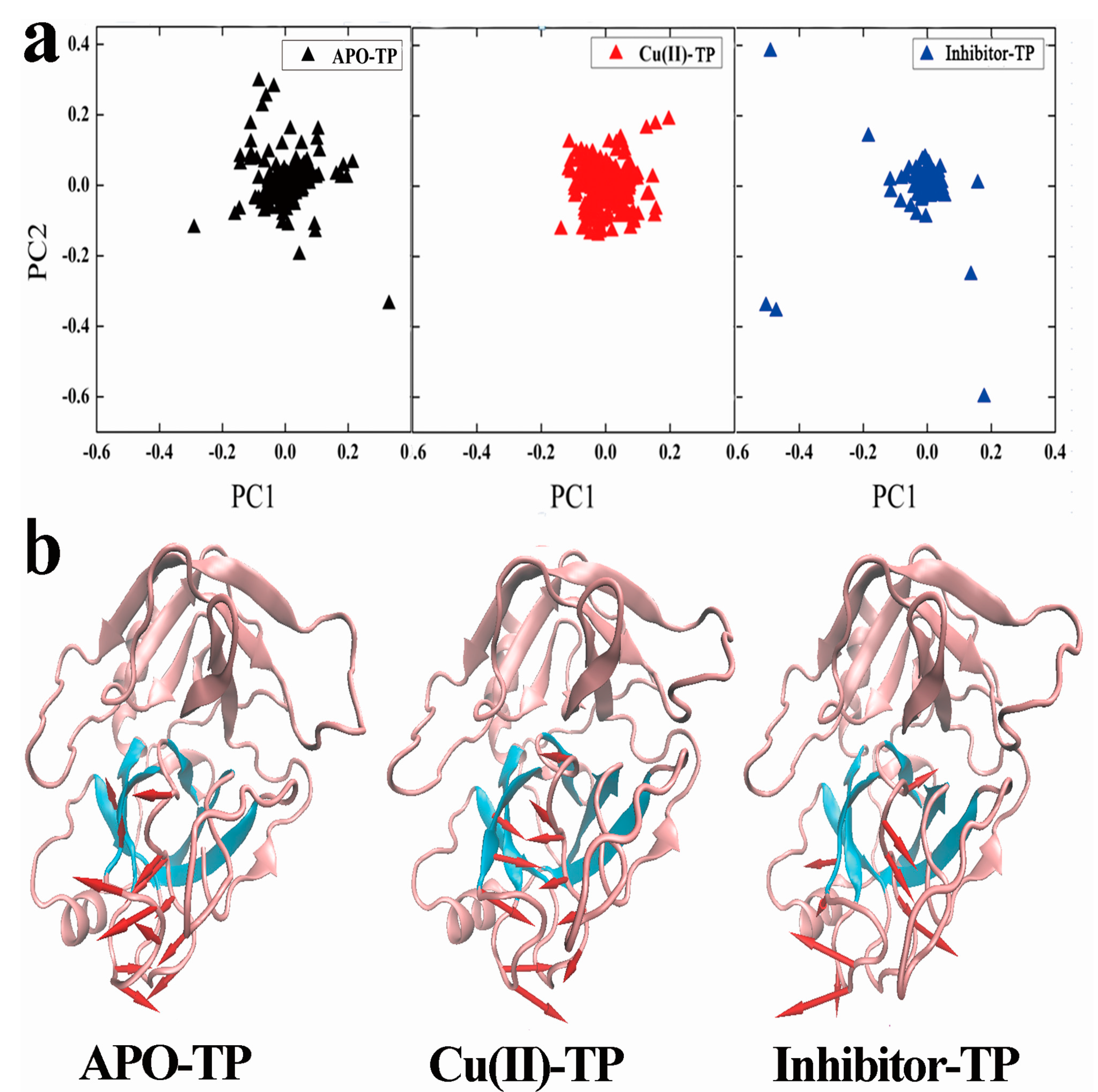
3.6. Principal Component Analysis (PCA) and Porcupine Plots
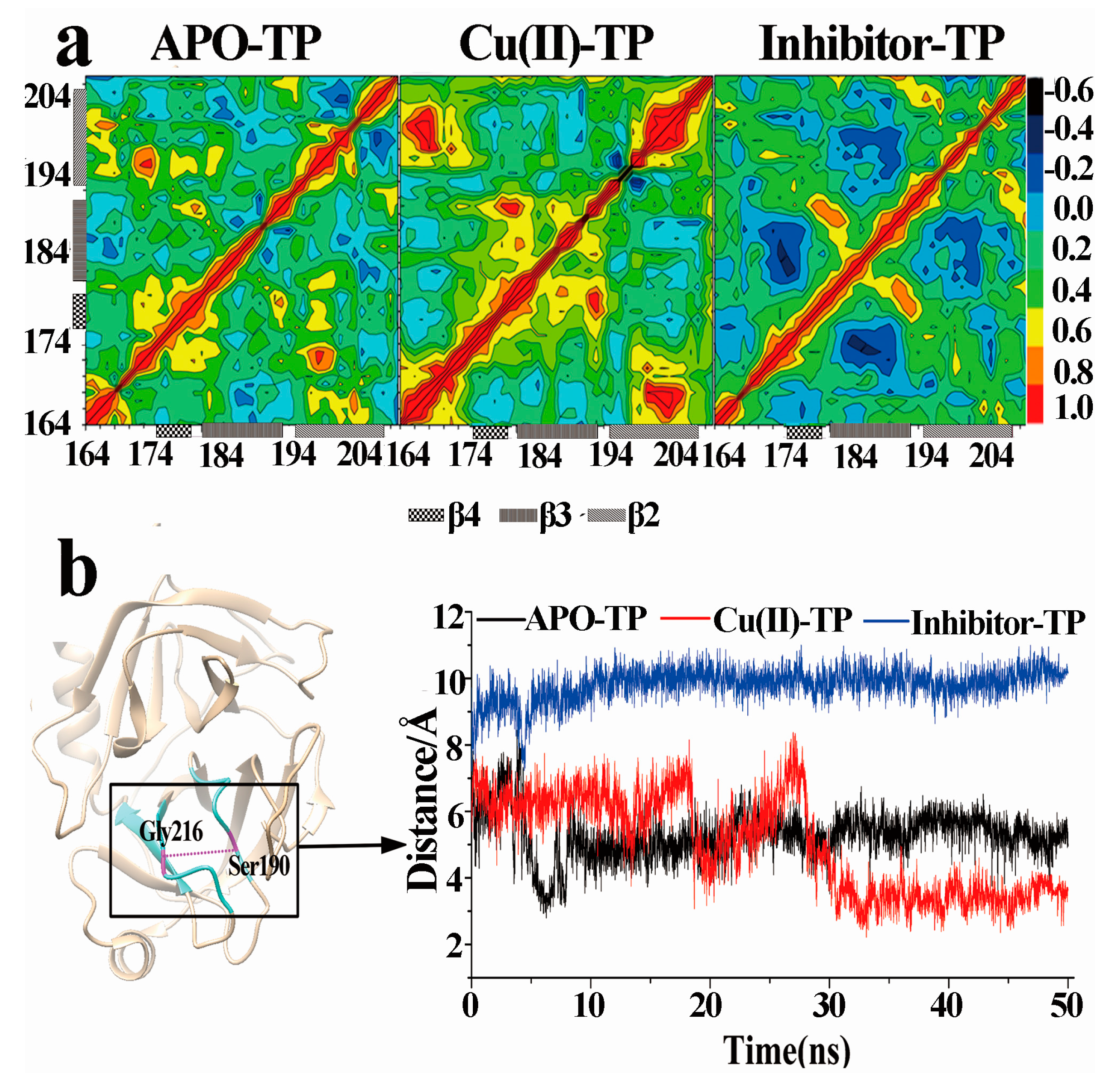
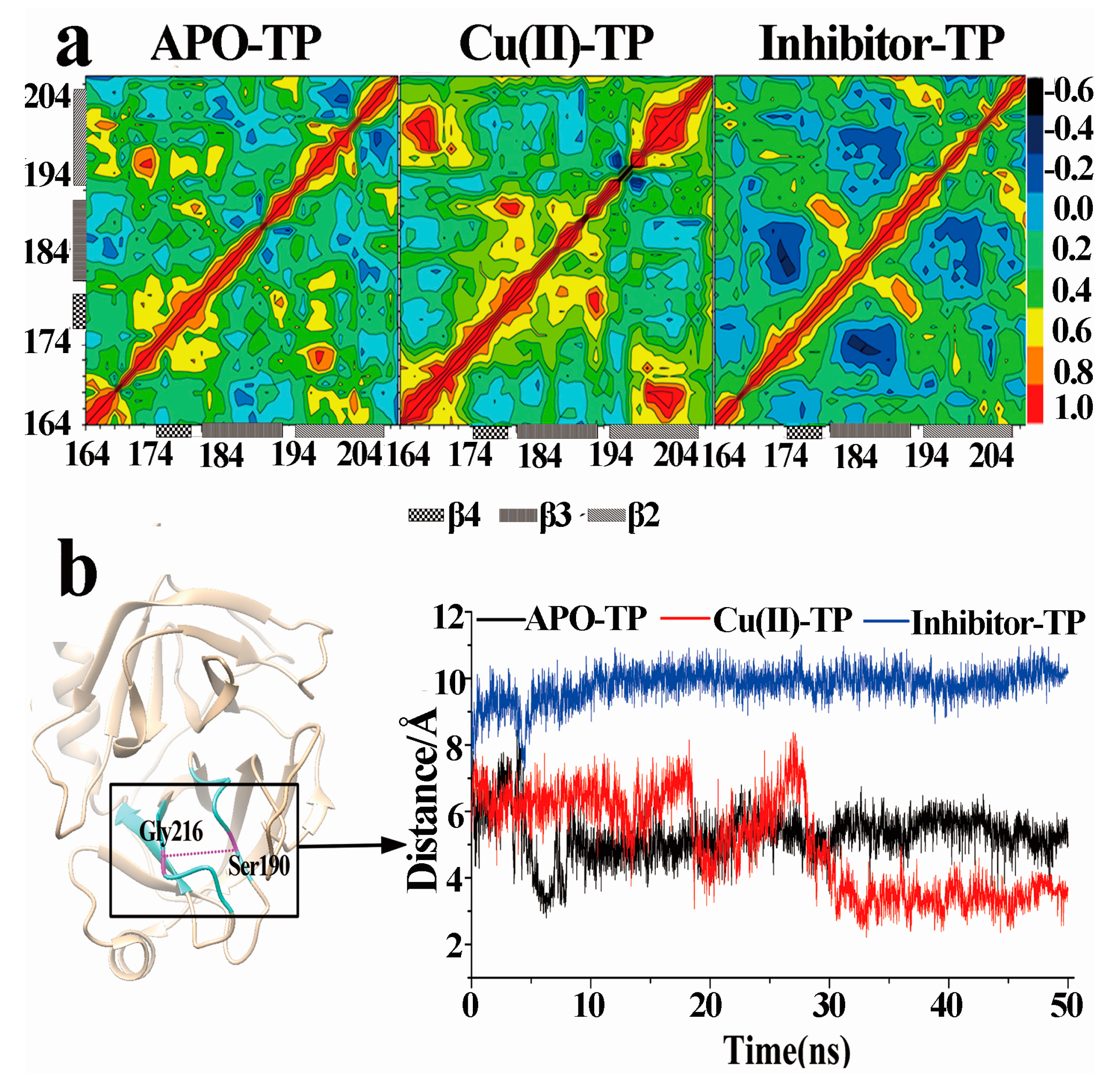
3.7. Dynamical Cross-Correlation Maps and Distance Analysis
4. Discussion
- (1)
- The activities of superoxide scavenging of trypsin were significantly accelerated by Cu ions.
- (2)
- A new β7 sheet transited from a random coil in the Cu(II)-TP system, which was favorable for forming more contacts with other sheets, including hydrogen-bond interactions. It indicated that trypsin underwent conformational changes which made the structure of trypsin much compact and tight upon binding with Cu ions.
- (3)
- Residue-residue network analysis demonstrated that many more contacts formed during the simulation process in the Cu(II)-TP system. In addition, the decreased Rg and increased SASA of the Cu(II)-TP system showed that more stable interactions gradually formed. It proved that Cu ions in trypsin strengthened some native interactions among residues, which finally resulted in much greater stability of the Cu(II)-TP system.
- (4)
- PCA analysis and the porcupine plot for the Cu(II)-TP system showed that the projection distribution of Cu(II)-TP was more concentrated in a smaller range, with the movement of residues of S1 sites toward the center of the pocket. This led to the formation of a much more compact S1 pocket together with a smaller distance for Ser190 and Gly216.
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barth, C.; Moeder, W.; Klessig, D.F.; Conklin, P.L. The timing of senescence and response to pathogens is altered in the ascorbate-deficient Arabidopsis mutant vitamin c-1. Plant Physiol. 2004, 134, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Hoidal, J.R. Reactive oxygen species and cell signaling. Am. J. Respir. Cell Mol. Biol. 2001, 25, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Demidchik, V.; Shabala, S.N.; Coutts, K.B.; Tester, M.A.; Davies, J.M. Free oxygen radicals regulate plasma membrane Ca2+- and K+-permeable channels in plant root cells. J. Cell Sci. 2003, 116, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Huycke, M.M.; Joyce, W.; Wack, M.F. Augmented production of extracellular superoxide by blood isolates of Enterococcus faecalis. J. Infect. Dis. 1996, 173, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Ratn, A.; Awasthi, Y.; Kumar, M.; Singh, S.K.; Tripathi, R.; Trivedi, S.P. Phorate induced oxidative stress, DNA damage and differential expression of p53, apaf-1 and cat genes in fish, Channa punctatus (Bloch, 1793). Chemosphere 2017, 182, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.C.; Meagher, M.; Kassouf, N.; Hafezparast, M.; McKinnon, P.J.; Haywood, R.; El-Khamisy, S.F. Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Sci. Adv. 2017, 3, e1602506. [Google Scholar] [CrossRef] [PubMed]
- Katsuwon, J.; Anderson, A.J. Catalase and superoxide dismutase of root-colonizing saprophytic fluorescent pseudomonads. Appl. Environ. Microb. 1990, 56, 3576–3582. [Google Scholar]
- Li, Q.; Wei, Q.Y.; Yuan, E.D.; Yang, J.G.; Ning, Z.X. Interaction between four flavonoids and trypsin: Effect on the characteristics of trypsin and antioxidant activity of flavonoids. Int. J. Food Sci. Technol. 2014, 49, 1063–1069. [Google Scholar] [CrossRef]
- Spreafico, F.; Bongarzone, I.; Pizzamiglio, S.; Magni, R.; Taverna, E.; De Bortoli, M.; Ciniselli, C.M.; Barzano, E.; Biassoni, V.; Luchini, A.; et al. Proteomic analysis of cerebrospinal fluid from children with central nervous system tumors identifies candidate proteins relating to tumor metastatic spread. Oncotarget 2017, 8, 46177–46190. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, E.; Sahin-Toth, M. Genetic risk in chronic pancreatitis: The trypsin-dependent pathway. Dig. Dis. Sci. 2017, 62, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tang, Z.; Pang, X.; Zhao, C.; Li, X.; Liu, Y. Trypsin slows the aging of mice due to its novel superoxide scavenging activity. Appl. Biochem. Biotechnol. 2017, 181, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pang, X.; Tang, Z.; Xiang, J.; Liu, Y.; Qiao, J. Trypsin: A novel scavenger of superoxide anion. Res. Rev. J. Pharm. Sci. 2016, 5, 1–5. [Google Scholar]
- Viles, J.H.; Klewpatinond, M.; Nadal, R.C. Copper and the structural biology of the prion protein. Biochem. Soc. 2008, 36, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Preferential Cu2+ coordination by His96 and His111 induces β-sheet formation in the unstructured amyloidogenic region of the prion protein. J. Biol. Chem. 2004, 279, 32018–32027. [Google Scholar] [CrossRef] [PubMed]
- Pagel, K.; Vagt, T.; Kohajda, T.; Koksch, B. From α-helix to β-sheet—A reversible metal ion induced peptide secondary structure switch. Org. Biomol. Chem. 2005, 3, 2500–2502. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Suzuki, K.; Kohata, N.; Takeuchi, H. Metal binding modes of Alzheimer’s amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry 2000, 39, 7024–7031. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Kelly, J.W. Synthesis and efficacy of square planar copper complexes designed to nucleate .beta.-sheet structure. J. Am. Chem. Soc. 1995, 117, 2533–2546. [Google Scholar] [CrossRef]
- Brinen, L.S.; Willett, W.S.; Craik, C.S.; Fletterick, R.J. X-ray structures of a designed binding site in trypsin show metal-dependent geometry. Biochemistry 1996, 35, 5999–6009. [Google Scholar] [CrossRef] [PubMed]
- Wachsmann, J.; Peng, F.Y. Molecular imaging and therapy targeting copper metabolism in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.T.; Illes, J.; Szekely, Z.; Forrai, E.; Gere, A. Effect of different metal ions on the oxidative damage and antioxidant capacity of hyaluronic acid. Arch. Biochem. Biophys. 2003, 410, 76–82. [Google Scholar] [CrossRef]
- Roy, A.S.; Tripathy, D.R.; Chatterjee, A.; Dasgupta, S. The influence of common metal ions on the interactions of the isoflavone genistein with bovine serum albumin. Spectrochim. Acta. A 2013, 102, 393–402. [Google Scholar] [CrossRef]
- Wang, Q.; Werstiuk, N.H.; Kramer, J.R.; Bell, R.A. Effects of Cu ions and explicit water molecules on the copper binding domain of amyloid precursor protein APP(131-189): A molecular dynamics study. J. Phys. Chem. B 2011, 115, 9224–9235. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Granillo, A.; Wittung-Stafshede, P. Differential roles of Met10, Thr11, and Lys60 in structural dynamics of human copper chaperone Atox1. Biochemistry 2009, 48, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Raffa, D.F.; Rauk, A. Molecular dynamics study of the beta amyloid peptide of Alzheimer’s disease and its divalent copper complexes. J. Phys. Chem. B 2007, 111, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Whitlow, M.; Arnaiz, D.O.; Buckman, B.O.; Davey, D.D.; Griedel, B.; Guilford, W.J.; Koovakkat, S.K.; Liang, A.; Mohan, R.; Phillips, G.B.; et al. Crystallographic analysis of potent and selective factor Xa inhibitors complexed to bovine trypsin. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Mapuskar, K.A.; Marek, R.F.; Xu, W.; Lehmler, H.J.; Robertson, L.W.; Hornbuckle, K.C.; Spitz, D.R.; Aykin-Burns, N. A new player in environmentally induced oxidative stress: Polychlorinated biphenyl congener, 3,3′-dichlorobiphenyl (PCB11). Toxicol. Sci. 2013, 136, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Von Göenner, M.; Schlösser, E.; Neubacher, H. Evidence from electron-spin resonance for the formation of free radicals during infection of Avena sativa by Drechslera spp. Physiol. Mol. Plant Pathol. 1993, 42, 405–412. [Google Scholar] [CrossRef]
- Li, X.; Pang, X.Y.; Zhi, D.J.; Wang, J.S.; Li, M.Q.; Li, H.Y. Extracellular superoxide anion production contributes to virulence of Xanthomonas oryzae pv. oryzae. Can. J. Microbiol. 2009, 55, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Takahama, U.; Shimizu-Takahama, M.; Egashira, T. Reduction of exogenous cytochrome c by Neurosporacrassaconidia: Effects of superoxide dismutase and blue light. J. Microbiol. 1982, 152, 151–156. [Google Scholar]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.X.; Lin, Z.X.; Wang, J.; Liu, H.Y. Refining the description of peptide backbone conformations improves protein simulations using the GROMOS 53A6 force field. J. Comput. Chem. 2009, 30, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Husby, J.; Todd, A.K.; Haider, S.M.; Zinzalla, G.; Thurston, D.E.; Neidle, S. Molecular dynamics studies of the STAT3 homodimer:DNA complex: Relationships between STAT3 mutations and protein-DNA recognition. J. Chem. Inf. Model. 2012, 52, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.Z.; Tang, C.; Lai, L.H. Specificity of trypsin and chymotrypsin: Loop-motion-controlled dynamic correlation as a determinant. Biophys. J. 2005, 89, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.W.; Xiu, Z.L.; Zeng, A.P. Discovery of intramolecular signal transduction network based on a new protein dynamics model of energy dissipation. PLoS ONE 2012, 7, e31529. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, X.; Shao, Z.; Huang, Y.; Knight, D.P. Effect of metallic ions on silk formation in the Mulberry silkworm, Bombyx mori. J. Phys. Chem. B 2005, 109, 16937–16945. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.A.; Nemethy, G.; Gibson, K.D.; Scheraga, H.A. Conformational energy studies of β-sheets of model silk fibroin peptides. I. Sheets of poly(Ala-Gly) chains. Biopolymers 1991, 31, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lu, D.N.; Liu, Z. How PEGylation enhances the stability and potency of insulin: Amoleculardynamics simulation. Biochemistry 2011, 50, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, E.; Kurbanov, R. Effect of ligand binding on the dynamics of trypsin. Comparison of different approaches. J. Mol. Graph. Model. 2014, 49, 99–109. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SASA (Å2) | APO-TP | Cu(II)-TP | Inhibitor-TP |
|---|---|---|---|
| All residues | 104.87 | 167.29 | 76.92 |
| Hydrophobic Residues | 53.68 | 85.36 | 32.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhong, Y.; Zhao, C. Trypsin Binding with Copper Ions Scavenges Superoxide: Molecular Dynamics-Based Mechanism Investigation. Int. J. Environ. Res. Public Health 2018, 15, 139. https://doi.org/10.3390/ijerph15010139
Li X, Zhong Y, Zhao C. Trypsin Binding with Copper Ions Scavenges Superoxide: Molecular Dynamics-Based Mechanism Investigation. International Journal of Environmental Research and Public Health. 2018; 15(1):139. https://doi.org/10.3390/ijerph15010139
Chicago/Turabian StyleLi, Xin, Yongliang Zhong, and Chunyan Zhao. 2018. "Trypsin Binding with Copper Ions Scavenges Superoxide: Molecular Dynamics-Based Mechanism Investigation" International Journal of Environmental Research and Public Health 15, no. 1: 139. https://doi.org/10.3390/ijerph15010139