Multi-Walled Carbon Nanotubes Supported Pd(II) Complexes: A Supramolecular Approach towards Single-Ion Oxygen Reduction Reaction Catalysts

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
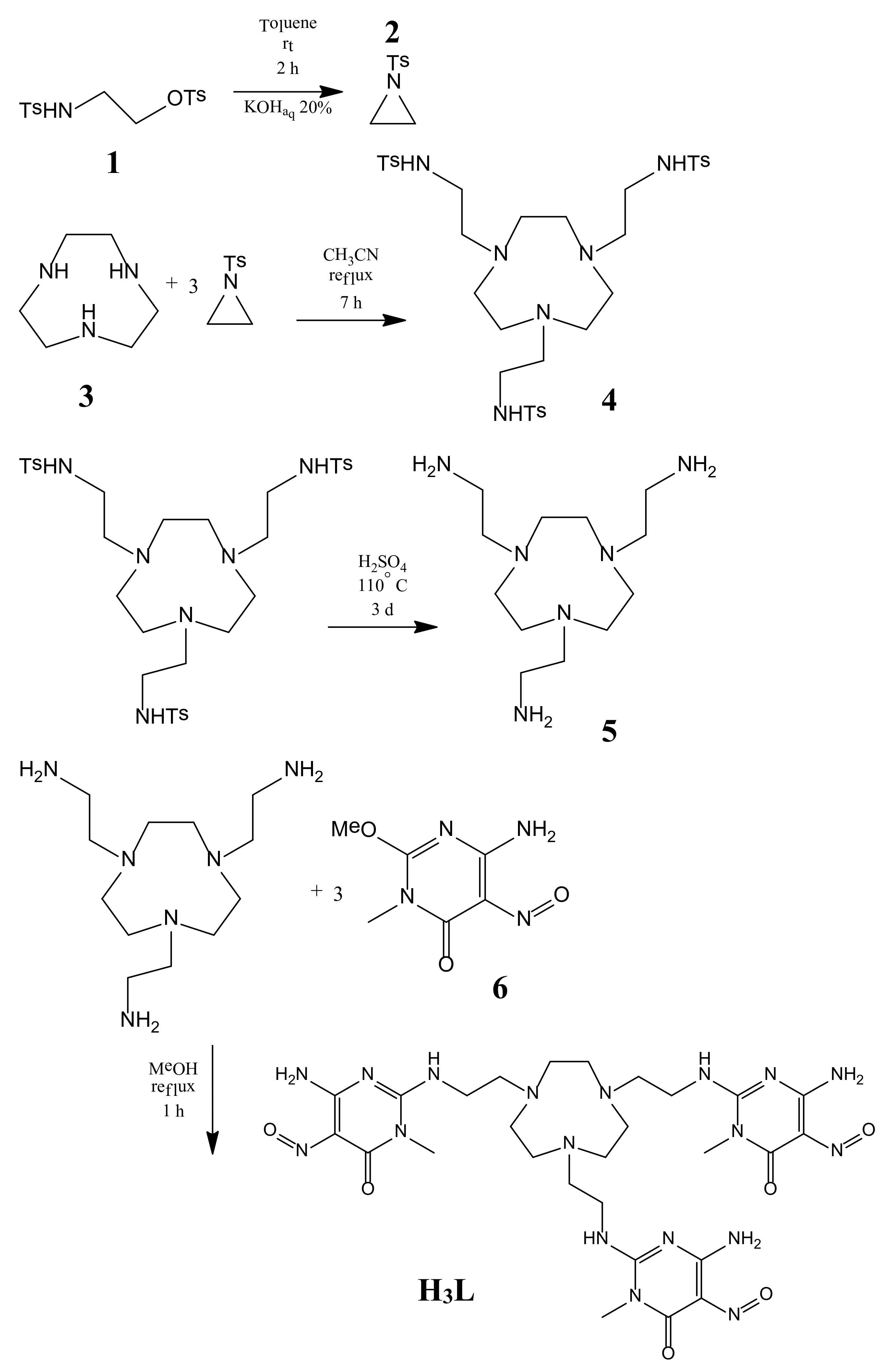
2.1. Synthesis of H3L
2.2. Potentiometric Measurements
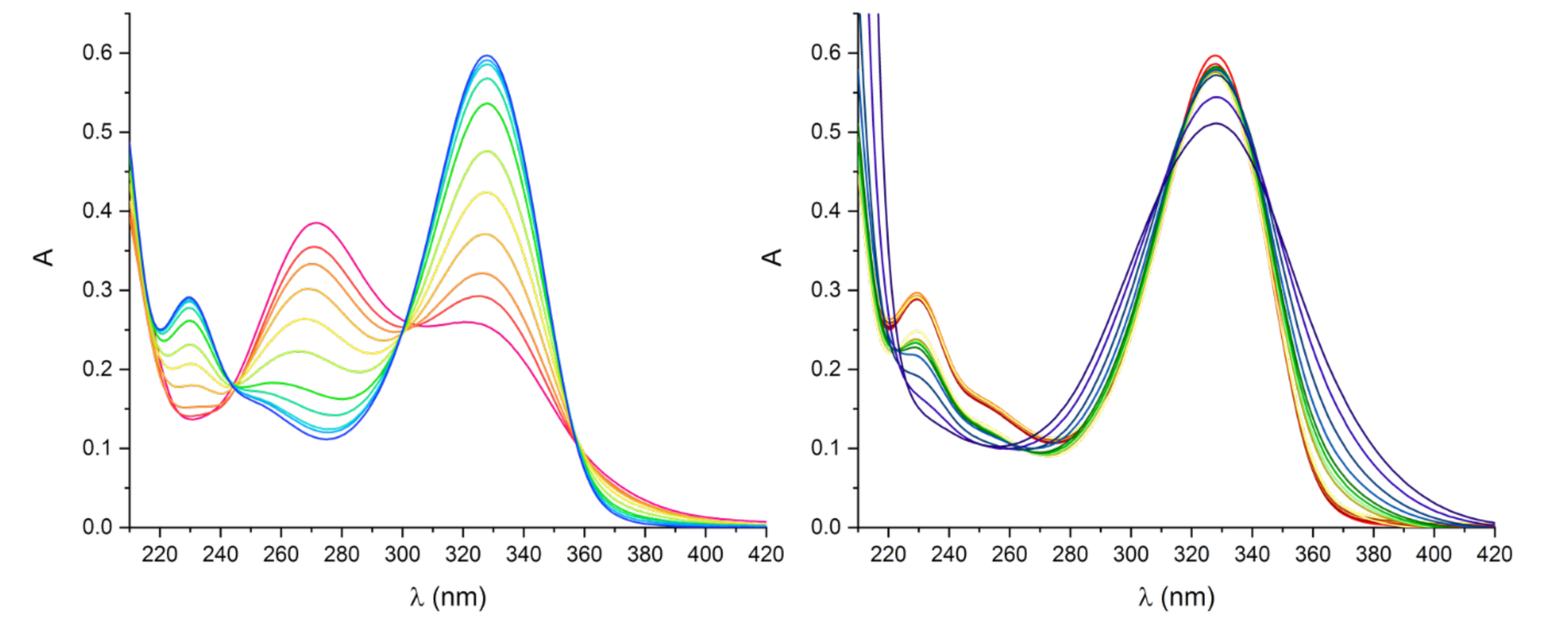
2.3. Spectroscopic Measurements
2.4. Preparation of MWCNTs/H3L and MWCNTS/H3L-Pd(II) Catalysts
2.5. Preparation of the Modified Electrodes
2.6. Electrochemical Characterization
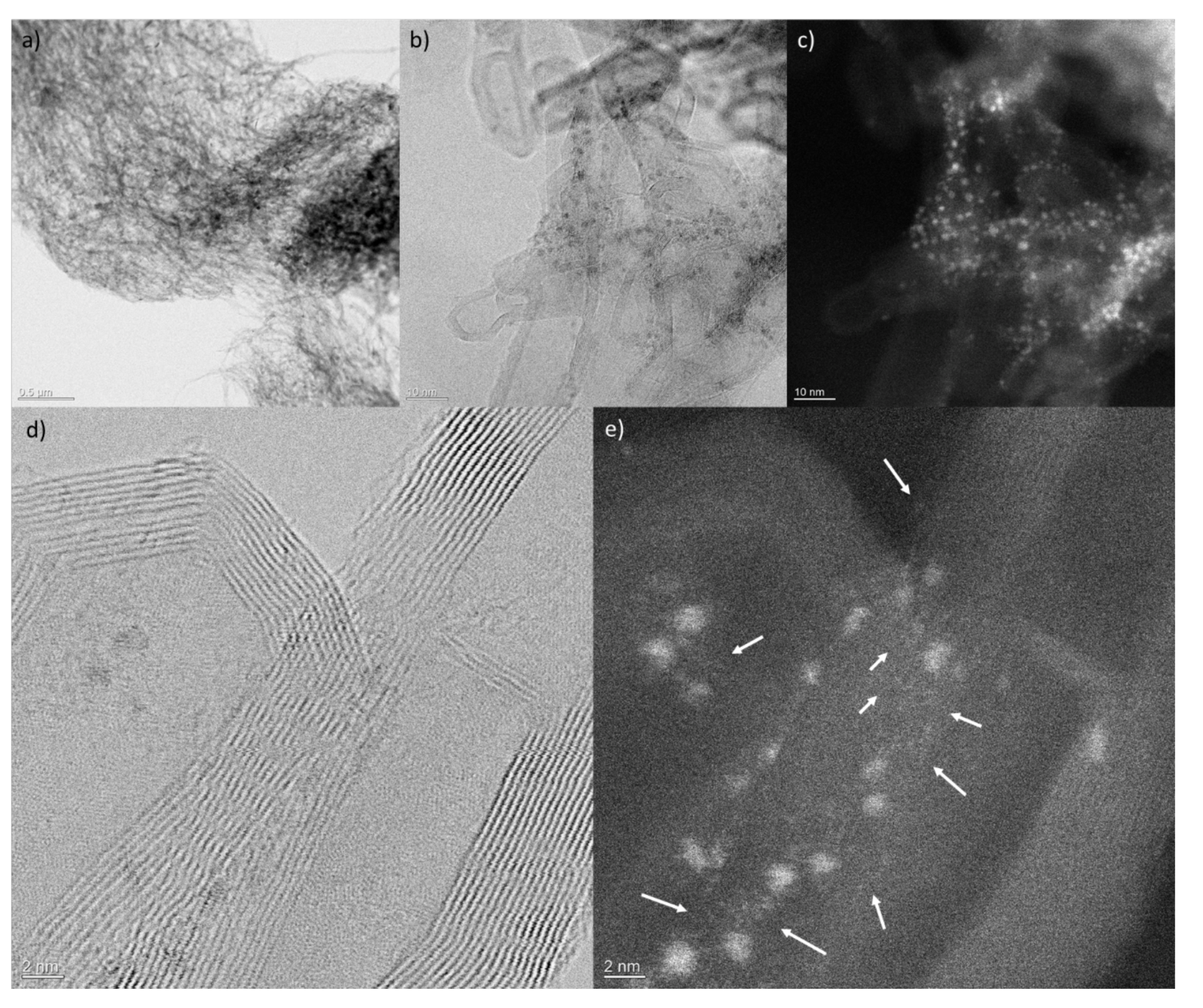
2.7. Scanning Transmission Electron Microscopy (STEM) Characterization
3. Results and Discussion
3.1. Ligand Protonation Equilibria
3.2. Pd(II) Complexes Formation
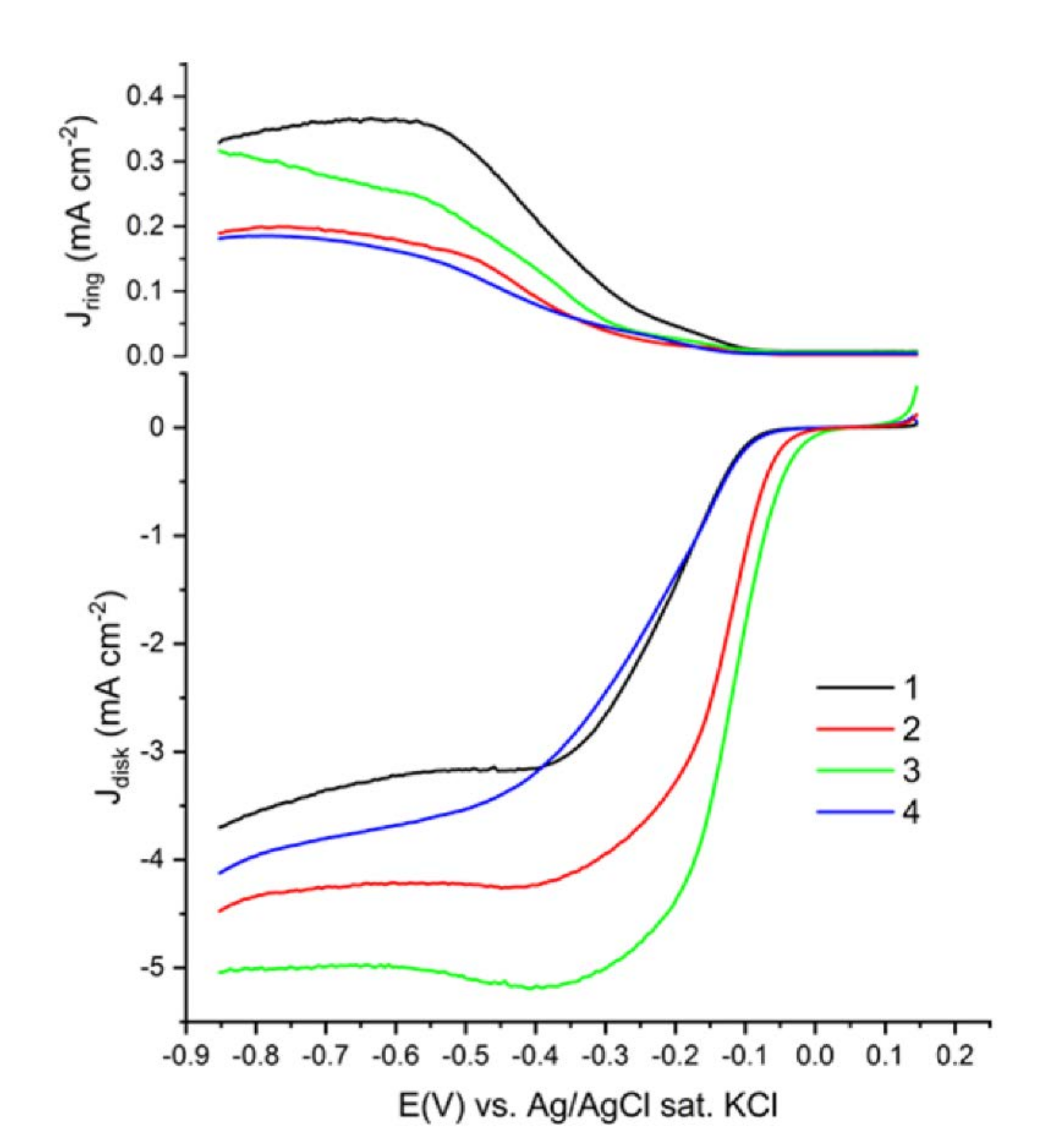
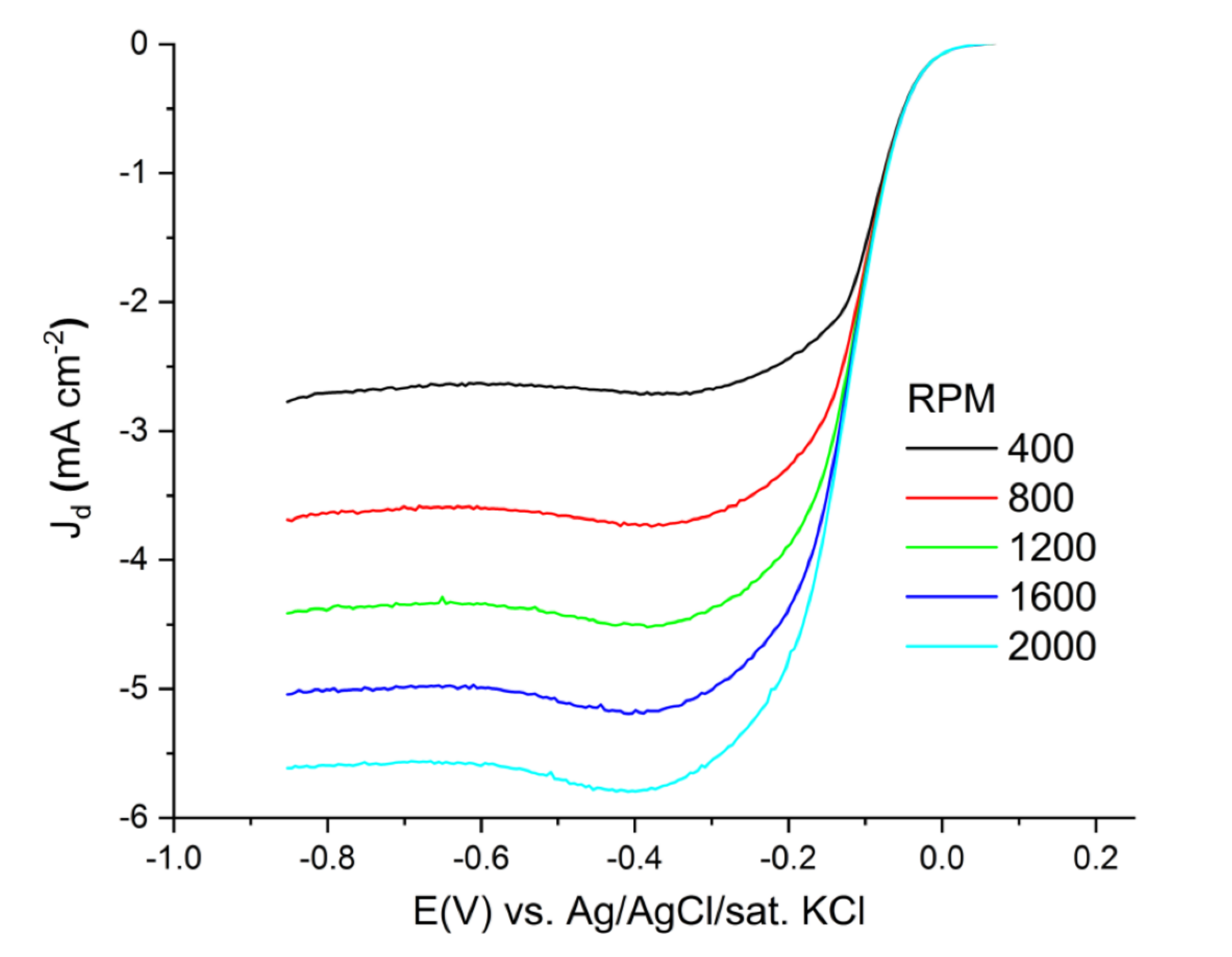
3.3. Electrochemical Evaluation of Catalytic Performance
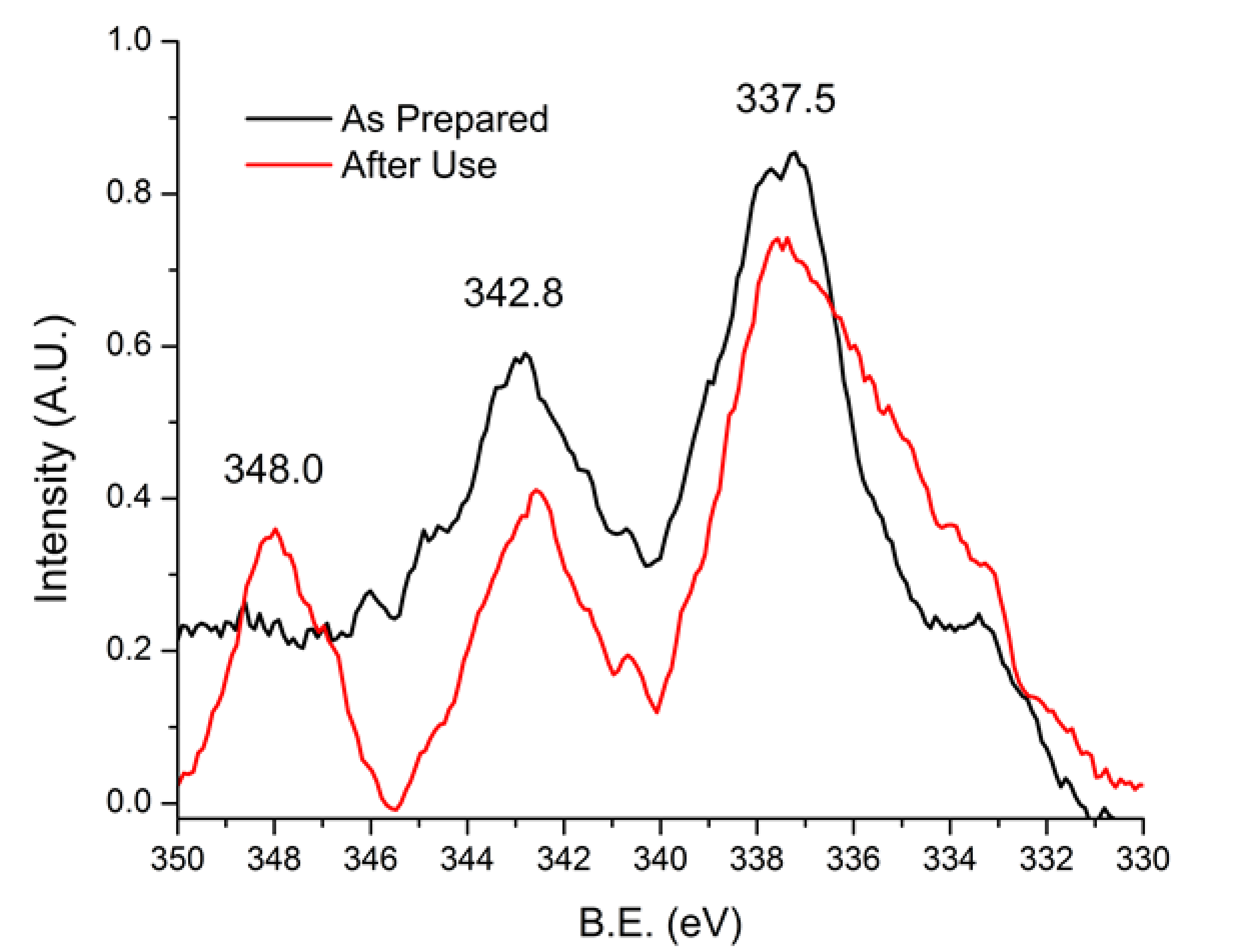
3.4. Before and After-Use Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations: List of Used Abbreviations (Alphabetical Order)
| CV | Cyclic Voltammogram |
| E1/2 | Half-Wave Potential |
| Eon | Onset Potential |
| GC | Glassy Carbon |
| HAADF | High Angle Annular Dark Field |
| KL | Koutechy-Levich |
| LSV | Linear Sweep Voltammogram |
| MWCNTs | Multi-Walled Carbon Nanotubes |
| N | Collection Efficiency Number |
| n | Number of Exchanged Electrons |
| ORR | Oxygen Reduction Reaction |
| PGM | Platinum Group Metals |
| RDE | Rotating Disk Electrode |
| RRDE | Rotating Ring Disk Electrode |
| SAC | Single Atom Catalyst |
| STEM | Scanning Transmission Electron Microscopy |
| XPS | X-ray Photoelectron Spectroscopy |
References
- Shindell, D.; Smith, C.J. Climate and air-quality benefits of a realistic phase-out of fossil fuels. Nature 2019, 573, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Air Quality in Europe 2019—European Environment Agency. Available online: https://www.eea.europa.eu/publications/air-quality-in-europe-2019 (accessed on 6 July 2020).
- Mahato, S.; Pal, S.; Ghosh, K.G. Effect of lockdown amid COVID-19 pandemic on air quality of the megacity Delhi, India. Sci. Total Environ. 2020, 730, 139086. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.; Basile, A.; Veziroğlu, T.N. (Eds.) Compendium of Hydrogen Energy; Woodhead Publishing Series in Energy; Woodhead Publishing: Oxford, UK, 2016; ISBN 978-1-78242-364-5. [Google Scholar]
- Hoogers, G. Fuel Cell Technology Handbook; CRC Press: Boca Raton, FL, USA, 2002; ISBN 978-1-4200-4155-2. [Google Scholar]
- Brockway, P.E.; Owen, A.; Brand-Correa, L.I.; Hardt, L. Estimation of global final-stage energy-return-on-investment for fossil fuels with comparison to renewable energy sources. Nat. Energy 2019, 4, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov, J.K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef]
- Stacy, J.; Regmi, Y.N.; Leonard, B.; Fan, M. The recent progress and future of oxygen reduction reaction catalysis: A review. Renew. Sustain. Energy Rev. 2017, 69, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wang, Y.; Li, W.; Hou, Y. Noble metal-free catalysts for oxygen reduction reaction. Sci. China Chem. 2017, 60, 1494–1507. [Google Scholar] [CrossRef]
- Miller, H.A.; Bevilacqua, M.; Filippi, J.; Lavacchi, A.; Marchionni, A.; Marelli, M.; Moneti, S.; Oberhauser, W.; Vesselli, E.; Innocenti, M.; et al. Nanostructured Fe–Ag electrocatalysts for the oxygen reduction reaction in alkaline media. J. Mater. Chem. A 2013, 1, 13337–13347. [Google Scholar] [CrossRef]
- Fu, Y.; Wei, Z.D.; Chen, S.G.; Li, L.; Feng, Y.C.; Wang, Y.Q.; Ma, X.L.; Liao, M.J.; Shen, P.K.; Jiang, S.P. Synthesis of Pd/TiO2 nanotubes/Ti for oxygen reduction reaction in acidic solution. J. Power Sources 2009, 189, 982–987. [Google Scholar] [CrossRef]
- Wu, R.; Tsiakaras, P.; Shen, P.K. Facile synthesis of bimetallic Pt-Pd symmetry-broken concave nanocubes and their enhanced activity toward oxygen reduction reaction. Appl. Catal. B Environ. 2019, 251, 49–56. [Google Scholar] [CrossRef]
- Tzorbatzoglou, F.; Brouzgou, A.; Tsiakaras, P. Electrocatalytic activity of Vulcan-XC-72 supported Pd, Rh and PdxRhy toward HOR and ORR. Appl. Catal. B Environ. 2015, 174–175, 203–211. [Google Scholar] [CrossRef]
- Tzorbatzoglou, F.; Brouzgou, A.; Jing, S.; Wang, Y.; Song, S.; Tsiakaras, P. Oxygen reduction and hydrogen oxidation reaction on novel carbon supported PdxIry electrocatalysts. Int. J. Hydrog. Energy 2018, 43, 11766–11777. [Google Scholar] [CrossRef]
- Bhatt, M.D.; Lee, J.Y. Advancement of Platinum (Pt)-Free (Non-Pt Precious Metals) and/or Metal-Free (Non-Precious-Metals) Electrocatalysts in Energy Applications: A Review and Perspectives. Energy Fuels 2020, 34, 6634–6695. [Google Scholar] [CrossRef]
- Iglesias, D.; Giuliani, A.; Melchionna, M.; Marchesan, S.; Criado, A.; Nasi, L.; Bevilacqua, M.; Tavagnacco, C.; Vizza, F.; Prato, M.; et al. N-Doped Graphitized Carbon Nanohorns as a Forefront Electrocatalyst in Highly Selective O2 Reduction to H2O2. Chem 2018, 4, 106–123. [Google Scholar] [CrossRef]
- Tuci, G.; Zafferoni, C.; Rossin, A.; Milella, A.; Luconi, L.; Innocenti, M.; Truong Phuoc, L.; Duong-Viet, C.; Pham-Huu, C.; Giambastiani, G. Chemically Functionalized Carbon Nanotubes with Pyridine Groups as Easily Tunable N-Decorated Nanomaterials for the Oxygen Reduction Reaction in Alkaline Medium. Chem. Mater. 2014, 26, 3460–3470. [Google Scholar] [CrossRef]
- Yan, Z.; Gao, L.; Dai, C.; Zhang, M.; Lv, X.; Shen, P.K. Metal-free mesoporous carbon with higher contents of active N and S codoping by template method for superior ORR efficiency to Pt/C. Int. J. Hydrogen Energy 2018, 43, 3705–3715. [Google Scholar] [CrossRef]
- Passaponti, M.; Rosi, L.; Savastano, M.; Giurlani, W.; Miller, H.A.; Lavacchi, A.; Filippi, J.; Zangari, G.; Vizza, F.; Innocenti, M. Recycling of waste automobile tires: Transforming char in oxygen reduction reaction catalysts for alkaline fuel cells. J. Power Sources 2019, 427, 85–90. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Zhuang, Z.; Lu, S.; Duan, F.; Du, M. Single-atom catalysts for electrochemical clean energy conversion: recent progress and perspectives. Sustain. Energy Fuels 2020, 4, 996–1011. [Google Scholar] [CrossRef]
- Zhao, D.; Zhuang, Z.; Cao, X.; Zhang, C.; Peng, Q.; Chen, C.; Li, Y. Atomic site electrocatalysts for water splitting, oxygen reduction and selective oxidation. Chem. Soc. Rev. 2020, 49, 2215–2264. [Google Scholar] [CrossRef]
- Liu, Q.; Peng, Y.; Li, Q.; He, T.; Morris, D.; Nichols, F.; Mercado, R.; Zhang, P.; Chen, S. Atomic Dispersion and Surface Enrichment of Palladium in Nitrogen-Doped Porous Carbon Cages Lead to High-Performance Electrocatalytic Reduction of Oxygen. ACS Appl. Mater. Interfaces 2020, 12, 17641–17650. [Google Scholar] [CrossRef]
- Passaponti, M.; Savastano, M.; Clares, M.P.; Inclán, M.; Lavacchi, A.; Bianchi, A.; García-España, E.; Innocenti, M. MWCNTs-Supported Pd(II) Complexes with High Catalytic Efficiency in Oxygen Reduction Reaction in Alkaline Media. Inorg. Chem. 2018, 57, 14484–14488. [Google Scholar] [CrossRef] [Green Version]
- Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R.; Arranz-Mascarós, P.; Santiago-Medina, A.; Melguizo, M.; Domingo-García, M.; López-Garzón, F.J.; Abdelkader-Fernández, V.K.; de Lecea, C.S.-M.; et al. New hybrid materials based on the grafting of Pd(II)-amino complexes on the graphitic surface of AC: preparation, structures and catalytic properties. RSC Adv. 2016, 6, 58247–58259. [Google Scholar] [CrossRef] [Green Version]
- López-Garzón, R.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; Melguizo, M.; García, C.; Domingo-García, M.; López-Garzón, F.J. Supramolecular assembling of molecular ion-ligands on graphite-based solid materials directed to specific binding of metal ions. Inorg. Chim. Acta 2014, 417, 208–221. [Google Scholar] [CrossRef]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Clares, M.P.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Inclán, M.; Bianchi, A.; García-España, E.; López-Garzón, R. Construction of green nanostructured heterogeneous catalysts via non-covalent surface decoration of multi-walled carbon nanotubes with Pd(II) complexes of azamacrocycles. J. Catal. 2017, 353, 239–249. [Google Scholar] [CrossRef]
- Savastano, M.; Arranz-Mascarós, P.; Clares, M.P.; Cuesta, R.; Godino-Salido, M.L.; Guijarro, L.; Gutiérrez-Valero, M.D.; Inclán, M.; Bianchi, A.; García-España, E.; et al. A New Heterogeneous Catalyst Obtained via Supramolecular Decoration of Graphene with a Pd2+ Azamacrocyclic Complex. Molecules 2019, 24, 2714. [Google Scholar] [CrossRef] [Green Version]
- García-Martín, J.; López-Garzón, R.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; Cuesta, R.; Carrasco-Marín, F. Ligand Adsorption on an Activated Carbon for the Removal of Chromate Ions from Aqueous Solutions. Langmuir 2005, 21, 6908–6914. [Google Scholar] [CrossRef]
- Gutiérrez-Valero, M.D.; Godino-Salido, M.L.; Arranz-Mascarós, P.; López-Garzón, R.; Cuesta, R.; García-Martín, J. Adsorption of Designed Pyrimidine Derivative Ligands on an Activated Carbon for the Removal of Cu(II) Ions from Aqueous Solution. Langmuir 2007, 23, 5995–6003. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Clares, M.P.; Godino-Salido, M.L.; Guijarro, L.; Gutiérrez-Valero, M.D.; Bianchi, A.; García-España, E.; López-Garzón, R. Polyfunctional Tetraaza-Macrocyclic Ligands: Zn(II), Cu(II) Binding and Formation of Hybrid Materials with Multiwalled Carbon Nanotubes. ACS Omega 2017, 2, 3868–3877. [Google Scholar] [CrossRef]
- Godino-Salido, M.L.; Santiago-Medina, A.; López-Garzón, R.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; López de la Torre, M.D.; Domingo-García, M.; López-Garzón, F.J. Preparation and characterization of trihydroxamic acid functionalized carbon materials for the removal of Cu(II) ions from aqueous solution. Appl. Surf. Sci. 2016, 387, 128–138. [Google Scholar] [CrossRef]
- Luz Godino-Salido, M.; Santiago-Medina, A.; Arranz-Mascarós, P.; López-Garzón, R.; Gutiérrez-Valero, M.D.; Melguizo, M.; Javier López-Garzón, F. Novel active carbon/crown ether derivative hybrid material for the selective removal of Cu(II) ions: The crucial role of the surface chemical functions. Chem. Eng. Sci. 2014, 114, 94–104. [Google Scholar] [CrossRef]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R. Binding and removal of octahedral, tetrahedral, square planar and linear anions in water by means of activated carbon functionalized with a pyrimidine-based anion receptor. RSC Adv. 2014, 4, 58505–58513. [Google Scholar] [CrossRef]
- Arranz, P.; Bianchi, A.; Cuesta, R.; Giorgi, C.; Godino, M.L.; Gutiérrez, M.D.; López, R.; Santiago, A. Binding and Removal of Sulfate, Phosphate, Arsenate, Tetrachloromercurate, and Chromate in Aqueous Solution by Means of an Activated Carbon Functionalized with a Pyrimidine-Based Anion Receptor (HL). Crystal Structures of [H3L(HgCl4)]·H2O and [H3L(HgBr4)]·H2O Showing Anion−π Interactions. Inorg. Chem. 2010, 49, 9321–9332. [Google Scholar] [PubMed]
- Savastano, M.; Zoppi, C.; Bianchi, A.; Bazzicalupi, C. Synthesis and coordination properties of a new ligand designed for surface functionalization of carbon substrates. Inorg. Chim. Acta 2020, 511, 119793. [Google Scholar] [CrossRef]
- Dietrich, B.; Hosseini, M.W.; Lehn, J.-M.; Sessions, R.B. Synthesis of Macrobicyclic Polyamines by Direct Macrobicyclisation via Tripode-Tripode Coupling. Helv. Chim. Acta 1985, 68, 289–299. [Google Scholar] [CrossRef]
- Tei, L.; Bencini, A.; Blake, A.J.; Lippolis, V.; Perra, A.; Valtancoli, B.; Wilson, C.; Schröder, M. Co-ordination chemistry of amino pendant arm derivatives of 1,4,7-triazacyclononane. Dalton Trans. 2004, 1934–1944. [Google Scholar] [CrossRef]
- Engelmann, M. Ueber eine Synthese des 1-Methylxanthins. Ber. der Dtsch. Chem. Ges. 1909, 42, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Bazzicalupi, C.; Bianchi, A.; Biver, T.; Giorgi, C.; Santarelli, S.; Savastano, M. Formation of Double-Strand Dimetallic Helicates with a Terpyridine-Based Macrocycle. Inorg. Chem. 2014, 53, 12215–12224. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; de la Torre, M.D.L.; Bianchi, A.; Melguizo, M. Supramolecular forces and their interplay in stabilizing complexes of organic anions: Tuning binding selectivity in water. Org. Chem. Front. 2018, 6, 75–86. [Google Scholar] [CrossRef]
- Fontanelli, M.; Micheloni, M. Proceedings of the I Spanish-Italian Congress on Thermodynamics of Metal Complexes; Diputación de Castellón: Castellón, Spain, 1990; pp. 41–43. [Google Scholar]
- Savastano, M.; Fiaschi, M.; Ferraro, G.; Gratteri, P.; Mariani, P.; Bianchi, A.; Bazzicalupi, C. Sensing Zn2+ in Aqueous Solution with a Fluorescent Scorpiand Macrocyclic Ligand Decorated with an Anthracene Bearing Tail. Molecules 2020, 25, 1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gran, G. Determination of the equivalence point in potentiometric titrations. Part II. Analyst 1952, 77, 661–671. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Borri, C.; Calisi, N.; Galvanetto, E.; Falsini, N.; Biccari, F.; Vinattieri, A.; Cucinotta, G.; Caporali, S. First Proof-of-Principle of Inorganic Lead Halide Perovskites Deposition by Magnetron-Sputtering. Nanomaterials 2020, 10, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calisi, N.; Caporali, S.; Milanesi, A.; Innocenti, M.; Salvietti, E.; Bardi, U. Composition-Dependent Degradation of Hybrid and Inorganic Lead Perovskites in Ambient Conditions. Top. Catal. 2018, 61, 1201–1208. [Google Scholar] [CrossRef]
- Muniz-Miranda, M.; Muniz-Miranda, F.; Caporali, S.; Calisi, N.; Pedone, A. SERS, XPS and DFT investigation on palladium surfaces coated with 2,2′-bipyridine monolayers. Appl. Surf. Sci. 2018, 457, 98–103. [Google Scholar] [CrossRef]
- Anderegg, G.; Wanner, H. Pyridine derivatives as complexing agents. XIII. The stability of the palladium(II) complexes with pyridine, 2,2′-bipyridyl, and 1,10-phenanthroline. Inorg. Chim. Acta 1986, 113, 101–108. [Google Scholar] [CrossRef]
- Lari, L.; Nuttall, C.J.; Copley, M.P.; Potter, R.J.; Simon, J.; Mingo, N.; Ozkaya, D. Characterization of nanoembedded alloyed thermoelectrics. J. Phys. Conf. Ser. 2014, 522, 012040. [Google Scholar] [CrossRef] [Green Version]
- López-Garzón, R.; Arranz-Mascarós, P.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Cuesta, R.; Moreno, J.M. Bifunctional pyrimidine-amino-acid ligands: Solution study and crystal structure of a Mn(II) chain alternating six- and sevenfold coordination environments. Inorg. Chim. Acta 2003, 355, 41–48. [Google Scholar] [CrossRef]
- López-Garzón, R.; Godino-Salido, M.L.; Arranz-Mascarós, P.; Fontecha-Cámara, M.A.; Gutiérrez-Valero, M.D.; Cuesta, R.; Moreno, J.M.; Stoeckli-Evans, H. Protonation and Zn(II) complexation with versatile valine and glycylglycine N-pyrimidines derivatives: crystal structures of layered {[Zn(HL1)2]·2H2O}n and [Zn(HL2)2(H2O)4]. Inorg. Chim. Acta 2004, 357, 2007–2014. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equilibrium | log K |
|---|---|
| HL2− + 2H+ = H3L | 21.65(1) |
| H3L + H+ = H4L+ | 9.84(1) |
| H4L+ + H+ = H5L2+ | 3.42(3) |
| H5L2+ + H+ = H6L3+ | 2.45(3) |
| H6L3+ + 2H+ = H8L5+ | 4.54 (4) |
| Sample | Ink Loading (mg) | ntheo (−0.5 V) a | nexp (−0.5 V) b | Nexp c | Eon (V) d | E1/2 (V) e |
|---|---|---|---|---|---|---|
| 1 | 2.4 | 4.00 | 4.00 | 0.229 | −0.0670 | −0.19 |
| 2 | 3.3 | 3.55 | 3.93 | 0.200 | −0.0288 | −0.11 |
| 3 | 8.7 | 3.68 | 3.96 | 0.233 | −0.0066 | −0.11 |
| 4 | 12.5 | 3.69 | 3.88 | 0.197 | −0.0605 | −0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savastano, M.; Passaponti, M.; Giurlani, W.; Lari, L.; Bianchi, A.; Innocenti, M. Multi-Walled Carbon Nanotubes Supported Pd(II) Complexes: A Supramolecular Approach towards Single-Ion Oxygen Reduction Reaction Catalysts. Energies 2020, 13, 5539. https://doi.org/10.3390/en13215539
Savastano M, Passaponti M, Giurlani W, Lari L, Bianchi A, Innocenti M. Multi-Walled Carbon Nanotubes Supported Pd(II) Complexes: A Supramolecular Approach towards Single-Ion Oxygen Reduction Reaction Catalysts. Energies. 2020; 13(21):5539. https://doi.org/10.3390/en13215539
Chicago/Turabian StyleSavastano, Matteo, Maurizio Passaponti, Walter Giurlani, Leonardo Lari, Antonio Bianchi, and Massimo Innocenti. 2020. "Multi-Walled Carbon Nanotubes Supported Pd(II) Complexes: A Supramolecular Approach towards Single-Ion Oxygen Reduction Reaction Catalysts" Energies 13, no. 21: 5539. https://doi.org/10.3390/en13215539