Isoperibolic Titration Calorimetry as a Tool for the Prediction of Thermodynamic Properties of Cyclodextrins



Abstract
:
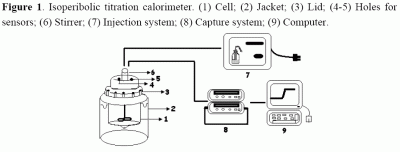{kind=link}
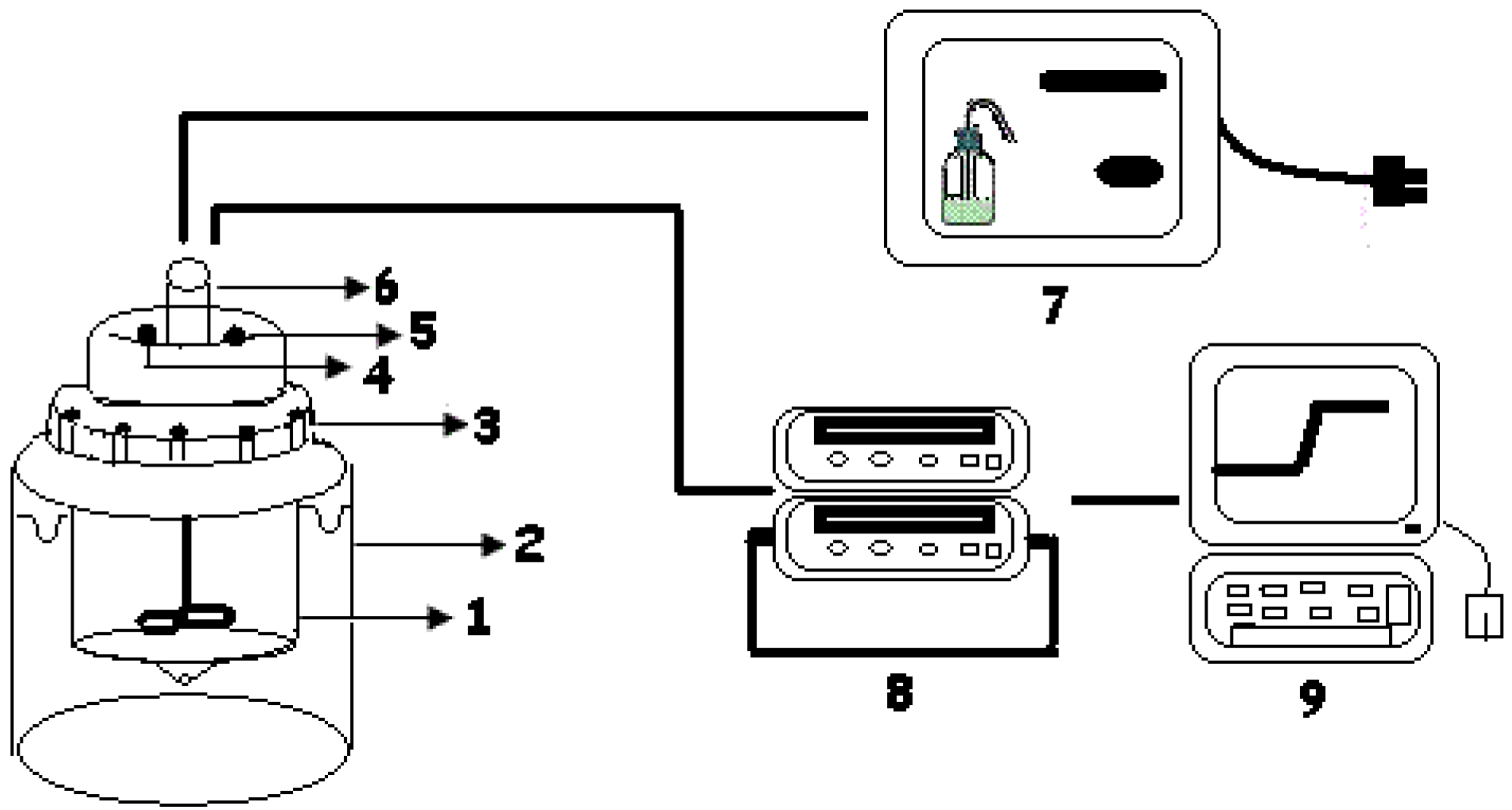{kind=link}
{kind=link}
{kind=link}
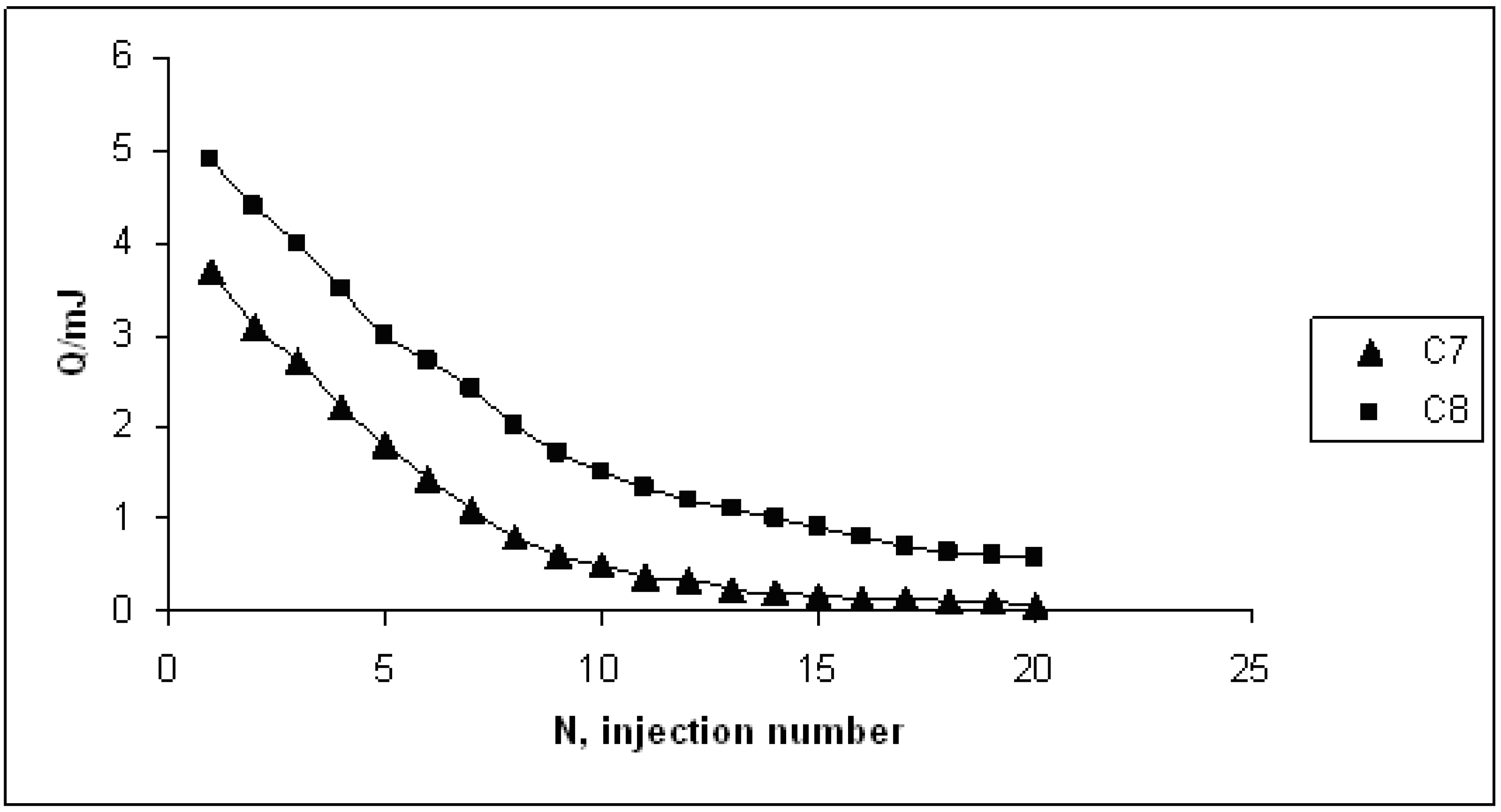{kind=link}
{kind=link}
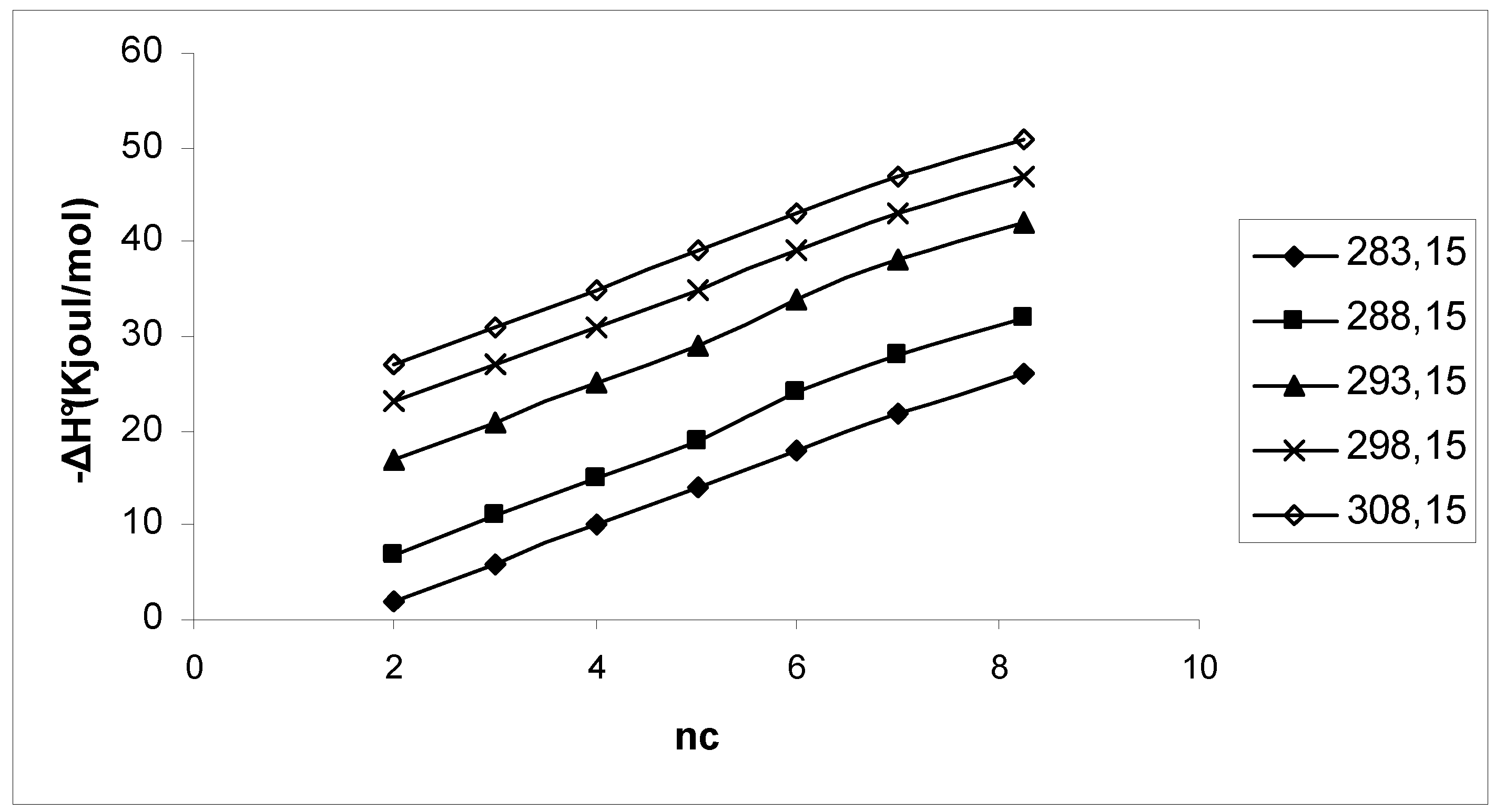{kind=link}
{kind=link}
{kind=link}
1. Introduction
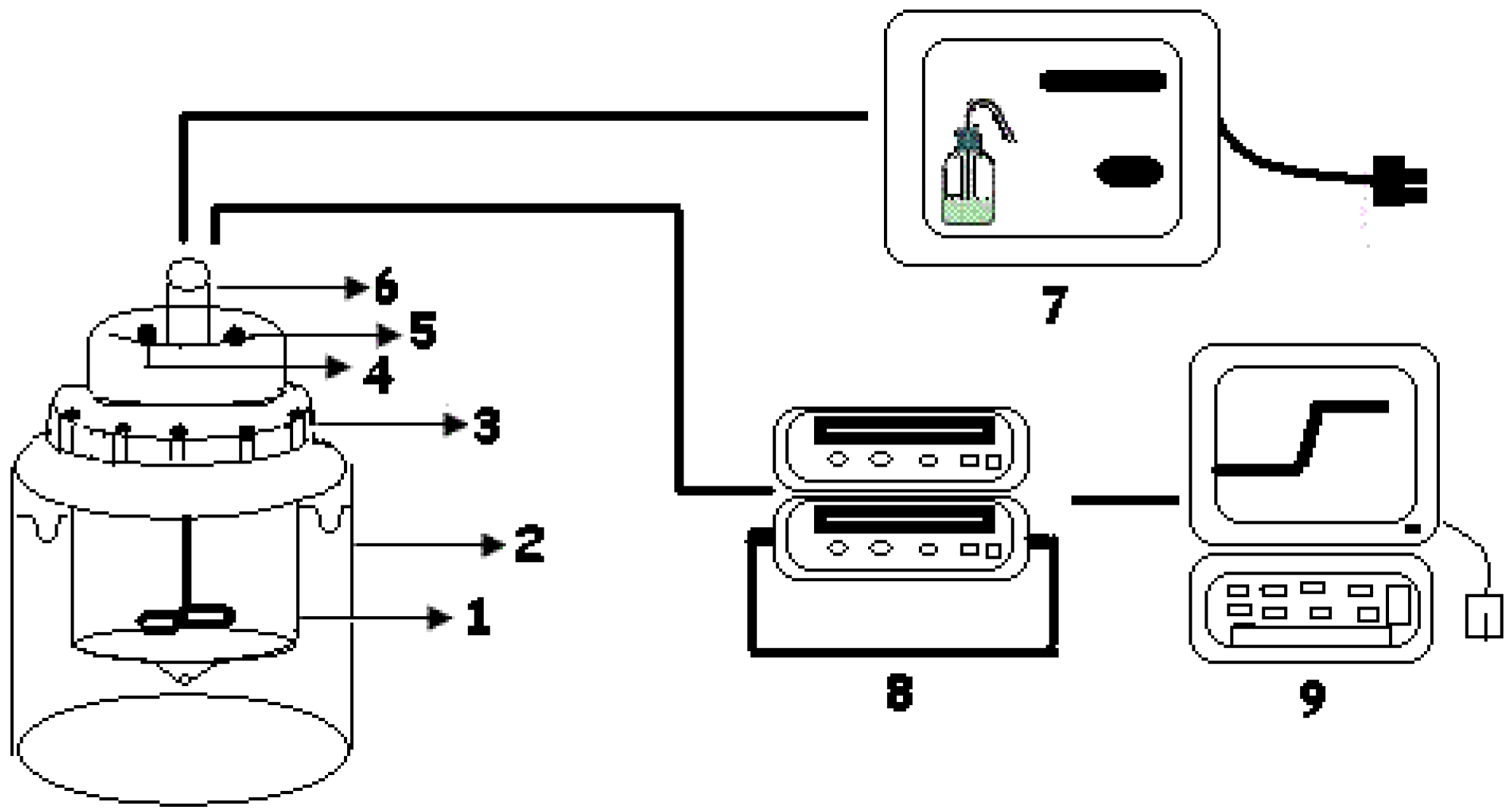
2. Experimental Section
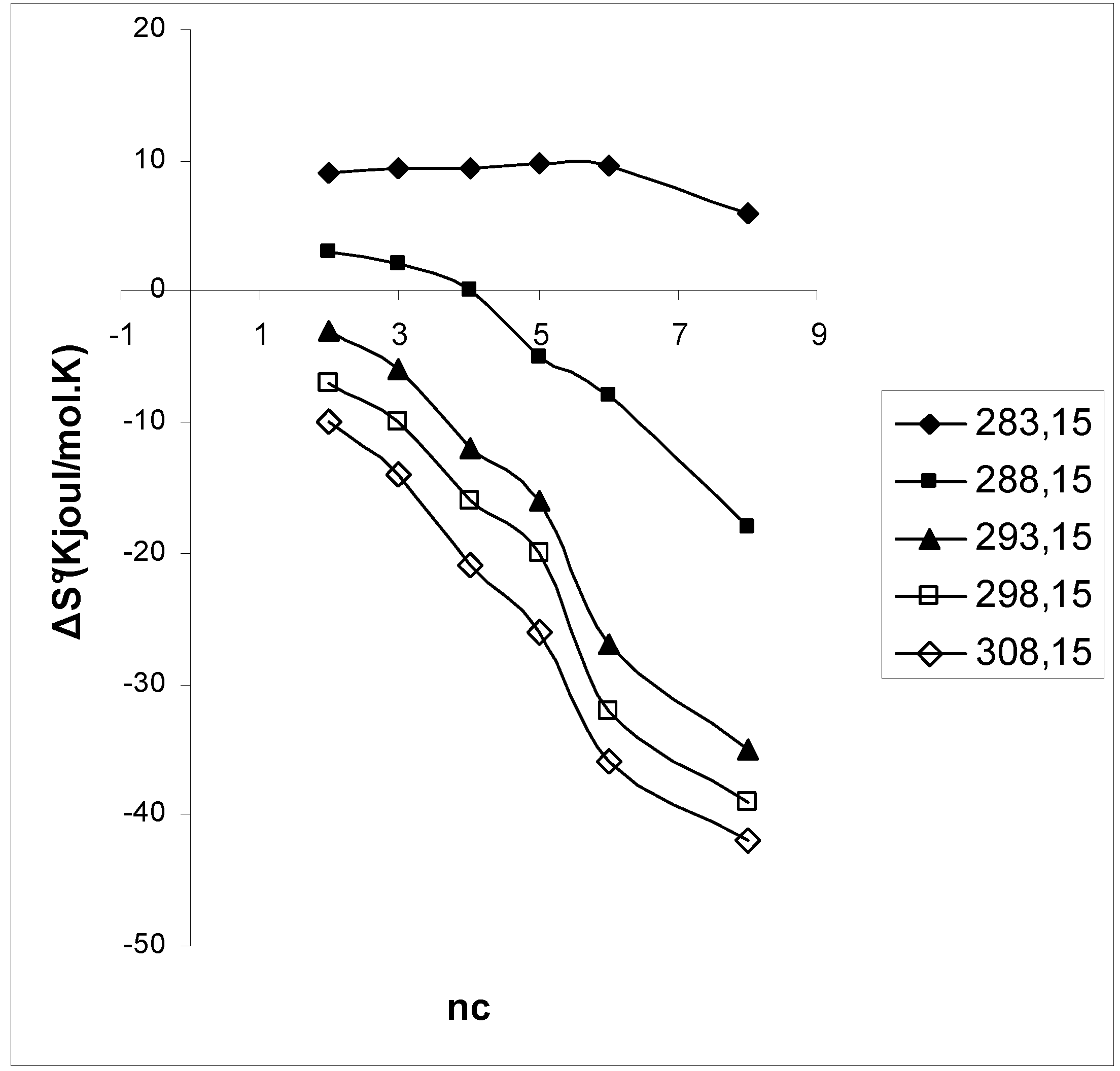
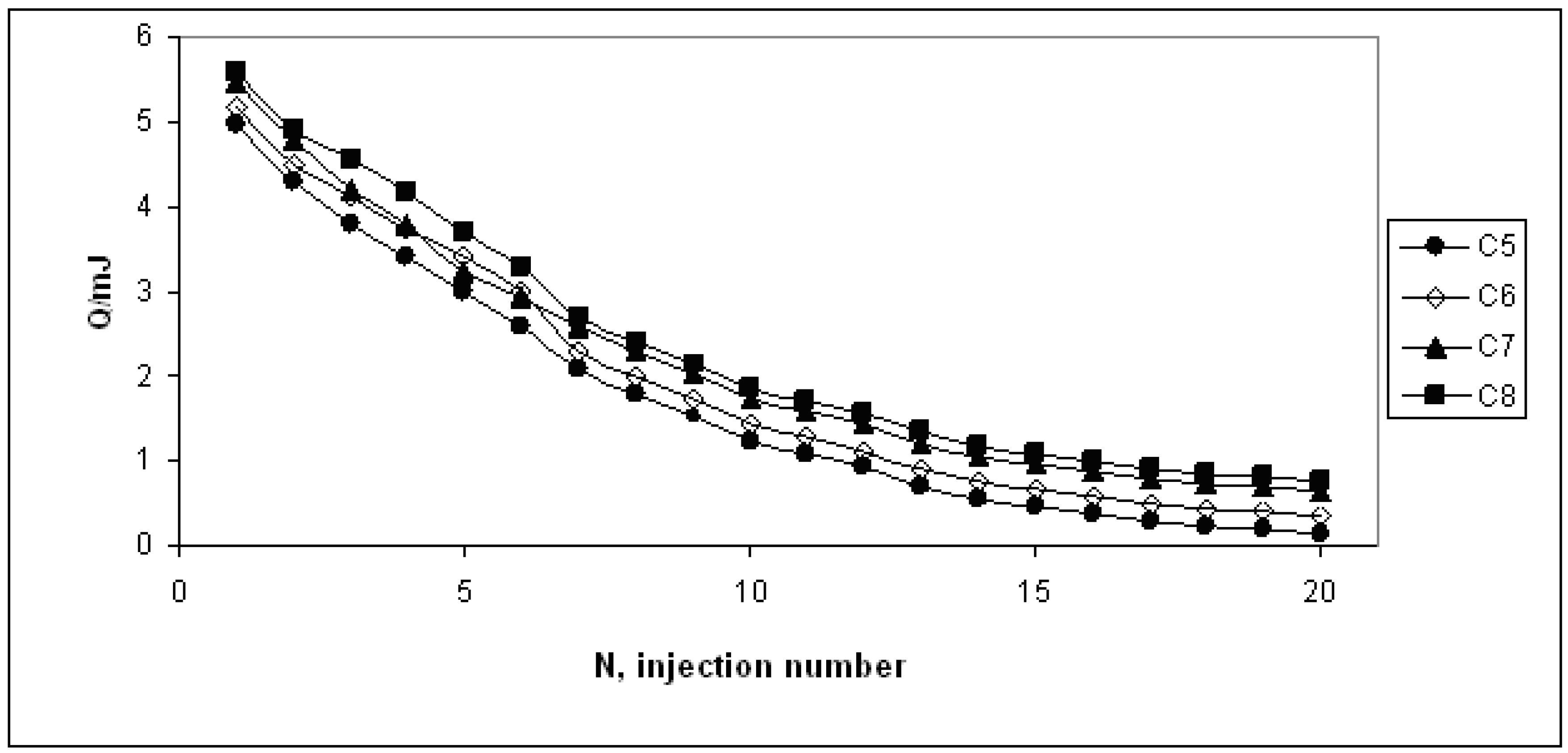
3. Results and Discussion

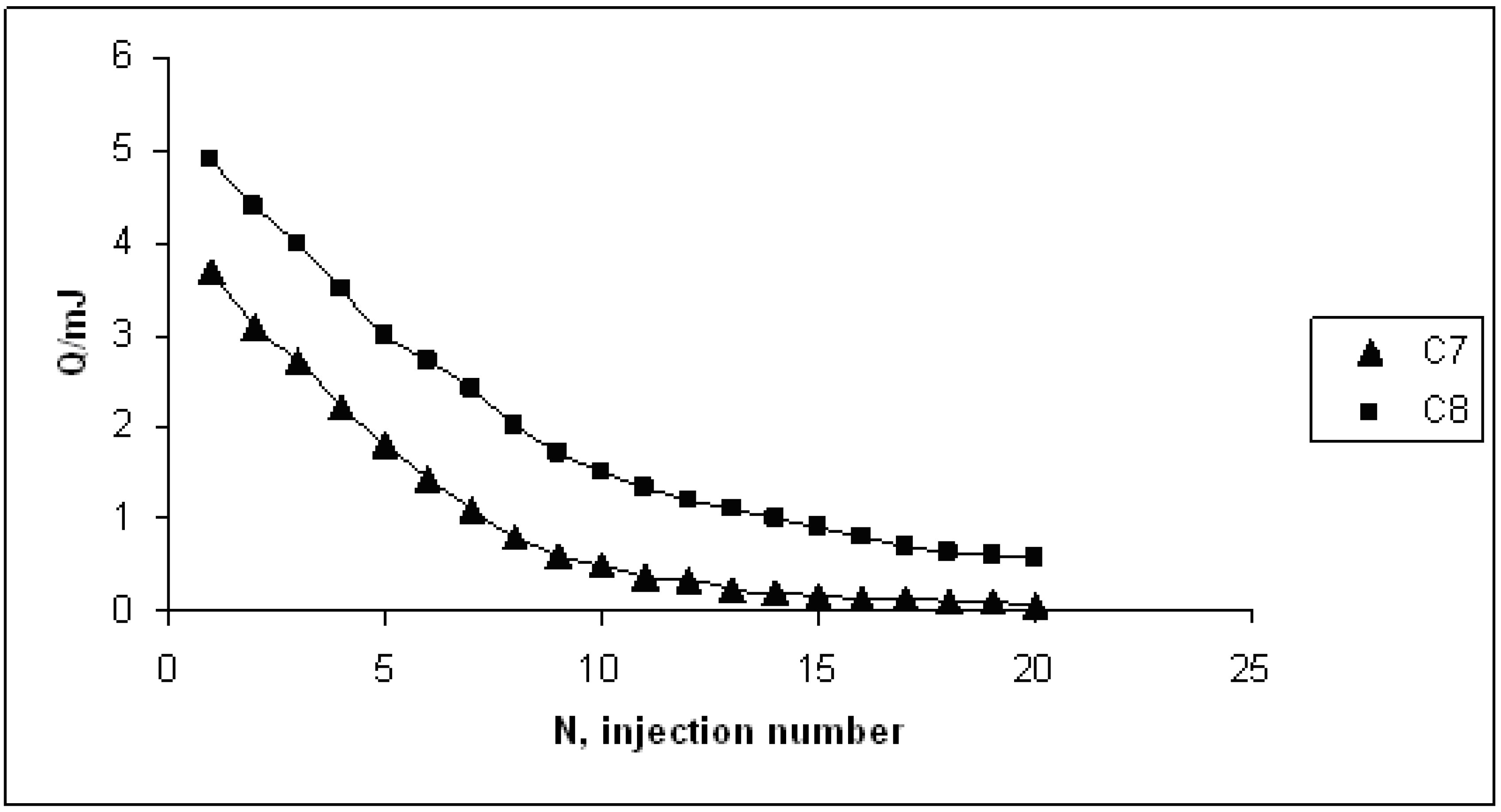
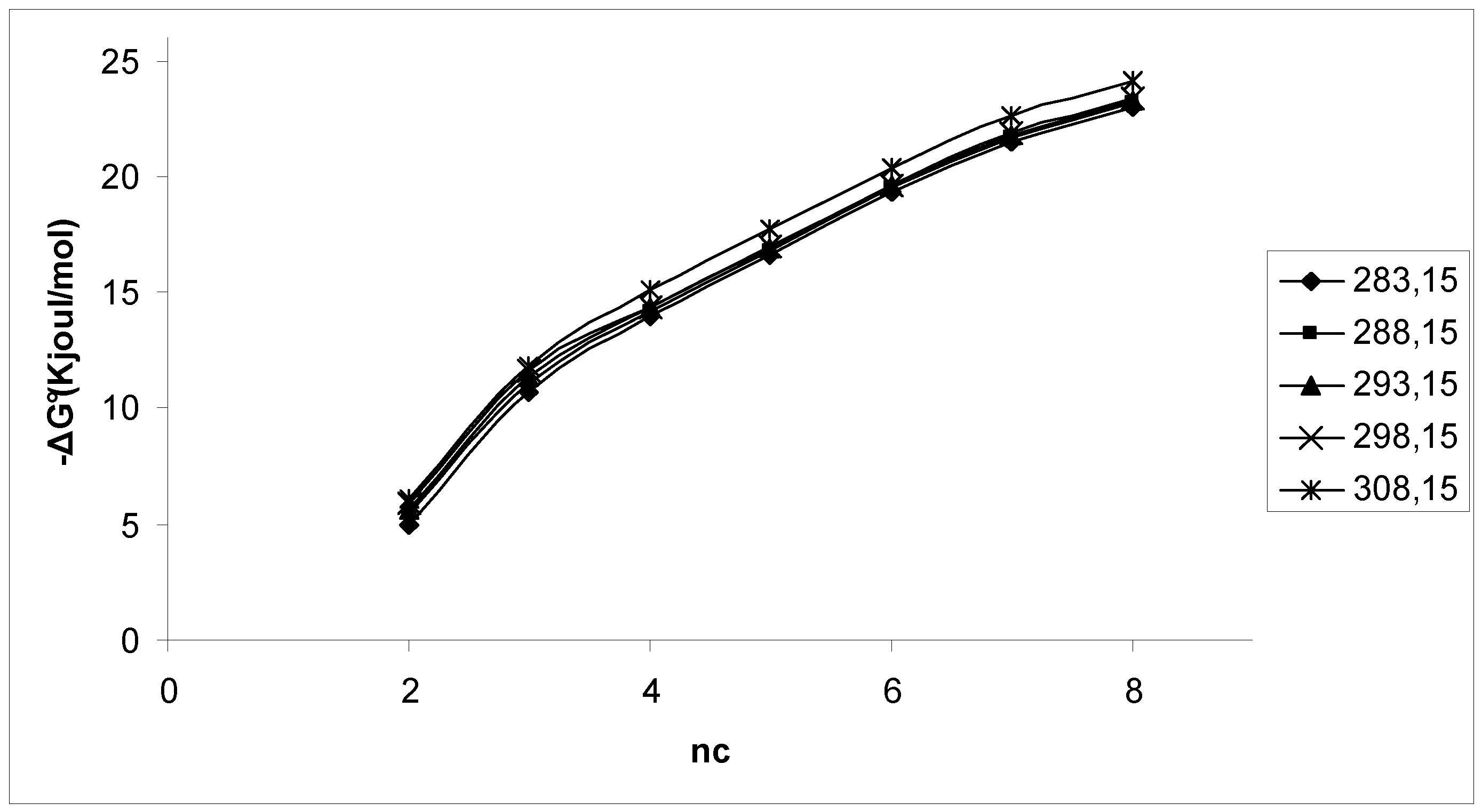
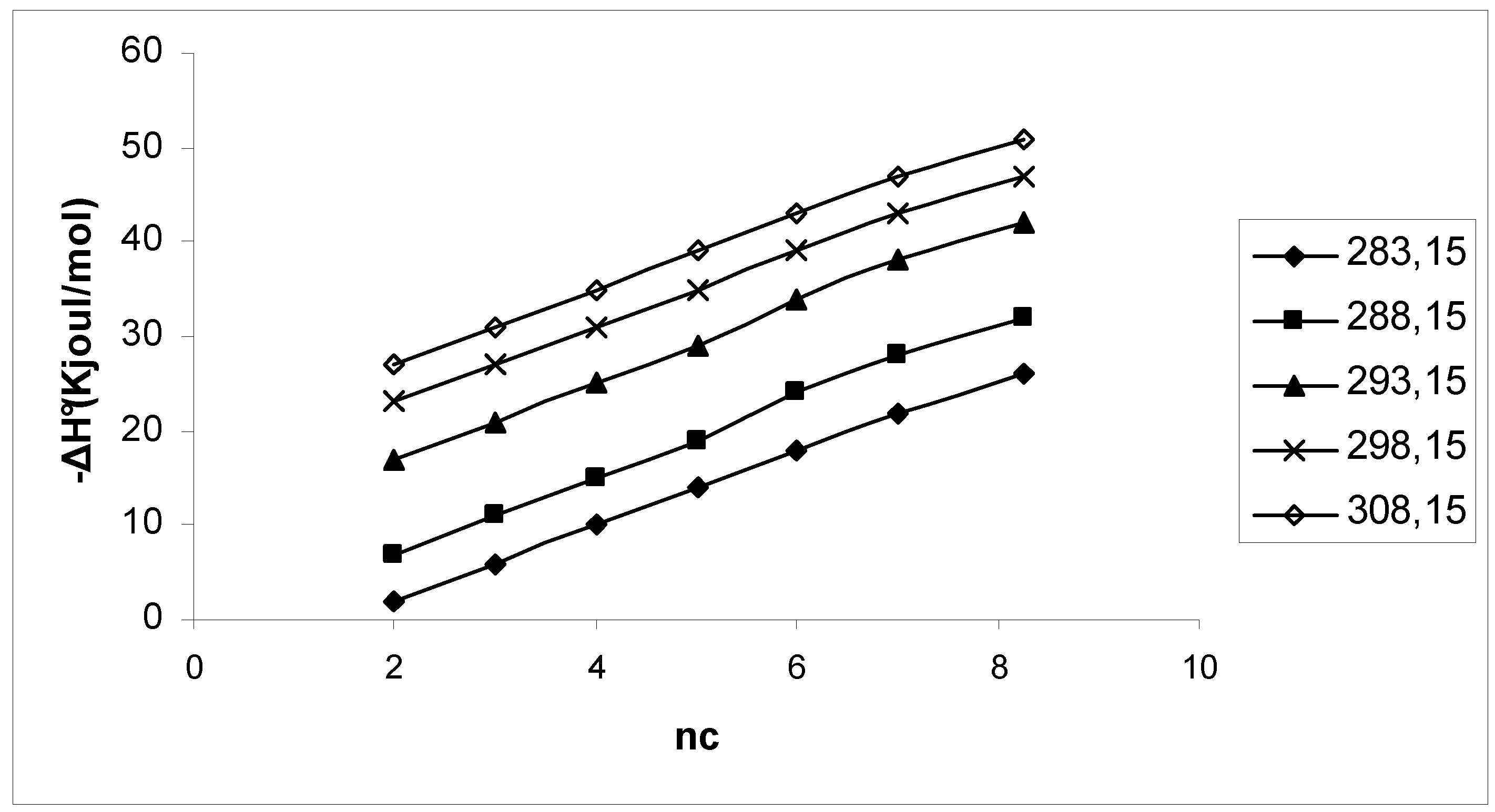
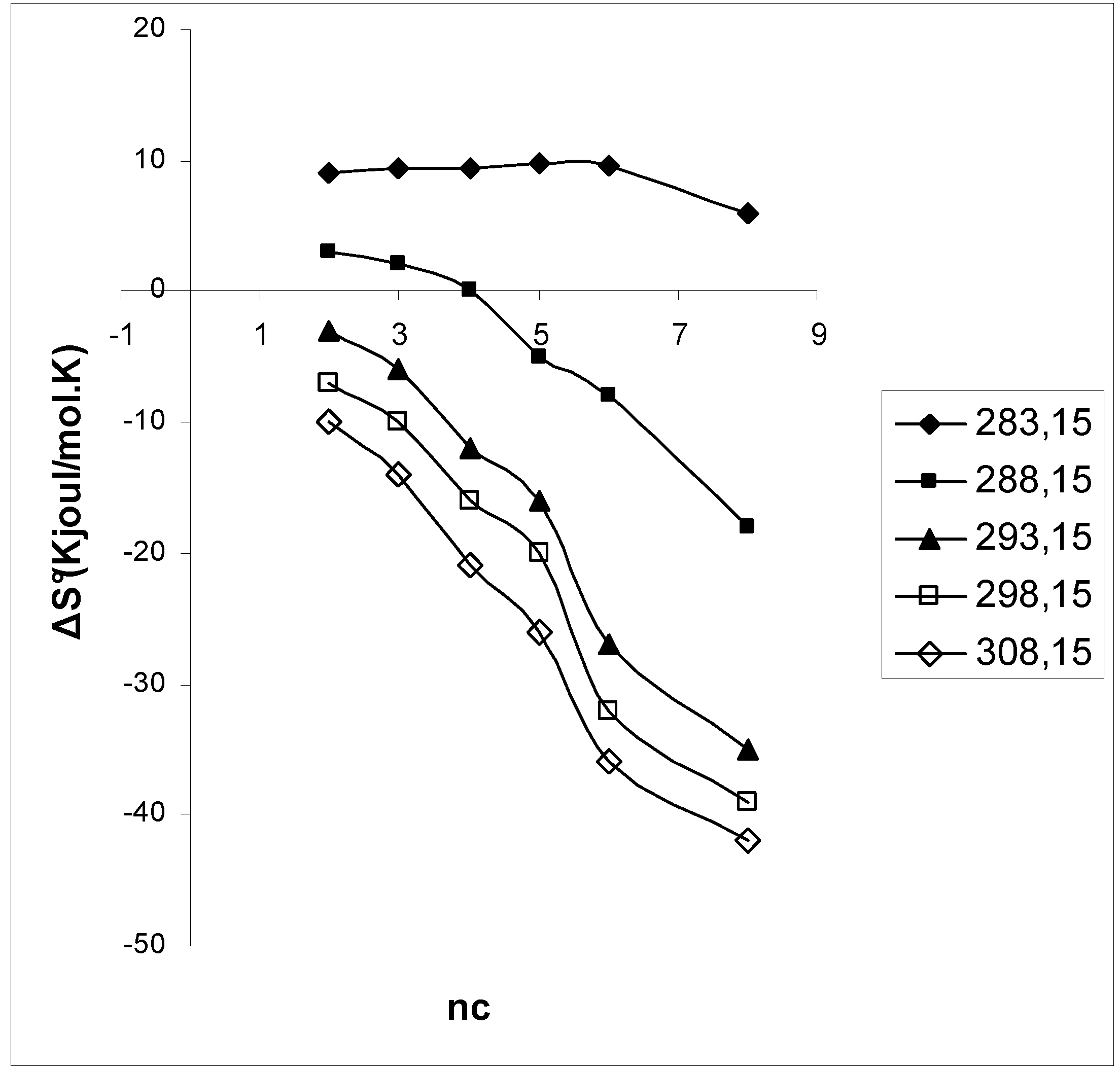

4. Conclusions
Acknowledgements
References
- Cramer, F.; Saenger, W.; Spatz, H.C. Inclusion Compounds. XIX.1a The Formation of Inclusion Compounds of α-Cyclodextrin in Aqueous Solutions. Thermodynamics and Kinetics. J. Am. Chem. Soc. 1967, 89, 14–20. [Google Scholar] [CrossRef]
- van Etten, R.L.; Sebastion, J.F.; Clowes, G.A.; Bender, M.L. Acceleration of phenyl ester cleavage by cycloamyloses. A model for enzymic specificity. J. Am. Chem. Soc. 1967, 89, 3242–3253. [Google Scholar]
- Straub, T.S.; Bender, M.L. Cycloamyloses as enzyme models. Decarboxylation of benzoylacetic acids. J. Am. Chem. Soc. 1972, 94, 8881–8892. [Google Scholar] [CrossRef]
- Griffths, D.W.; Bender, M.L. Substrate binding to cyclodextrins in aqueous solution. Adv. Catalysis 1973, 23, 209–214. [Google Scholar]
- Bergeron, R.J.; Channing, M.A.; Gibeily, G.J.; Pillor, D.M. Disposition requirements for binding in aqueous solution of polar substrates in the cyclohexaamylose cavity. J. Am. Chem. Soc. 1977, 99, 5146–5151. [Google Scholar] [CrossRef]
- Komiyama, M.; Bender, M.L. Importance of apolar binding in complex formation of cyclodextrins with adamantanecarboxylate. J. Am. Chem. Soc. 1978, 100, 2259–2267. [Google Scholar] [CrossRef]
- Tabushi, I.; Kiyosuka, Y.; Sugimoto, T.; Yamamura, K. Approach to the aspects of driving force of inclusion by .alpha.-cyclodextrin. J. Am. Chem. Soc. 1978, 100, 916–919. [Google Scholar] [CrossRef]
- Mcmillan, R.K.; Saenger, W.; Fayos, J.; Mootz, D. Topography of cyclodextrin inclusion complexes: Part I. Classification of crystallographic data of α-cyclodextrin inclusion complexes. Carbohydr. Res. 1973, 31, 37–46. [Google Scholar] [CrossRef]
- Hingerty, B.; Saenger., W. Disorder in a hydrophobic cage illustrated by X-ray structure of α-cyclodextrin ·methanol pentahydrate adduct. Nature 1975, 255, 395–397. [Google Scholar] [CrossRef]
- Manor, P.C.; Saenger, W. Topography of cyclodextrin inclusion complexes. III. Crystal and molecular structure of cyclohexaamylose hexahydrate, the water dimer inclusion complex. J. Am. Chem. Soc. 1974, 96, 3630–3639. [Google Scholar] [CrossRef]
- Saenger, W.; Steiner, T. Cyclodextrin Inclusion Complexes: Host-guest Interactions and Hydrogen-Bonding Networks. Acta Crystallogr. 1998, A54, 798–805. [Google Scholar] [CrossRef]
- Hingerty, B.; Saenger, W. Dimeric -cyclodextrin complexes may mimic membrane diffusion transport. Nature (London) 1975, 255, 396–407. [Google Scholar] [CrossRef]
- Hingerty, B.; Saenger, W. Topography of cyclodextrin inclusion complexes. 8. Crystal and molecular structure of the .alpha.-cyclodextrin-methanol-pentahydrate complex. Disorder in a hydrophobic cage. J. Am. Chem. Soc. 1976, 98, 3357–3365. [Google Scholar] [CrossRef]
- Lindner, K.; Saenger, W. Crystal structure of the γ-cyclodextrin n-propanol inclusion complex; Correlation of α-, β-, γ-cyclodextrin geometries Biochem. Biophys. Res. Commun. 1980, 92, 933–942. [Google Scholar] [CrossRef]
- Stezowsky, J.J.; Jogun, K.H.; Ekkle, E.; Bartels, I.S. Dimeric -cyclodextrin complexes may mimic membrane diffusion transport. Nature (London) 1978, 274, 617–625. [Google Scholar] [CrossRef]
- MacLennan, J.M.; Stezowski, J.J. The crystal structure of uncomplexed-hydrated cyclooctaamylose. Biochem. Biophys. Res. Commun. 1980, 92, 926–937. [Google Scholar] [CrossRef]
- Barone, G.; Castronuovo, G.; Elia, V.; Muscetta, M. Interaction of α-cyclodextrin with non electrolytes in water at 298.15 K. Thermochim. Acta 1985, 85, 447–450. [Google Scholar] [CrossRef]
- Barone, G.; Castronuovo, G.; Elia, V.; Muscetta, M. Interaction of urea and urea derivatives with cyclohexamylose in aqueous solutions at 25°C. J. Solut. Chem. 1986, 15, 129–140. [Google Scholar] [CrossRef]
- Lewis, E.A.; Hansen, L.D. Thermodynamics of binding of guest molecules to α- and β-cyclodextrins. J. Chem. Soc., Perkin Trans. 2 1973, 2081–2085. [Google Scholar]
- Maeda, M.; Tokagi, S. Calorimetric studies on molecular inclusion. Gibbs energies and entropies of inclusion of 1-propanol and 1-pentanol into cyclohexaamylose and cycloheptaamylose in aqueous solutions at 298.15K. Netsusokutei 1983, 10, 103–118. [Google Scholar]
- McMillan Jr., W.G.; Mayer, J.E. The Statistical Thermodynamics of Multicomponent Systems. J. Chem. Phys. 1945, 13, 276–287. [Google Scholar] [CrossRef]
- Kozak, J.J.; Knight, W.S.; Kauanann, W. Solute-Solute Interactions in Aqueous Solutions. J. Chem. Phys. 1945, 48, 675–690. [Google Scholar] [CrossRef]
- Friedman, H.L.; Krishnan, C.V. Studies of hydrophobic bonding in aqueous alcohols: Enthalpy measurements and model calculations. J. Solut. Chem. 1973, 2, 119–140. [Google Scholar] [CrossRef]
- Lilley, T.H.; Scott, R.P. Aqueous solutions containing amino-acids and peptides. Part 2.—Gibbs function and enthalpy behaviour of the systems urea + glycine, urea +-alanine, urea +-aminobutyric acid and urea + glycylglycine at 298.15 K. J. Chem. Soc., Faraday Trans. 1 1976, 72, 184–193. [Google Scholar] [CrossRef]
- Franks, F.; Pedley, M.D.; Reid, D.S. Solute interactions in dilute aqueous solutions. Part 1.—Microcalorimetric study of the hydrophobic interaction. J. Chem. Soc., Faraday Trans. 1 1976, 72, 359–362. [Google Scholar] [CrossRef]
- Desnoyers, J.E.; Perron, G.; Avedikian, L.; Morel, J-P. Enthalpies of the urea-tert-butanol-water system at 25°C. J. Solut. Chem. 1976, 5, 631–644. [Google Scholar] [CrossRef]
- Savage, J.J.; Wood, R.H. Enthalpy of dilution of aqueous mixtures of amides, sugars, urea, ethylene glycol, and pentaerythritol at 25°C: Enthalpy of interaction of the hydrocarbon, amide, and hydroxyl functional groups in dilute aqueous solutions. J. Solut. Chem. 1976, 5, 733–750. [Google Scholar] [CrossRef]
- Barone, G.; Cacace, P.; Castronuovo, G.; Elia, V. Interactions in aqueous solutions of urea-like compounds. Heats of mixing of urea, monomethylurea and thiourea at 298.15 K. J. Chem. Soc., Faraday Trans. I 1988, 77, 1569–1574. [Google Scholar] [CrossRef]
- King, E.J. Acid Base Equilibria, 1rd Ed. ed; Pergamon Press: Oxford, England, 1965; pp. 184–217. [Google Scholar]
- Sigurskjold, B.W.; Berland, C.R.; Svensson, B. Thermodynamics of Inhibitor Binding to the Catalytic Site of Glucoamylase from Aspergillus niger Determined by Displacement Titration Calorimetry. Biochemistry 1994, 33, 101–110. [Google Scholar] [CrossRef]
- Danil de Namor, A.F.; Traboulssi, R.; Lewis, D.F.V. Host properties of cyclodextrins towards anion constituents of antigenic determinants. A thermodynamic study in water and in N,N-dimethylformamide. J. Am. Chem. Soc. 1990, 112, 8442–8447. [Google Scholar] [CrossRef]
- Hallen, D.; Wadso, I. Solution microcalorimetry. Pure Appl. Chem. 1989, 61, 123–132. [Google Scholar] [CrossRef]
- Hallen, D.; Nilsson, S.O.; Wadso, I. A new flow-microcalorimetric vessel for dissolution of small quantities of easily or slightly soluble liquids. J. Chem. Thermodyn. 1989, 21, 529–534. [Google Scholar] [CrossRef]
- Dec, S.F.; Gill, S.J. Steady-state gas dissolution flow microcalorimeter for determination of heats of solution of slightly soluble gases in water. Rev. Sci. Instrum. 1984, 55, 765–772. [Google Scholar] [CrossRef]
- Hallen, D.; Wadso, I. A new microcalorimetric vessel for dissolution of slightly soluble gases enthalpies of solution in water of carbon tetrafluoride and sulphur hexafluoride at 288.15, 298.15, and 308.15 K. J. Chem. Thermodyn. 1989, 21, 519–528. [Google Scholar] [CrossRef]
- Nilsson, S.O.; Wadso, I. A flow-microcalorimetric vessel for solution of slightly soluble solids. J. Chem. Thermodyn. 1986, 18, 1125–1133. [Google Scholar] [CrossRef]
- Murphy, K.P.; Gill, S.J. Calorimetric measurement of the enthalpy of dissolution of diketopiperazine in water as a function of temperature. Thermochim. Acta 1989, 139, 279–285. [Google Scholar] [CrossRef]
- Wadso, I. Isothermal microcalorimetry for the characterization of interactions between drugs and biological materials. Thermochim. Acta 1995, 267, 45–49. [Google Scholar] [CrossRef]
- Suurkuusk, J.; Wadso, I. Multichannel Microcalorimetry. Chem. Scripta 1982, 20, 155–161. [Google Scholar]
- Hallen, D.; Schbn, A.; Shehatta, I.; Wadso, I. Microcalorimetric titration of -cyclodextrin with some straight-chain alkan-1-ols at 288.15, 298.15 and 308.15 K. J. Chem. Soc. Faraday Trans. 1992, 88, 2859–2866. [Google Scholar] [CrossRef]
- Bastos, M.; Briggner, L.-E.; Shehatta, I.; Wadso, I. The binding of alkane-α,ω-diols to α-cyclodextrin. A microcalorimetric study. J. Chem. Thermodyn. 1990, 22, 1181–1190. [Google Scholar] [CrossRef]
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moreno-Piraján, J.C.; Giraldo, L. Isoperibolic Titration Calorimetry as a Tool for the Prediction of Thermodynamic Properties of Cyclodextrins. Energies 2008, 1, 93-104. https://doi.org/10.3390/en1030093
Moreno-Piraján JC, Giraldo L. Isoperibolic Titration Calorimetry as a Tool for the Prediction of Thermodynamic Properties of Cyclodextrins. Energies. 2008; 1(3):93-104. https://doi.org/10.3390/en1030093
Chicago/Turabian StyleMoreno-Piraján, Juan Carlos, and Liliana Giraldo. 2008. "Isoperibolic Titration Calorimetry as a Tool for the Prediction of Thermodynamic Properties of Cyclodextrins" Energies 1, no. 3: 93-104. https://doi.org/10.3390/en1030093