Investigation of Incidents and Trends of Antimicrobial Resistance in Foodborne Pathogens in Eight Countries from Historical Sample Data

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source: NCBI Pathogen Isolates Browsers
2.2. Statistical Analysis
3. Results
3.1. Identification of Common Highly-Occurring AMR Genes in the Studied Countries
3.1.1. Common Frequently-Occurring Antimicrobial-Resistance Genes for Individual Countries for All Years
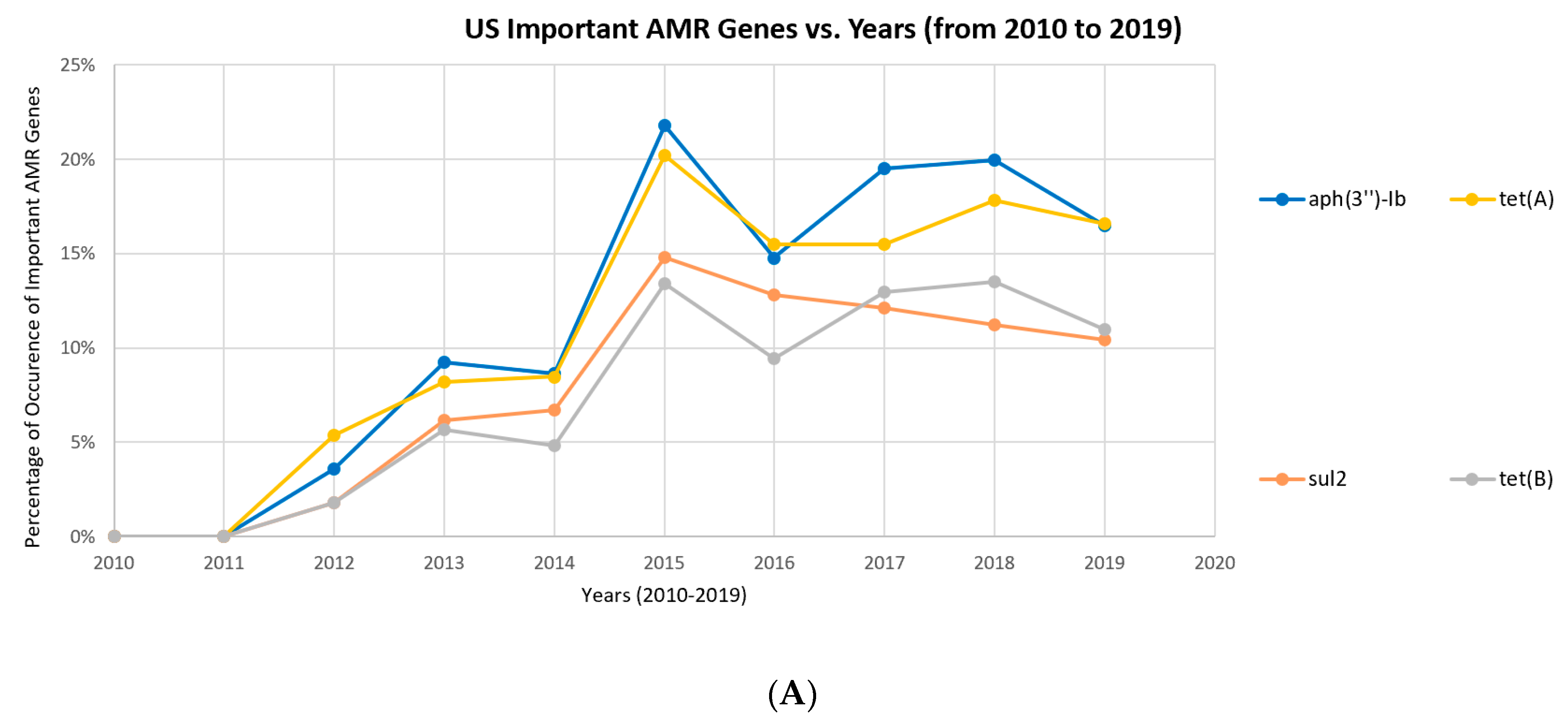
3.1.2. Historical Profiles of Highly-Occurring AMR Gene Occurrence from 2010 to 2019
3.2. Identification of Common, Highly Occurring Pathogens in the Studied Countries
3.2.1. Common, Highly Occurring Pathogens of Individual Countries for All Years
3.2.2. Historical Profiles of Highly-Occurring Pathogen Occurrence between 2010 and 2018
3.3. Major Meat Carriers Found in the Studied Countries
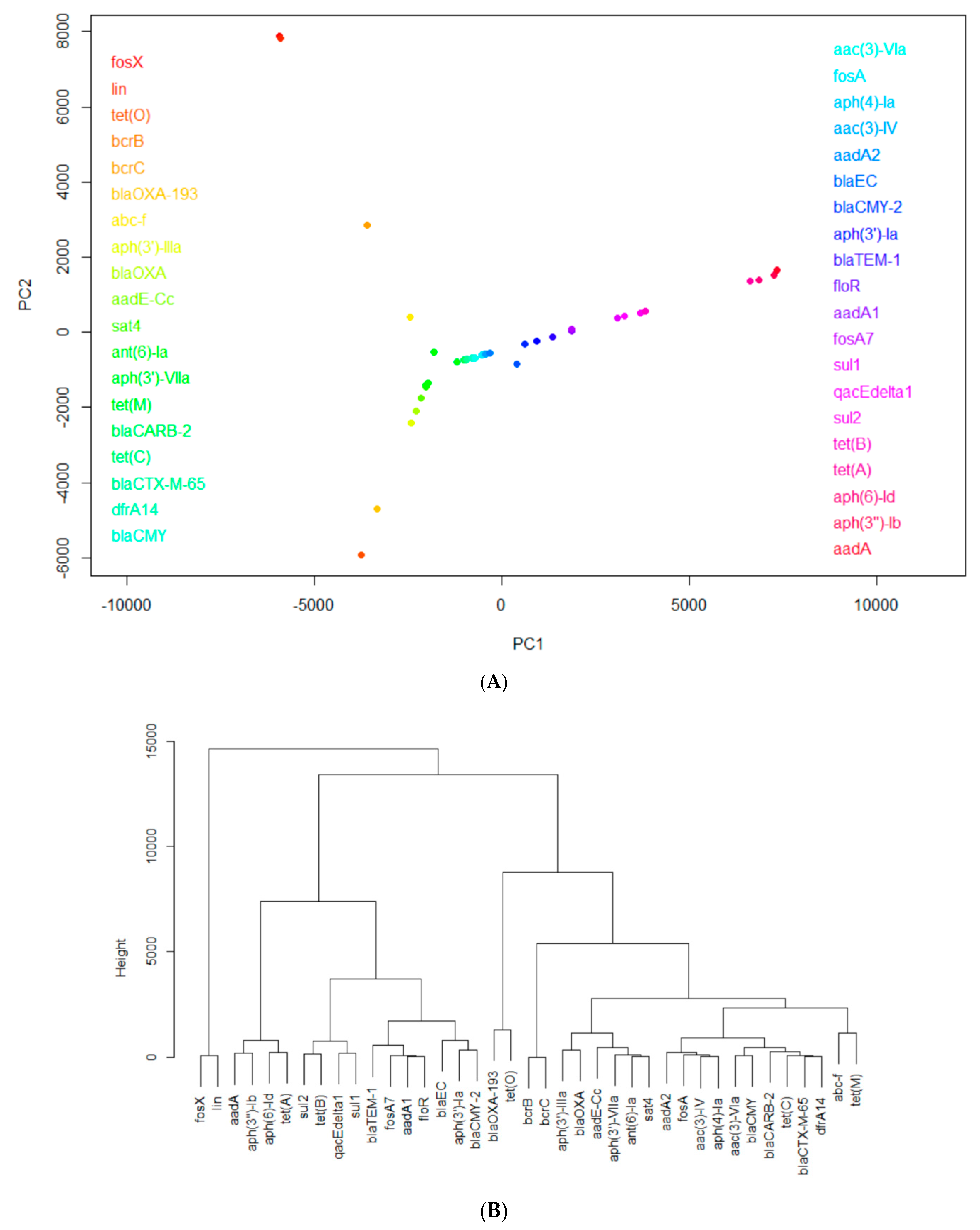
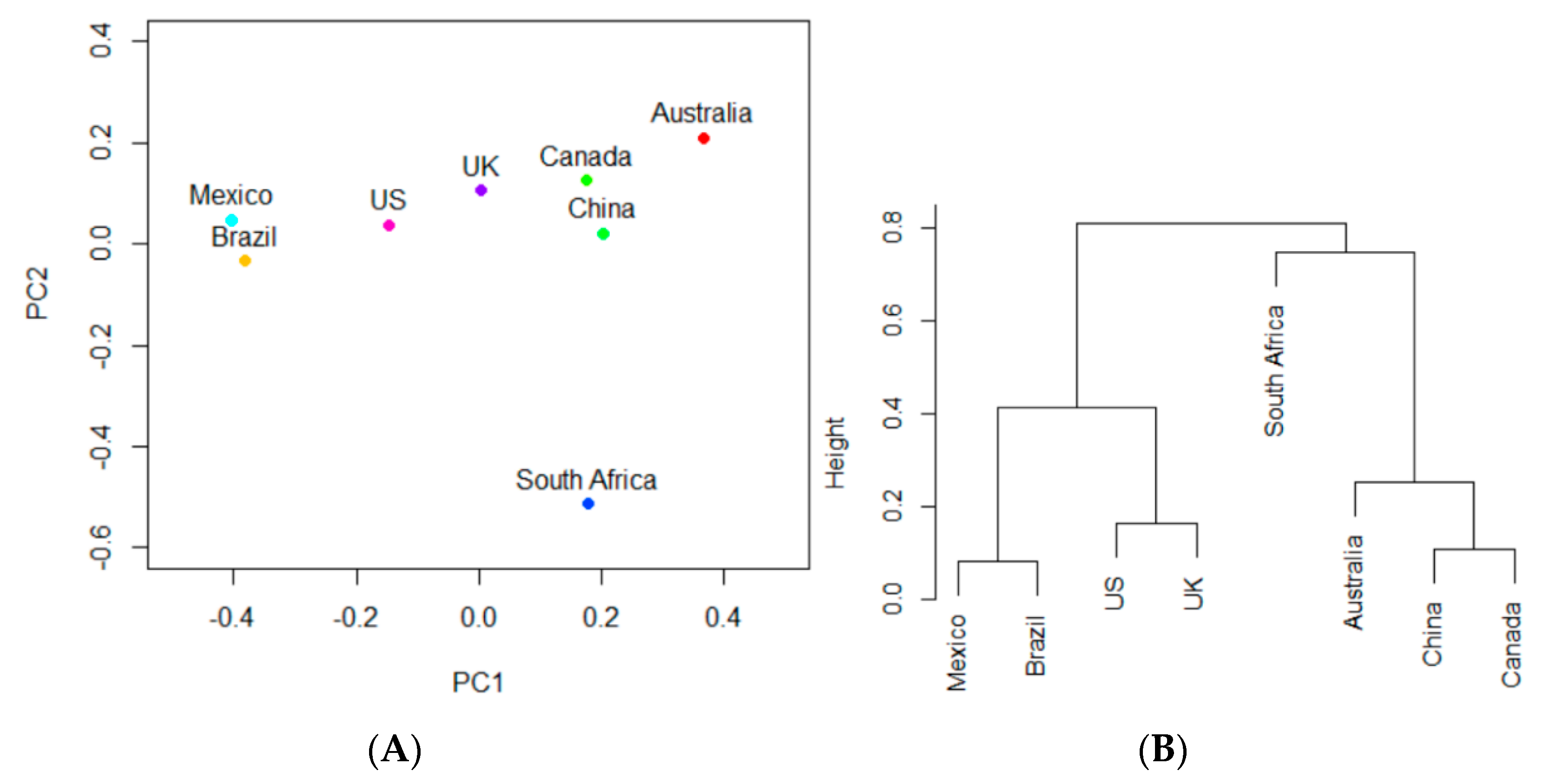
3.4. Identification of Similarity in Antimicrobial Resistance Occurrence in Studied Countries
4. Discussion
4.1. Absolute Occurrence versus the Relative Occurrence for Studying the Time Trend of Antimicrobial Resistance
4.2. Antimicrobial Usage and the Rising Overall Trend of AMR Genes
4.3. Geographic and Trade Relations between AMR Genes of Different Countries
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Velez, R.; Sloand, E. Combating antibiotic resistance, mitigating future threats and ongoing initiatives. J. Clin. Nurs. 2016, 25, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Michael, G.B.; Freitag, C.; Wendlandt, S.; Eidam, C.; Feßler, A.T.; Lopes, G.V.; Kadlec, K.; Schwarz, S. Emerging issues in antimicrobial resistance of bacteria from food-producing animals. Futur. Microbiol. 2015, 10, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Boil. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, S.L.; Oliver, K.B. Antibiotic resistance threats in the United States: Stepping back from the brink. Am. Fam. Phys. 2014, 89, 938–941. [Google Scholar]
- Friedrich, A.W. Control of hospital acquired infections and antimicrobial resistance in Europe: The way to go. Wien. Med. Wochenschr. 2019, 169 (Suppl. 1), 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.R.; Hota, B.; Ahmad, I.; Ii, R.D.S.; Foster, S.D.; Abbasi, F.; Schabowski, S.; Kampe, L.M.; Ciavarella, G.G.; Supino, M.; et al. Hospital and Societal Costs of Antimicrobial-Resistant Infections in a Chicago Teaching Hospital: Implications for Antibiotic Stewardship. Clin. Infect. Dis. 2009, 49, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Lekshmi, M.; Ammini, P.; Kumar, S.; Varela, M.F. The Food Production Environment and the Development of Antimicrobial Resistance in Human Pathogens of Animal Origin. Microorganisms 2017, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Suárez, E.J.; Selem-Mójica, N.; Ortiz-Lopez, R.; Gebreyes, W.A.; Allard, M.W.; Barona-Gómez, F.; Rubio-Lozano, M.S. Whole genome sequencing reveals widespread distribution of typhoidal toxin genes and VirB/D4 plasmids in bovine-associated nontyphoidal Salmonella. Sci. Rep. 2018, 8, 9864. [Google Scholar] [CrossRef] [PubMed]
- Davison, J. Genetic Exchange between Bacteria in the Environment. Plasmid 1999, 42, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, X.; Skogerbø, G.; Zhang, P.; Chen, R.; He, S.; Huang, D.-W. The human microbiome: A hot spot of microbial horizontal gene transfer. Genomics 2012, 100, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceric, O.; Tyson, G.H.; Goodman, L.B.; Mitchell, P.K.; Zhang, Y.; Prarat, M.; Cui, J.; Peak, L.; Scaria, J.; Antony, L.; et al. Enhancing the one health initiative by using whole genome sequencing to monitor antimicrobial resistance of animal pathogens: Vet-LIRN collaborative project with veterinary diagnostic laboratories in United States and Canada. BMC Veter-Res. 2019, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, E.A.; Gupta, S.K.; Williams, L.E.; Jové, T.; Hiott, L.M.; Woodley, T.A.; Barrett, J.B.; Jackson, C.R.; Wasilenko, J.L.; Simmons, M.; et al. Antimicrobial Resistance Genes, Cassettes, and Plasmids Present in Salmonella enterica Associated with United States Food Animals. Front. Microbiol. 2019, 10, 832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, E.; Tang, A.; Ye, M.C.; Wang, K.; Jia, Q.; Huang, Z. Data-Driven Analysis of Antimicrobial Resistance in Foodborne Pathogens from Six States within the US. Int. J. Environ. Res. Public Heal. 2019, 16, 1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.J. Statistical process monitoring: Basics and beyond. J. Chemom. 2003, 17, 480–502. [Google Scholar] [CrossRef]
- Kourti, T. Application of latent variable methods to process control and multivariate statistical process control in industry. Int. J. Adapt. Control. Signal Process. 2005, 19, 213–246. [Google Scholar] [CrossRef]
- Arnau, V.; Mars, S.; Marin, I. Iterative cluster analysis of protein interaction data. Bioinformatics 2005, 21, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Smith, K.; Hyndman, R.; Smith-Miles, K. Characteristic-Based Clustering for Time Series Data. Data Min. Knowl. Discov. 2006, 13, 335–364. [Google Scholar] [CrossRef]
- Bar-Joseph, Z.; Demaine, E.D.; Gifford, D.K.; Srebro, N.; Hamel, A.M.; Jaakkola, T.S. K-ary clustering with optimal leaf ordering for gene expression data. Bioinformatics 2003, 19, 1070–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299. [Google Scholar]
- USDA. Per Capita Consumption of Poultry and Livestock, 1960 to Forecast 2020. 2019. Available online: https://www.nationalchickencouncil.org/about-the-industry/statistics/per-capita-consumption-of-poultry-and-livestock-1965-to-estimated-2012-in-pounds/ (accessed on 16 September 2019).
- Farm Antibiotics. Available online: https://www.farmantibiotics.org/science-facts/antibiotic-infographs/ (accessed on 16 September 2019).
- WHO. Highest Priority Critically Important Antimicrobials. 2019. Available online: https://www.who.int/foodsafety/cia/en/ (accessed on 16 September 2019).
- FDA. Most Common Foodborne Illness. 2019. Available online: https://www.fda.gov/files/food/published/Most-Common-Foodborne-Illnesses-%28PDF%29.pdf (accessed on 16 September 2019).
- Australia, M.A.L. Know the Numbers: State of the Industry Report 2018. 2018. Available online: https://www.mla.com.au/prices-markets/market-news/know-the-numbers-state-of-the-industry-report-2018/ (accessed on 16 September 2019).
- CDC. Reports of Salmonella Outbreak Investigations from 2015. 2017. Available online: https://www.cdc.gov/salmonella/outbreaks-2015.html (accessed on 16 September 2019).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Genes | US | UK | South Africa | China | Australia | Canada | Mexico | Brazil |
|---|---|---|---|---|---|---|---|---|
| tet(A) | tet(A) | tet(A) | tet(A) | tet(A) | tet(A) | tet(A) | tet(A) | tet(A) |
| aph(3″)-Ib | aph(3″)-Ib | aph(3″)-Ib | aph(3″)-Ib | aph(3″)-Ib | aph(3″)-Ib | aph(3″)-Ib | aph(3″)-Ib | |
| aph(6)-Id | aph(6)-Id | aph(6)-Id | aph(6)-Id | aph(6)-Id | aph(6)-Id | aph(6)-Id | aph(6)-Id | |
| blaEC | blaEC | blaEC | blaEC | blaEC | blaEC | blaEC | blaEC | |
| blaTEM-1 | blaTEM-1 | blaTEM-1 | blaTEM-1 | blaTEM-1 | blaTEM-1 | blaTEM-1 | blaTEM-1 | |
| qacEdelta1 | qacEdelta1 | qacEdelta1 | qacEdelta1 | qacEdelta1 | qacEdelta1 | qacEdelta1 | qacEdelta1 | |
| sul1 | sul1 | sul1 | sul1 | sul1 | sul1 | sul1 | sul1 | |
| sul2 | sul2 | sul2 | sul2 | sul2 | sul2 | sul2 | sul2 | |
| aadA1 | aadA1 | aadA1 | aadA1 | aadA1 | aadA1 | aadA1 | ||
| tet(B) | tet(B) | tet(B) | tet(B) | tet(B) | tet(B) | |||
| aadA2 | aadA2 | aadA2 | aadA2 | aadA2 | aadA2 | |||
| fosX | fosX | fosX | fosX | fosX | fosX | |||
| lin | lin | lin | lin | lin | lin | |||
| aadA | aadA | aadA | aadA | aadA | ||||
| floR | floR | floR | floR | floR | ||||
| fosA7 | fosA7 | fosA7 | fosA7 | fosA7 | ||||
| aph(3′)-Ia | aph(3′)-Ia | aph(3′)-Ia | aph(3′)-Ia | |||||
| dfrA12 | dfrA12 | dfrA12 | dfrA12 | |||||
| blaCMY-2 | blaCMY-2 | blaCMY-2 | blaCMY-2 | |||||
| bcrB | bcrB | bcrB | ||||||
| bcrC | bcrC | bcrC | ||||||
| abc-f | abc-f | abc-f | ||||||
| sul3 | sul3 | sul3 | ||||||
| qnrB19 | qnrB19 | qnrB19 | ||||||
| blaCTX-M-55 | blaCTX-M-55 | |||||||
| blaOXA-193 | blaOXA-193 | |||||||
| catA1 | catA1 | |||||||
| mph(A) | mph(A) | |||||||
| tet(O) | tet(O) | |||||||
| aadA5 | aadA5 | |||||||
| ble | ble | |||||||
| bleO | bleO | |||||||
| fosA3 | fosA3 | |||||||
| mcr-1.1 | mcr-1.1 | |||||||
| oqxA | oqxA | |||||||
| oqxB | oqxB | |||||||
| aac(2′)-Ic | aac(2′)-Ic | |||||||
| blaA | blaA | |||||||
| erm(37) | erm(37) | |||||||
| cmlA1 | cmlA1 | |||||||
| dfrA5 | dfrA5 | |||||||
| qacL | qacL | |||||||
| fosA | fosA |
| US | UK | South Africa | China | Australia | Canada | Mexico | Brazil |
|---|---|---|---|---|---|---|---|
| aadA1 | aadA1 | aph(3″)-Ib | aadA2 | blaTEM-1 | aadA1 | aadA2 | aph(3″)-Ib |
| blaOXA-193 | aadA2 | aph(6)-Id | blaTEM-1 | dfrA5 | tet(B) | blaTEM-1 | aph(6)-Id |
| blaTEM-1 | abc.f | blaEC | ble | sul2 | dfrA12 | blaCMY-2 | |
| floR | aph(3″)-Ib | blaTEM-1 | floR | fosA | blaTEM-1 | ||
| qacEdelta1 | aph(6)-Id | dfrA5 | mph(A) | qacEdelta1 | floR | ||
| sul1 | blaCMY.2 | sul2 | oqxA | sul1 | fosA7 | ||
| tet(A) | blaTEM.1 | tet(A) | oqxB | sul2 | sul2 | ||
| tet(O) | fosA7 | tet(B) | qacEdelta1 | tet(A) | tet(A) | ||
| qacEdelta1 | sul1 | tet(B) | |||||
| sul1 | sul2 | ||||||
| sul2 | |||||||
| tet(A) |
| Countries | US | UK | South Africa | China | Australia | Canada | Mexico | Brazil | |
|---|---|---|---|---|---|---|---|---|---|
| Pathogens | |||||||||
| E. coli and Shigella | x | x | x | x | x | x | x | x | |
| Salmonella enterica | x | x | x | x | x | x | x | ||
| Listeria monocytogenes | x | x | x | x | x | ||||
| Klebsiella pneumoniae | x | x | |||||||
| Mycobacterium tuberculosis | x | ||||||||
| Campylobacter jejuni | x | ||||||||
| Pseudomonas aeruginosa | x | ||||||||
| Enterobacter | x | ||||||||
| Cronobacter | x | ||||||||
| US | UK | South Africa | China | Australia | Canada | Mexico | Brazil |
|---|---|---|---|---|---|---|---|
| Campylobacter jejuni | Listeria monocytogenes | E. coli and Shigella | Klebsiella pneumoniae | E. coli and Shigella | E. coli and Shigella | E. coli and Shigella | Salmonella enterica |
| Salmonella enterica | Salmonella enterica |
| Countries | US | UK | South Africa | China | Australia | Canada | Mexico | Brazil | Total Rank | |
|---|---|---|---|---|---|---|---|---|---|---|
| Meats | ||||||||||
| Beef | 2 | 4 | No Data | 4 | 1 | 1 | 1 | 4 | 17 | |
| Chicken | 1 | 1 | No Data | 1 | 4 | 2 | 2 | 1 | 12 | |
| Pork | 4 | 2 | No Data | 2 | 4 | 4 | 4 | 2 | 22 | |
| Turkey | 4 | 4 | No Data | 4 | 4 | 4 | 4 | 4 | 28 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Wang, A.; Fu, M.; Wang, A.; Chen, K.; Jia, Q.; Huang, Z. Investigation of Incidents and Trends of Antimicrobial Resistance in Foodborne Pathogens in Eight Countries from Historical Sample Data. Int. J. Environ. Res. Public Health 2020, 17, 472. https://doi.org/10.3390/ijerph17020472
Yang K, Wang A, Fu M, Wang A, Chen K, Jia Q, Huang Z. Investigation of Incidents and Trends of Antimicrobial Resistance in Foodborne Pathogens in Eight Countries from Historical Sample Data. International Journal of Environmental Research and Public Health. 2020; 17(2):472. https://doi.org/10.3390/ijerph17020472
Chicago/Turabian StyleYang, Katherine, Annie Wang, Matthew Fu, Aaron Wang, Kevin Chen, Qian Jia, and Zuyi Huang. 2020. "Investigation of Incidents and Trends of Antimicrobial Resistance in Foodborne Pathogens in Eight Countries from Historical Sample Data" International Journal of Environmental Research and Public Health 17, no. 2: 472. https://doi.org/10.3390/ijerph17020472