The L1 Retrotranspositional Stimulation by Particulate and Soluble Cadmium Exposure is Independent of the Generation of DNA Breaks

Abstract
:Introduction
Materials and Methods
Plasmids
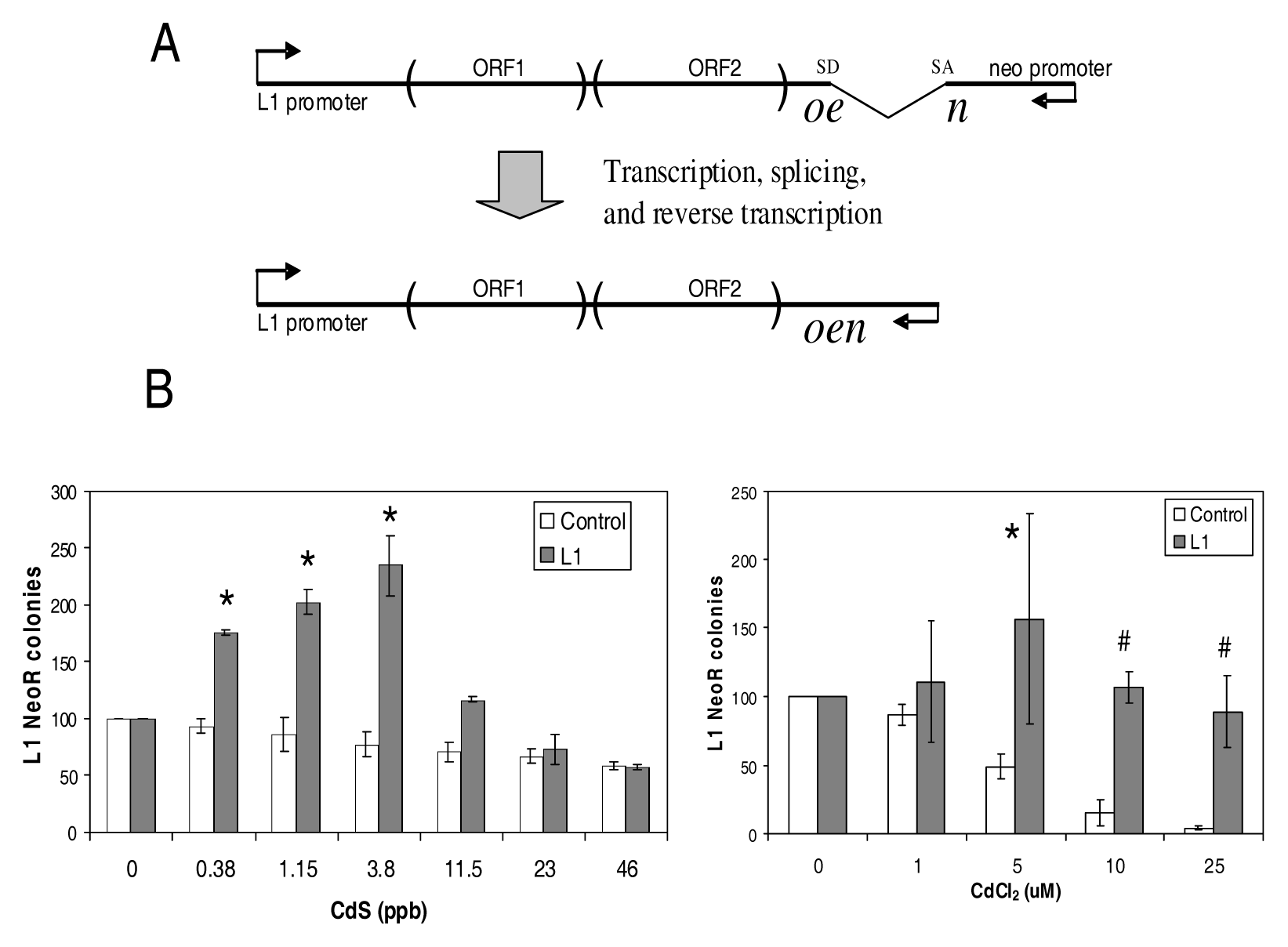
- Schematic of the L1 retrotransposition plasmid (top). L1 RNA is transcribed by the L1 internal promoter. A neomycin resistance gene (neo) or a blasticidin resistance gene is located at the 3′ end in the opposite strand that contains a disrupting intron. The intron interrupting the neo can only be removed by splicing from RNA generated from the L1 promoter. During the L1 retrotransposition process the spliced RNA is reverse transcribed, and the cDNA inserted into the genome (bottom). The new L1 insert now contains a functional neo gene. Only newly integrated copies that retrotransposed from the spliced L1 RNA will present neomycin resistance. Promoters and transcription orientations are indicated by arrows. SD: splice donor, SA: splice acceptor.
- Both CdS and CdCl2stimulate L1 retrotransposition in a dose dependent manner: NeoR colonies from separate L1 transfections (gray bars) treated with different doses of CdS, or CdCl2 (X axis) are shown. The no treatment (0 doses) for each experiment was defined as 100%. For toxicity control (white bars), cells were transfected in parallel with an unrelated plasmid with neomycin resistance and no L1 plasmid. Error bars indicate standard deviation. Statistically significant differences are indicated relative to the no treatment [t-test p<0.01(*)], or relative to no treatment after correcting for the cell death observed in the control [t-test p<0.01(*)].
L1 Retrotransposition Assay
Toxicity Assay
Chemical Compounds
Comet Assay
Results
Both Soluble and Particulate Cadmium Stimulate L1 Retrotransposition
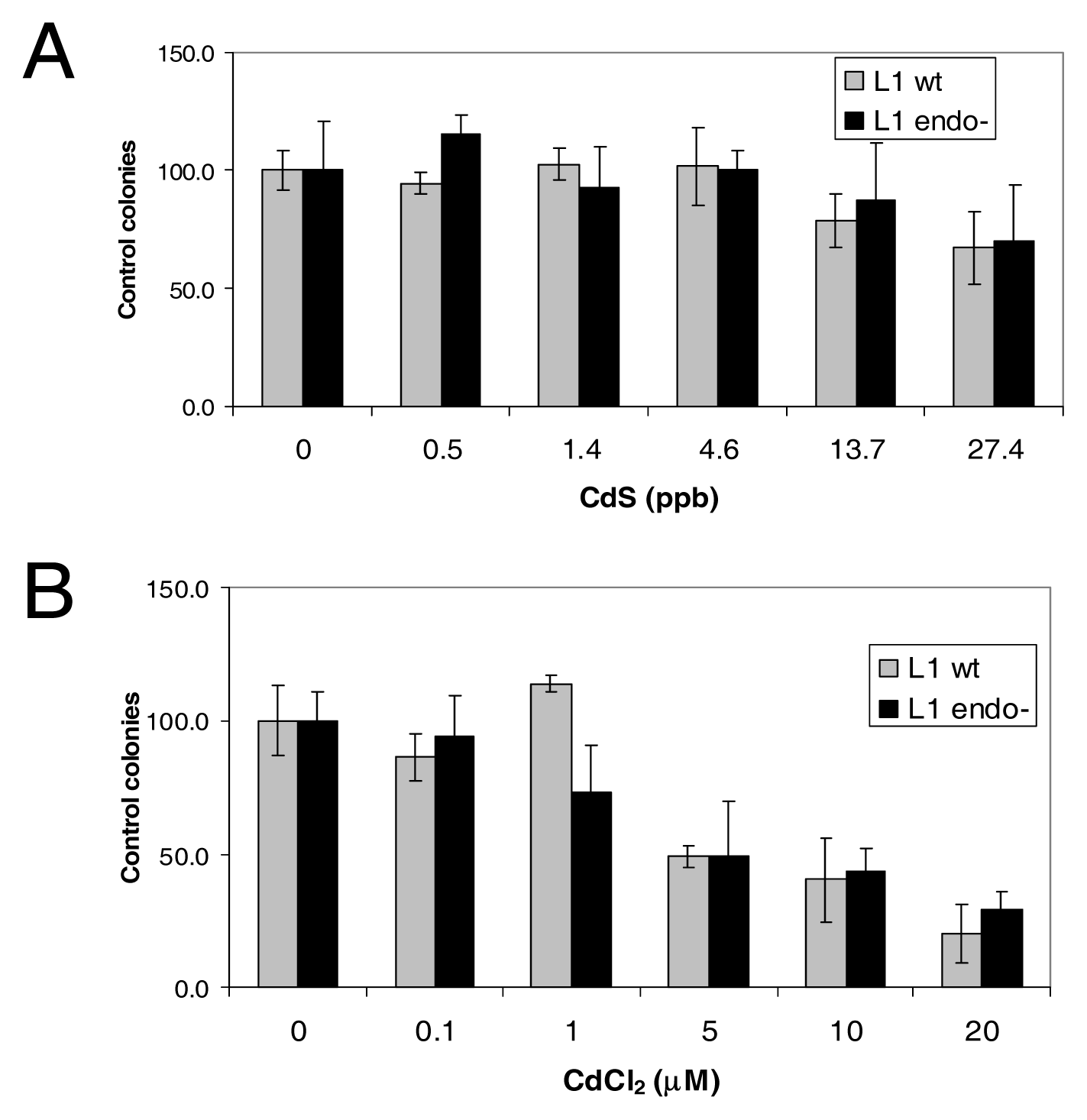
L1 Endonuclease Does Not Contribute to the Toxicity Observed
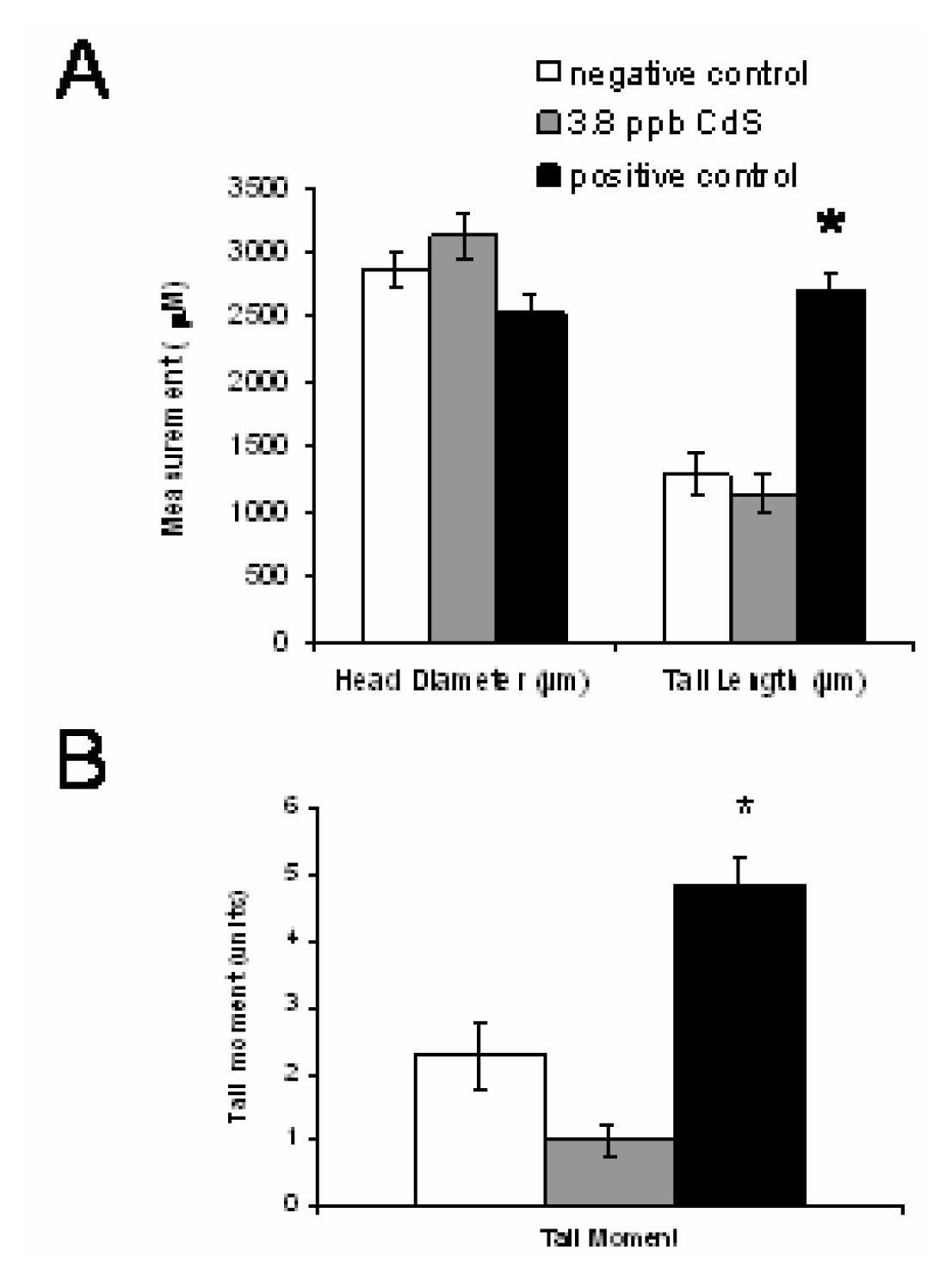
The L1 Stimulating Doses of Cds Do Not Contribute to DNA Breaks
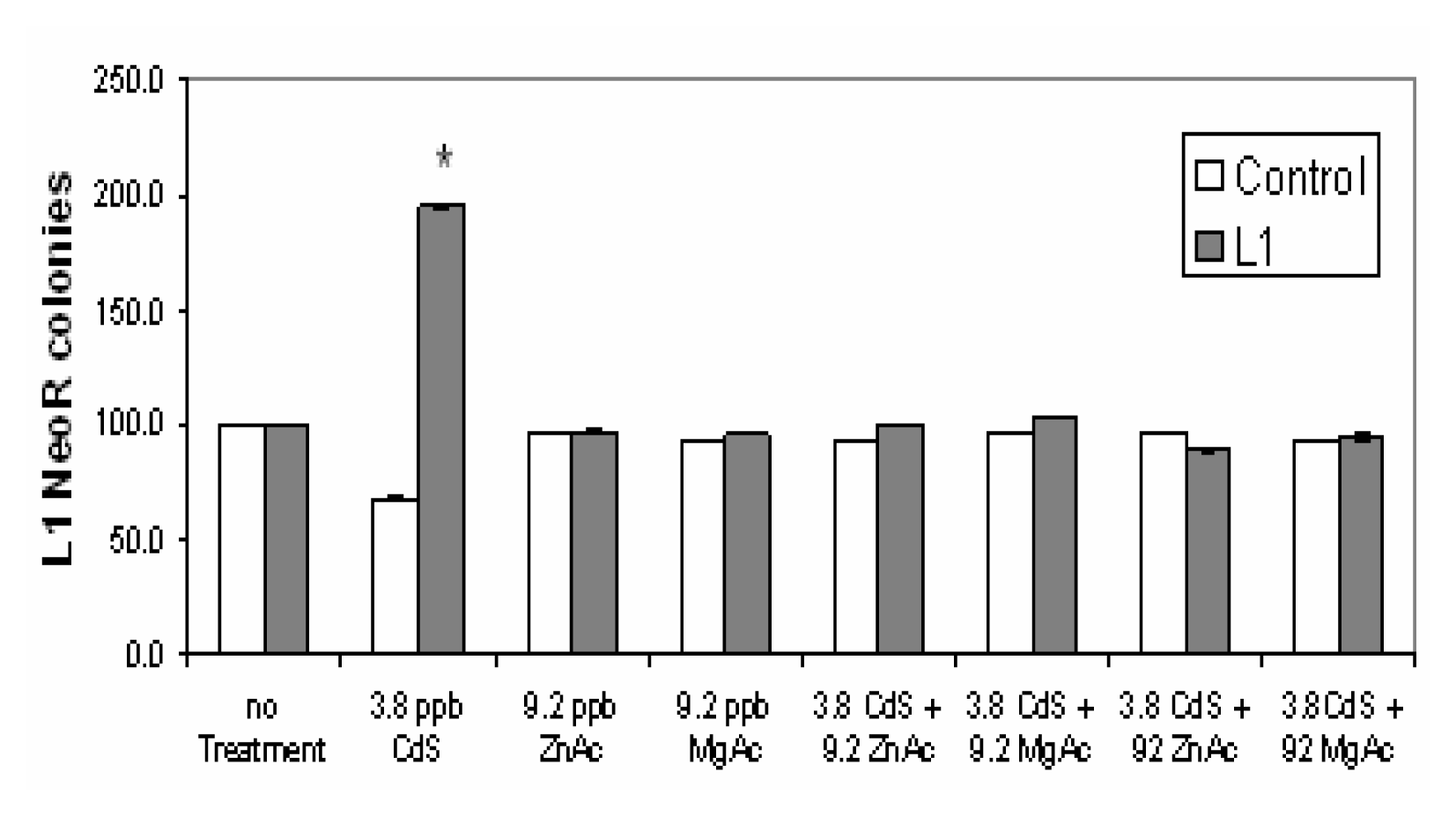
Magnesium and Zinc Reverse the L1 Stimulation Induced by Cds Exposure
Discussion





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heavy Metal | DNA Repair Pathway Affected | L1 Stimulation |
|---|---|---|
| Cadmium | NER-affect recognition step of DNA damage [55]; Strong inhibition of 8-oxo-dGTPase activity [16]; inhibition of APE-1 nuclease [56], inhibition of mismatch repair [57]. | Yes [44] (CdS, CdCl2§) |
| Nickel | NER-affect recognition step of DNA damage [55]; reduce the repair of DNA adducts [47]; weak inhibition of 8-oxo-dGTPase activity [16]. | Yes [44,54] (NiO, NiCl2) |
| Cobalt | NER-affect both incision and polymerization of repair patches [58]; weak inhibition of 8-oxo-dGTPase activity [16] | No [54] (CoCl2) |
| Mercury | Inhibition of single strand DNA repair [59], inhibition of dUTPase and DNA polymerase alpha [60]. | Yes [44] (HgS) |
| Copper (Cu II) | Strong inhibition of 8-oxo-dGTPase activity [16]; inhibition of single strand DNA repair [61]. | N.D. |
| Iron (Fe III) | Inhibition of APE-1 nuclease [56]. | Yes† |
| Arsenite (As III) | Low doses inhibit PARP [62], and impair incision step [63]. | Yes† |
Acknowledgements
References
- Meeting of the IARC working group on beryllium, cadmium, mercury and exposures in the glass manufacturing industry. Scand. J. Work Environ. Health 1993, 19(5), 360–363.
- Waalkes, M. P. Cadmium carcinogenesis in review. J. Inorg. Biochem 2000, 79(1–4), 241–244. [Google Scholar]
- Takenaka, S.; Oldiges, H.; Konig, H.; Hochrainer, D.; Oberdorster, G. Carcinogenicity of cadmium chloride aerosols in W rats. J. Natl. Cancer Inst 1983, 70(2), 367–373. [Google Scholar]
- Costa, M.; Sutherland, J. E.; Peng, W.; Salnikow, K.; Broday, L.; Kluz, T. Molecular biology of nickel carcinogenesis. Mol. Cell Biochem 2001, 222(1–2), 205–211. [Google Scholar]
- Szuster-Ciesielska, A.; et al. The inhibitory effect of zinc on cadmium-induced cell apoptosis and reactive oxygen species (ROS) production in cell cultures. Toxicology 2000, 145(2–3), 159–171. [Google Scholar]
- Littlefield, N. A.; Hass, B. S.; James, S. J.; Poirier, L. A. Protective effect of magnesium on DNA strand breaks induced by nickel or cadmium. Cell Biol. Toxicol 1994, 10(2), 127–135. [Google Scholar]
- Sunderman, F. W., Jr. Search for molecular mechanisms in the genotoxicity of nickel. Scand. J. Work Environ. Health 1993, 19 Suppl 1, 75–80. [Google Scholar]
- Kasprzak, K. S. Possible role of oxidative damage in metal-induced carcinogenesis. Cancer Invest 1995, 13(4), 411–430. [Google Scholar]
- Tully, D. Effects of arsenic, cadmium, chromium, and lead on gene expression regulated by a battery of 13 different promoters in recombinant HepG2 cells. Toxicol. Appl. Pharmacol 2000, 168(2), 79–90. [Google Scholar]
- Beyersmann, D.; Hechtenberg, S. Cadmium, gene regulation, and cellular signalling in mammalian cells. Toxicol. Appl. Pharmacol 1997, 144(2), 247–261. [Google Scholar]
- Asmuss, M.; Mullenders, L. H.; Hartwig, A. Interference by toxic metal compounds with isolated zinc finger DNA repair proteins. Toxicol. Lett 2000, 112–113, 227–231. [Google Scholar]
- Hartwig, A. Carcinogenicity of metal compounds: possible role of DNA repair inhibition. Toxicol. Lett 1998, 102–103, 235–239. [Google Scholar]
- Kopera, E.; Schwerdtle, T.; Hartwig, A.; Bal, W. Co(II) and Cd(II) substitute for Zn(II) in the zinc finger derived from the DNA repair protein XPA, demonstrating a variety of potential mechanisms of toxicity. Chem. Res. Toxicol 2004, 17(11), 1452–1458. [Google Scholar]
- Chen, X.; Chu, M.; Giedroc, D. P. Spectroscopic characterization of Co(II)-, Ni(II)-, and Cd(II)-substituted wild-type and non-native retroviral-type zinc finger peptides. J. Biol. Inorg. Chem 2000, 5(1), 93–101. [Google Scholar]
- Asmuss, M.; Mullenders, L. H.; Eker, A.; Hartwig, A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis 2000, 21(11), 2097–2104. [Google Scholar]
- Kasprzak, K. S.; Bialkowski, K. Inhibition of antimutagenic enzymes, 8-oxo-dGTPases, by carcinogenic metals. Recent developments. J. Inorg. Biochem 2000, 79(1–4), 231–236. [Google Scholar]
- Lander, E. S.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409(6822), 860–921. [Google Scholar]
- Grimaldi, G.; Skowronski, J.; Singer, M. F. Defining the beginning and end of KpnI family segments. Embo J 1984, 3(8), 1753–1759. [Google Scholar]
- Brouha, B.; Schustak, J.; Badge, R. M.; Lutz-Prigge, S.; Farley, A. H.; Moran, J. V.; Kazazian, H. H., Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. U. S. A 2003, 100(9), 5280–5285. [Google Scholar]
- Ostertag, E. M.; Kazazian, H. H., Jr. Biology of Mammalian l1 retrotransposons. Annu. Rev. Genet 2001, 355, 01–538. [Google Scholar]
- Trelogan, S. A.; Martin, S. L. Tightly regulated, developmentally specific expression of the first open reading frame from LINE-1 during mouse embryogenesis. Proc Natl Acad Sci U S A 1995, 92(5), 1520–1524. [Google Scholar]
- Ergun, S.; et al. Cell type-specific expression of LINE-1 open reading frames 1 and 2 in fetal and adult human tissues. J. Biol. Chem 2004, 279(26), 27753–27763. [Google Scholar]
- Muotri, A. R.; Chu, V. T.; Marchetto, M. C.; Deng, W.; Moran, J. V.; Gage, F. H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435(7044), 903–910. [Google Scholar]
- Singer, M. F.; Krek, V.; McMillan, J. P.; Swergold, G. D.; Thayer, R. E. LINE-1: a human transposable element. Gene 1993, 135(1–2), 183–188. [Google Scholar]
- Skowronski, J.; Fanning, T. G.; Singer, M. F. Unit-length line-1 transcripts in human teratocarcinoma cells. Mol. Cell Biol 1988, 8(4), 1385–1397. [Google Scholar]
- Asch, H. L.; Eliacin, E.; Fanning, T. G.; Connolly, J. L.; Bratthauer, G.; Asch, B. B. Comparative expression of the LINE-1 p40 protein in human breast carcinomas and normal breast tissues. Oncol. Res 1996, 8(6), 239–247. [Google Scholar]
- Bratthauer, G. L.; Cardiff, R. D.; Fanning, T. G. Expression of LINE-1 retrotransposons in human breast cancer. Cancer 1994, 73(9), 2333–2336. [Google Scholar]
- Bratthauer, G. L.; Fanning, T. G. Active LINE-1 retrotransposons in human testicular cancer. Oncogene 1992, 7(3), 507–510. [Google Scholar]
- Chalitchagorn, K.; et al. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene 2004, 23(54), 8841–8846. [Google Scholar]
- Roman-Gomez, J.; et al. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene 2005. epub. [Google Scholar]
- Tchenio, T.; Casella, J. F.; Heidmann, T. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res 2000, 28(2), 411–415. [Google Scholar]
- Perepelitsa-Belancio, V.; Deininger, P. L. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat Genet 2003, 35(4), 363–366. [Google Scholar]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K. W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res 1992, 52(3), 643–645. [Google Scholar]
- Morse, B.; Rotherg, P. G.; South, V. J.; Spandorfer, J. M.; Astrin, S. M. Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nature 1988, 333(6168), 87–90. [Google Scholar]
- Werle-Schneider, G.; von Brevern, M. C.; Sylla, B. S.; Hollstein, M. C. De novo retrotransposition of unbiased sequences in a human breast cancer cell clone. Genes Chromosomes. Cancer 1999, 26(1), 84–91. [Google Scholar]
- Luan, D. D.; Korman, M. H.; Jakubczak, J. L.; Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 1993, 72, 595–605. [Google Scholar]
- Martin, F.; Maranon, C.; Olivares, M.; Alonso, C.; Lopez, M. C. Characterization of a non-long terminal repeat retrotransposon cDNA (L1Tc) from Trypanosoma cruzi: homology of the first ORF with the ape family of DNA repairs enzymes. J. Mol. Biol 1995, 247(1), 49–59. [Google Scholar]
- Feng, Q.; Moran, J. V.; Kazazian, H. H., Jr; Boeke, J. D. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 1996, 87(5), 905–916. [Google Scholar]
- Scott, A. F.; et al. Origin of the human L1 elements: proposed progenitor genes deduced from a consensus DNA sequence. Genomics 1987, 1(2), 113–125. [Google Scholar]
- Mathias, S. L.; Scott, A. F.; Kazazian, H. H., Jr; Boeke, J. D.; Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science 1991, 254, 1808–1810. [Google Scholar]
- Morrish, T. A.; Gilbert, N.; Myers, J. S.; Vincent, B. J.; Stamato, T. D.; Taccioli, G. E.; Batzer, M. A.; Moran, J. V. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet 2002, 31(2), 159–165. [Google Scholar]
- Moore, J. K.; Haber, J. E. Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature 1996, 383(6601), 644–646. [Google Scholar]
- Moran, J. V.; DeBerardinis, R. J.; Kazazian, H. H., Jr. Exon shuffling by L1 retrotransposition. Science 1999, 283(5407), 1530–1534. [Google Scholar]
- Kale, S. P.; Moore, L.; Deininger, P. L.; Roy-Engel, A. M. Heavy metals stimulate human LINE-1 retrotransposition. Int. J. Env. Res. Public Health 2005, 2, 84–90. [Google Scholar]
- Costa, M. Perspectives on the mechanism of nickel carcinogenesis gained from models of in vitro carcinogenesis. Environ. Health Perspect 1989, 81, 73–76. [Google Scholar]
- Singh, J.; Carlisle, D. L.; Pritchard, D. E.; Patierno, S. R. Chromium-induced genotoxicity and apoptosis: relationship to chromium carcinogenesis (review). Oncol. Rep 1998, 5(6), 1307–1318. [Google Scholar]
- Schwerdtle, T.; Seidel, A.; Hartwig, A. Effect of soluble and particulate nickel compounds on the formation and repair of stable benzo[a]pyrene DNA adducts in human lung cells. Carcinogenesis 2002, 23(1), 47–53. [Google Scholar]
- Harrison, P. T.; Heath, J. C. Apparent synergy in lung carcinogenesis: interactions between N-nitrosoheptamethyleneimine, particulate cadmium and crocidolite asbestos fibres in rats. Carcinogenesis 1986, 7(11), 1903–1908. [Google Scholar]
- Hartwig, A.; Schwerdtle, T. Interactions by carcinogenic metal compounds with DNA repair processes: toxicological implications. Toxicol. Lett 2002, 127(1–3), 47–54. [Google Scholar]
- Kasprzak, K. S.; Sunderman, F. W., Jr. Mechanisms of dissolution of nickel subsulfide in rat serum. Res. Commun. Chem. Pathol. Pharmacol 1977, 16(1), 95–108. [Google Scholar]
- Klein, C. B.; Frenkel, K.; Costa, M. The role of oxidative processes in metal carcinogenesis. Chem. Res. Toxicol 1991, 4(6), 592–604. [Google Scholar]
- Coen, N.; Mothersill, C.; Kadhim, M.; Wright, E. G. Heavy metals of relevance to human health induce genomic instability. J. Pathol 2001, 195(3), 293–299. [Google Scholar]
- Misra, R. R.; Smith, G. T.; Waalkes, M. P. Evaluation of the direct genotoxic potential of cadmium in four different rodent cell lines. Toxicology 1998, 126(2), 103–114. [Google Scholar]
- El Sawy, M.; Kale, S. P.; Dugan, C.; Nguyen, T. Q.; Belancio, V.; Bruch, H.; Roy-Engel, A.; Deininger, P. L. Nickel stimulates L1 retrotransposition by a post-transcriptional mechanism. J Mol. Biol 2005, 354(2), 246–257. [Google Scholar]
- Hartmann, M.; Hartwig, A. Disturbance of DNA damage recognition after UV-irradiation by nickel(II) and cadmium(II) in mammalian cells. Carcinogenesis 1998, 19(4), 617–621. [Google Scholar]
- McNeill, D. R.; Narayana, A.; Wong, H. K.; Wilson, D. M., III. Inhibition of Ape1 nuclease activity by lead; iron; and cadmium. Environ. Health Perspect 2004, 112(7), 799–804. [Google Scholar]
- Banerjee, S.; Flores-Rozas, H. Cadmium inhibits mismatch repair by blocking the ATPase activity of the MSH2-MSH6 complex. Nucleic Acids Res 2005, 33(4), 1410–1419. [Google Scholar]
- Kasten, U.; Mullenders, L. H.; Hartwig, A. Cobalt(II) inhibits the incision and the polymerization step of nucleotide excision repair in human fibroblasts. Mutat. Res 1997, 383(1), 81–89. [Google Scholar]
- Cantoni, O.; Costa, M. Correlations of DNA strand breaks and their repair with cell survival following acute exposure to mercury(II) and X-rays. Mol. Pharmacol 1983, 24(1), 84–89. [Google Scholar]
- Williams, M. V.; Winters, T.; Waddell, K. S. In vivo effects of mercury (II) on deoxyuridine triphosphate nucleotidohydrolase, DNA polymerase (alpha; beta), and uracil-DNA glycosylase activities in cultured human cells: relationship to DNA damage, DNA repair, and cytotoxicity. Mol. Pharmacol 1987, 31(2), 200–207. [Google Scholar]
- Snyder, R. D.; Lachmann, P. J. Thiol involvement in the inhibition of DNA repairs by metals in mammalian cells. Mol. Toxicol 1989, 2(2), 117–128. [Google Scholar]
- Yager, J. W.; Wiencke, J. K. Inhibition of poly (ADP-ribose) polymerase by arsenite. Mutat. Res 1997, 386(3), 345–351. [Google Scholar]
- Hartwig, A.; Groblinghoff, U. D.; Beyersmann, D.; Natarajan, A. T.; Filon, R.; Mullenders, L. H. Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis 1997, 18(2), 399–405. [Google Scholar]
- Gasior, S. L.; Wakeman, T. P.; Xu, B.; Deininger, P. L. The human LINE-1 retrotransposon creates DNA double-strand breaks. In J. Mol. Biol; 2006; Volume 357, 5, pp. 1383–1393, * Personal communication was published after submission of manuscript. [Google Scholar]
© 2006 MDPI. All rights reserved.
Share and Cite
Kale, S.P.; Carmichael, M.C.; Harris, K.; Roy-Engel, A.M. The L1 Retrotranspositional Stimulation by Particulate and Soluble Cadmium Exposure is Independent of the Generation of DNA Breaks. Int. J. Environ. Res. Public Health 2006, 3, 121-128. https://doi.org/10.3390/ijerph2006030015
Kale SP, Carmichael MC, Harris K, Roy-Engel AM. The L1 Retrotranspositional Stimulation by Particulate and Soluble Cadmium Exposure is Independent of the Generation of DNA Breaks. International Journal of Environmental Research and Public Health. 2006; 3(2):121-128. https://doi.org/10.3390/ijerph2006030015
Chicago/Turabian StyleKale, Shubha P., Mary C. Carmichael, Kelley Harris, and Astrid M. Roy-Engel. 2006. "The L1 Retrotranspositional Stimulation by Particulate and Soluble Cadmium Exposure is Independent of the Generation of DNA Breaks" International Journal of Environmental Research and Public Health 3, no. 2: 121-128. https://doi.org/10.3390/ijerph2006030015