
Corrosion of Metal Alloys in Potassium Acetate Solutions for Liquid Desiccant Dehumidification and Air Conditioning

 , ,
, ,
Abstract
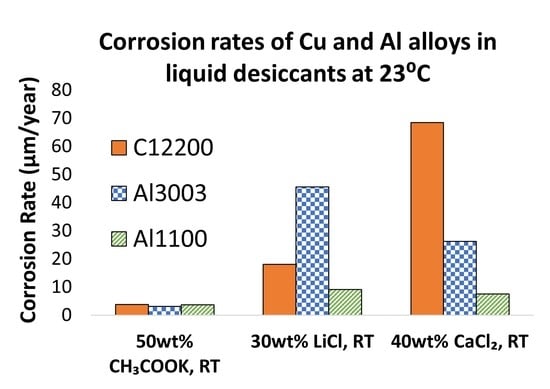
:
1. Introduction
2. Materials and Methods
2.1. Material Selection
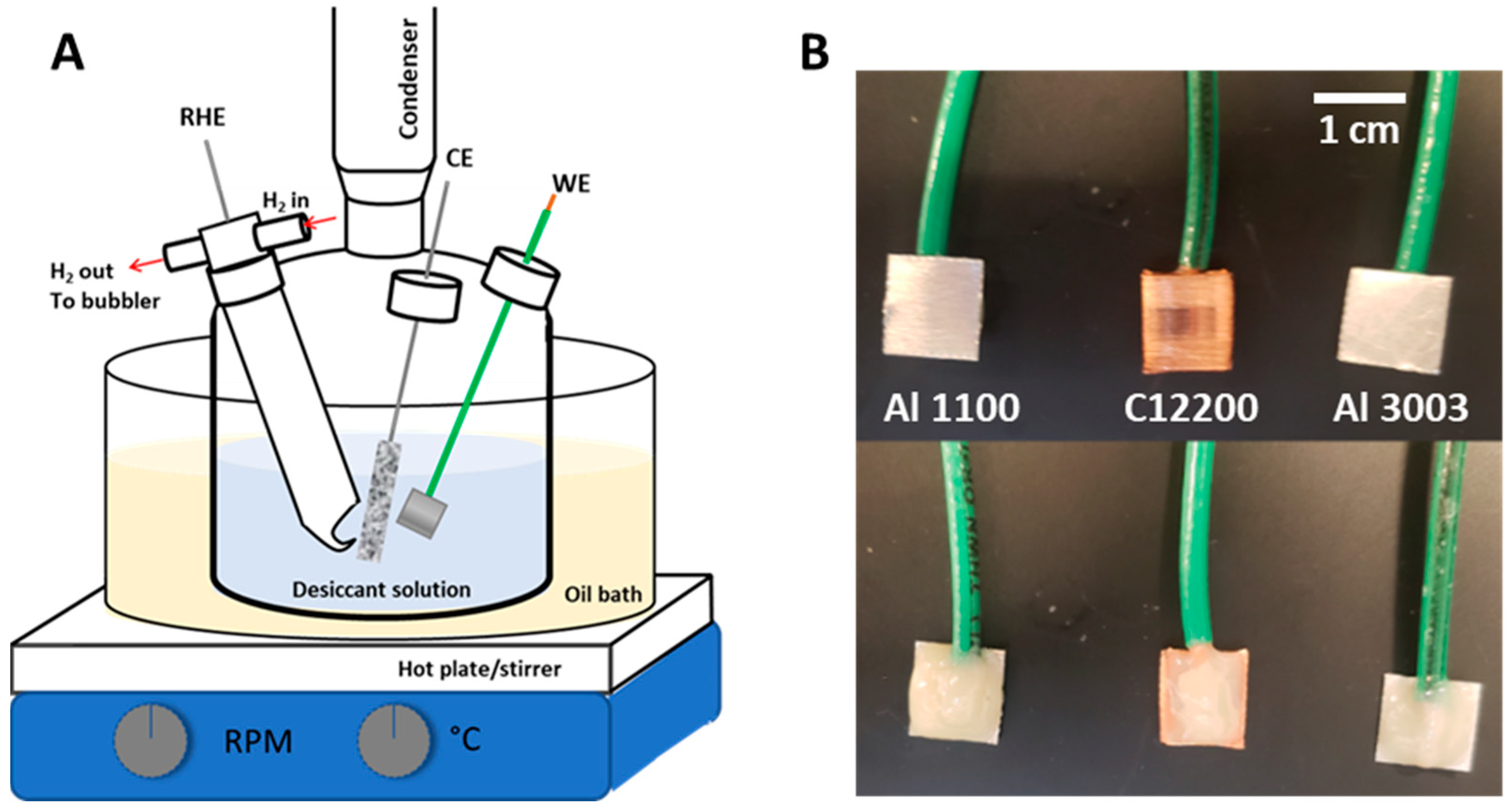
2.2. General Overview of Methods for Electrochemical Corrosion Evaluation
2.3. Electrolyte Preparation
2.4. Electrode Fabrication
2.5. OCP Determination
2.6. Electrochemical Corrosion Tests
2.7. Characterization of Corroded Samples and Salt Solutions
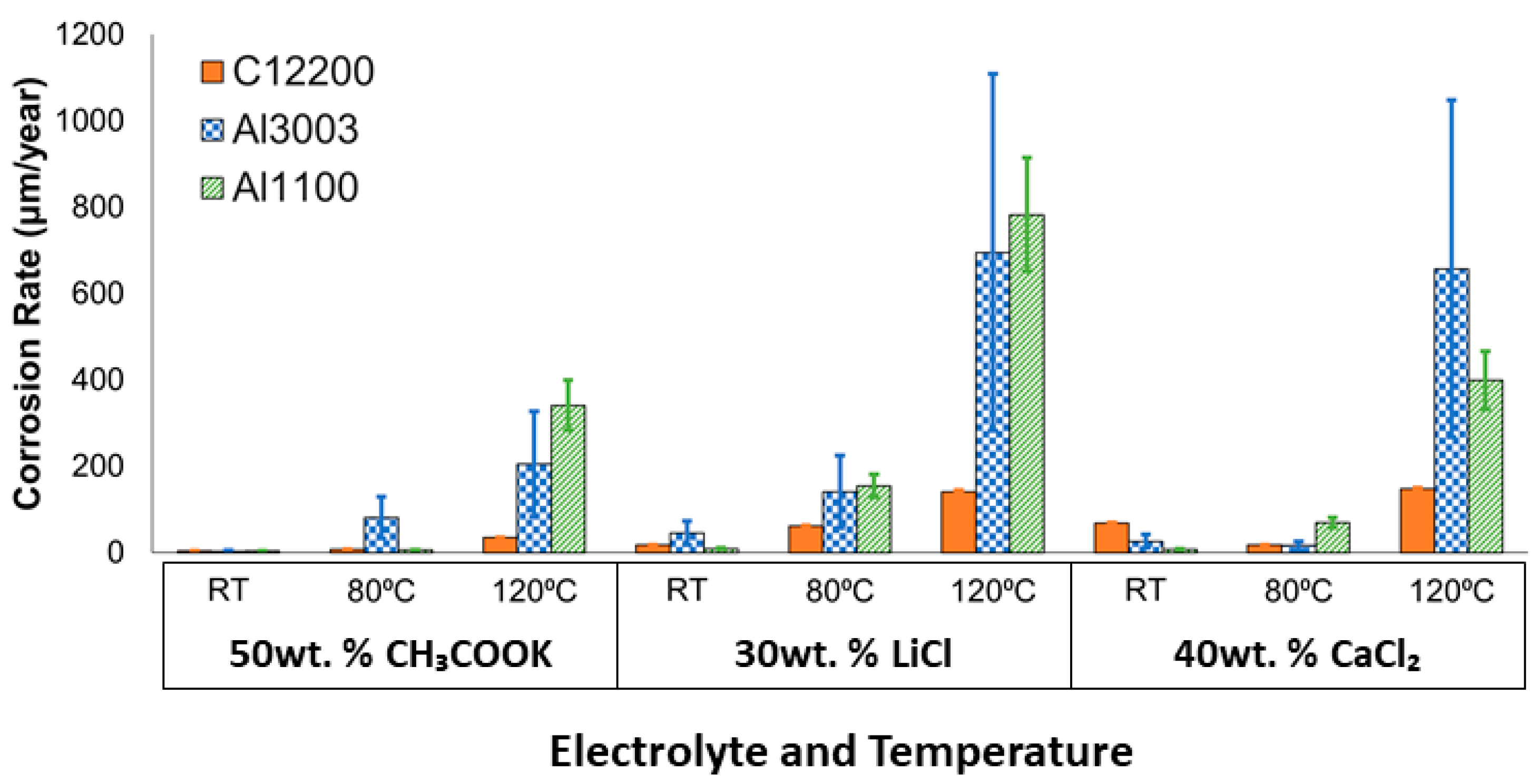
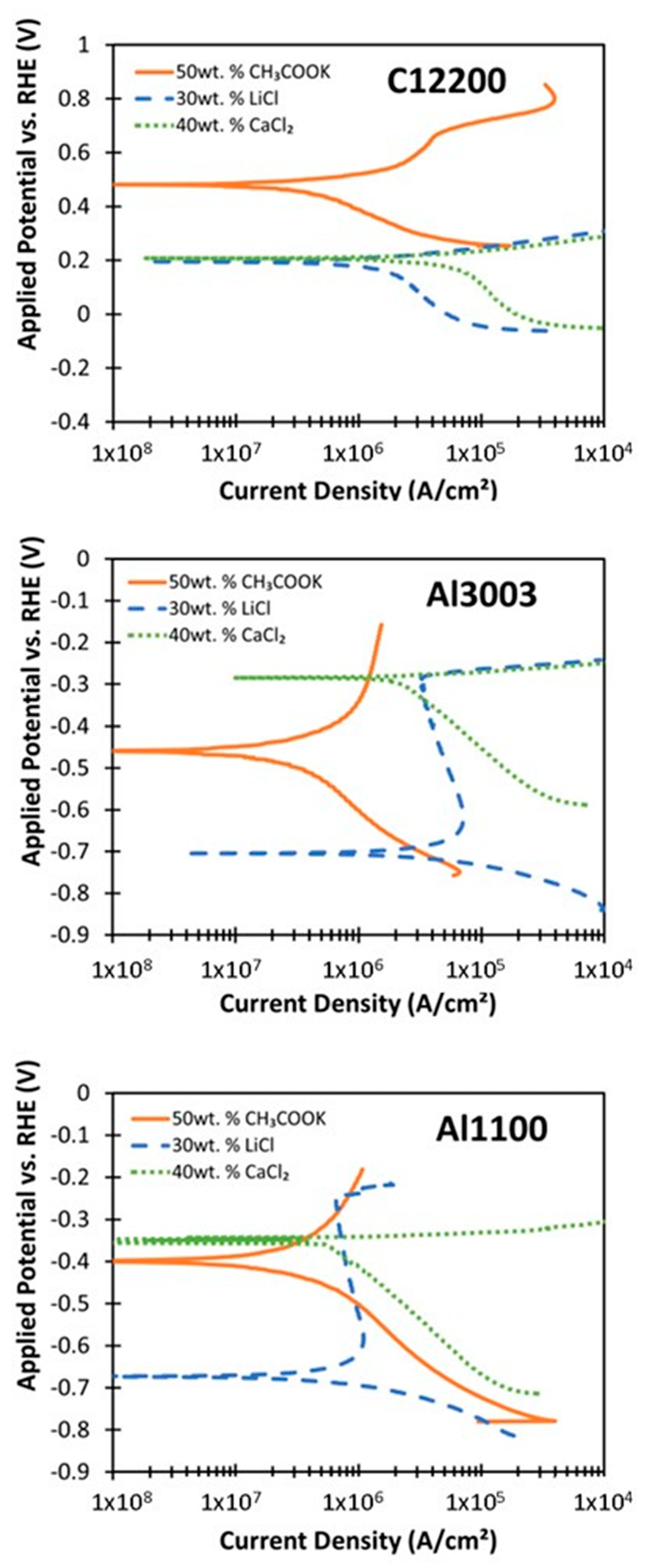
3. Results
4. Discussion
4.1. Corrosion Behavior
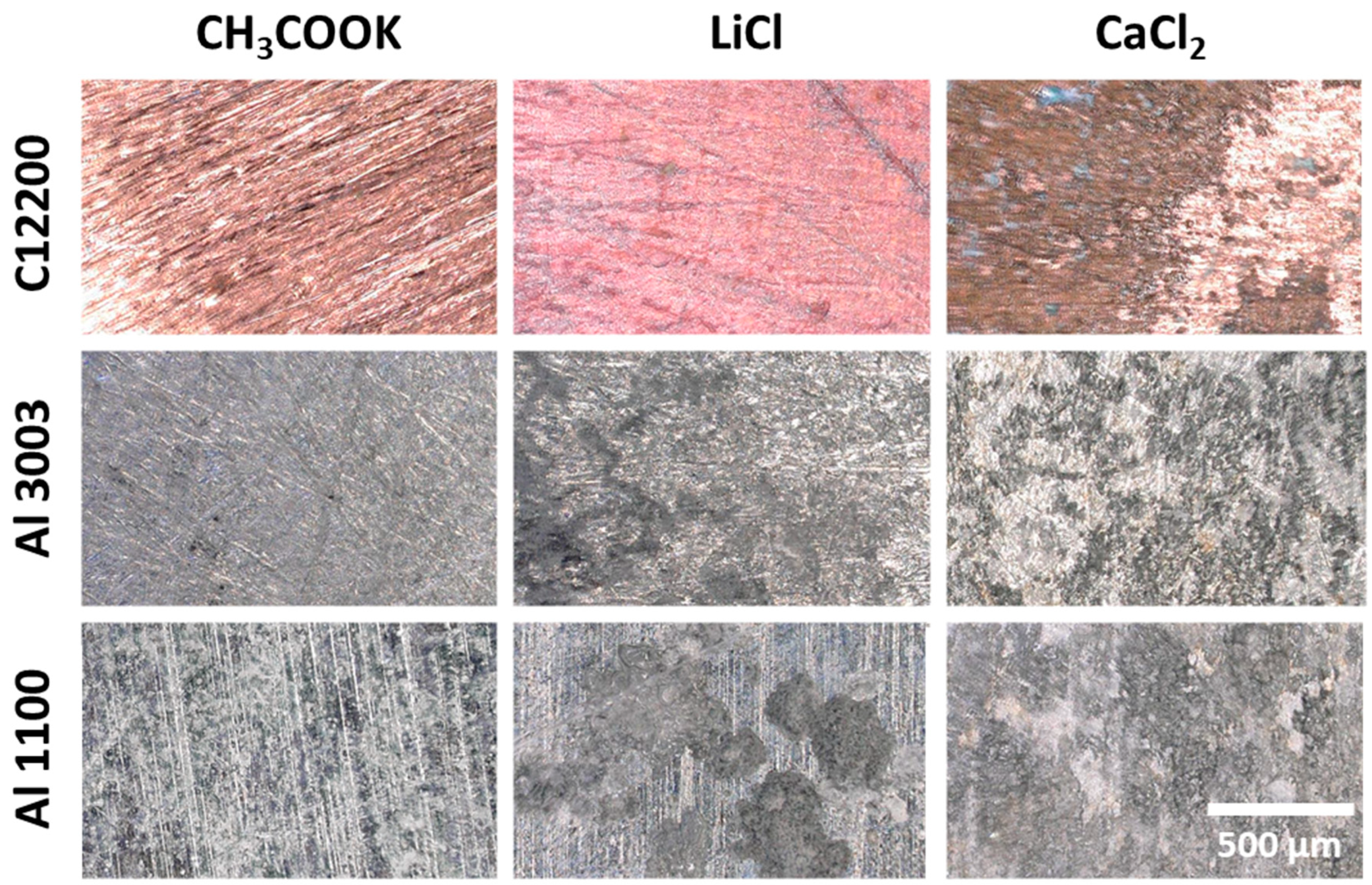
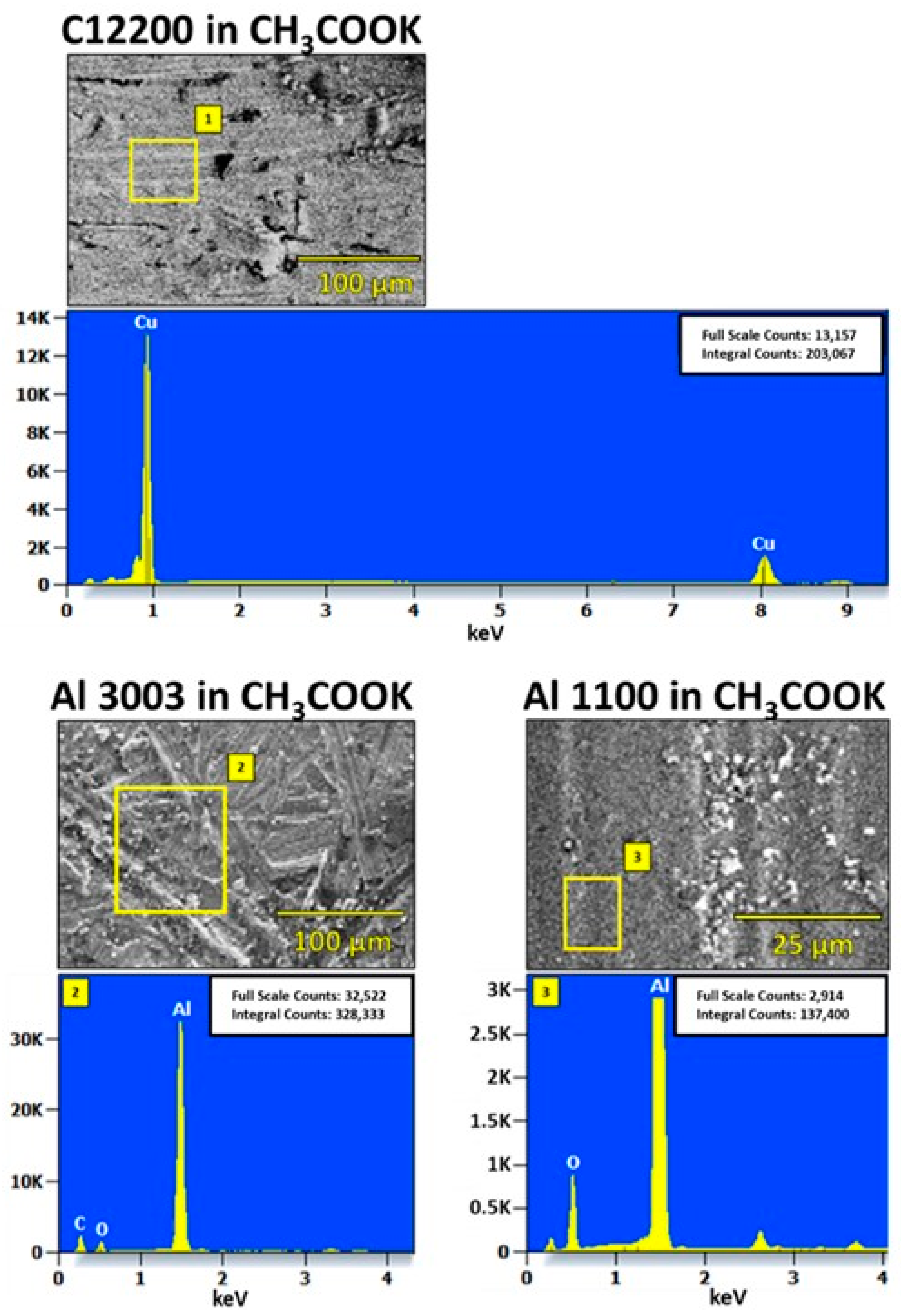
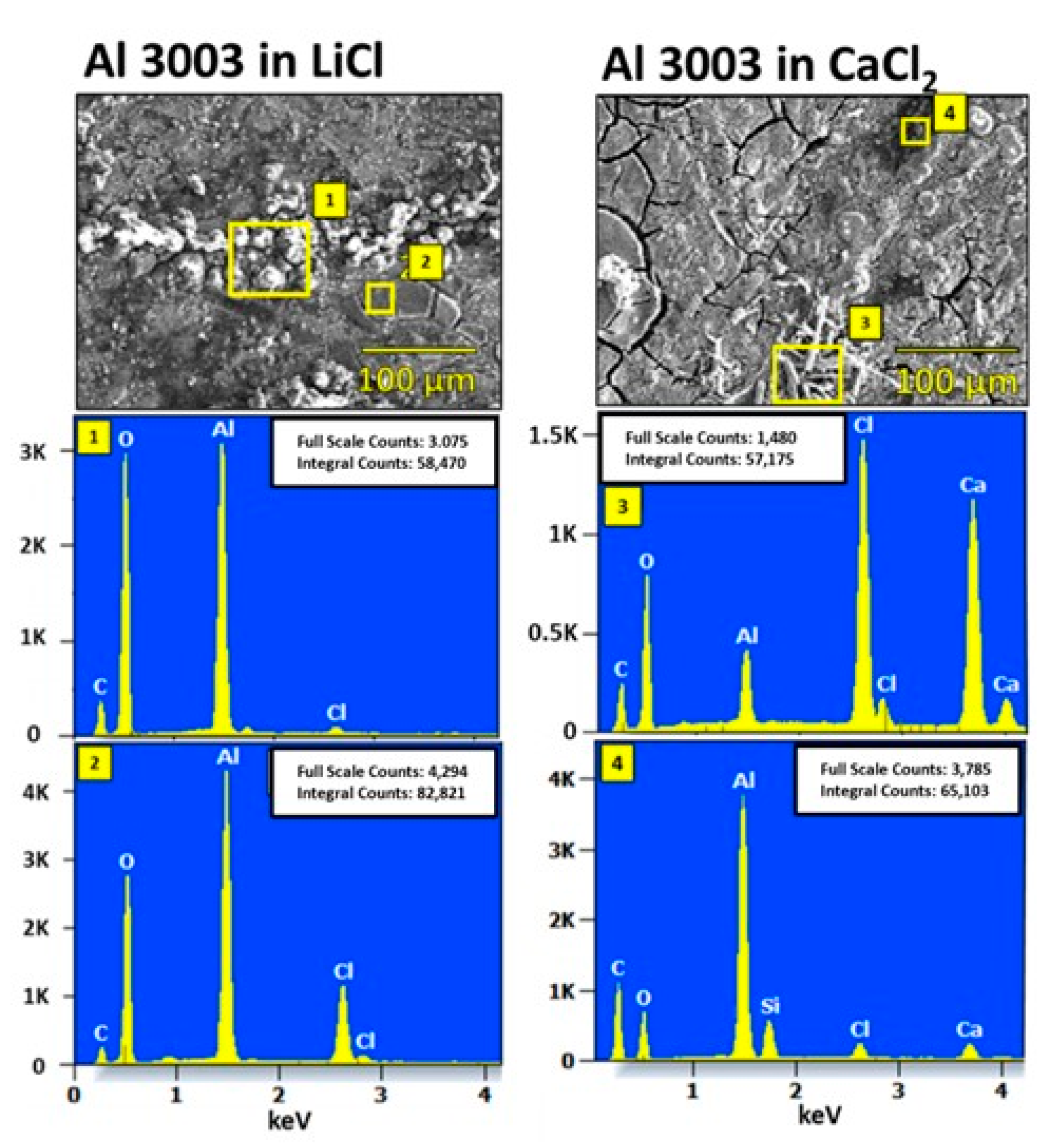
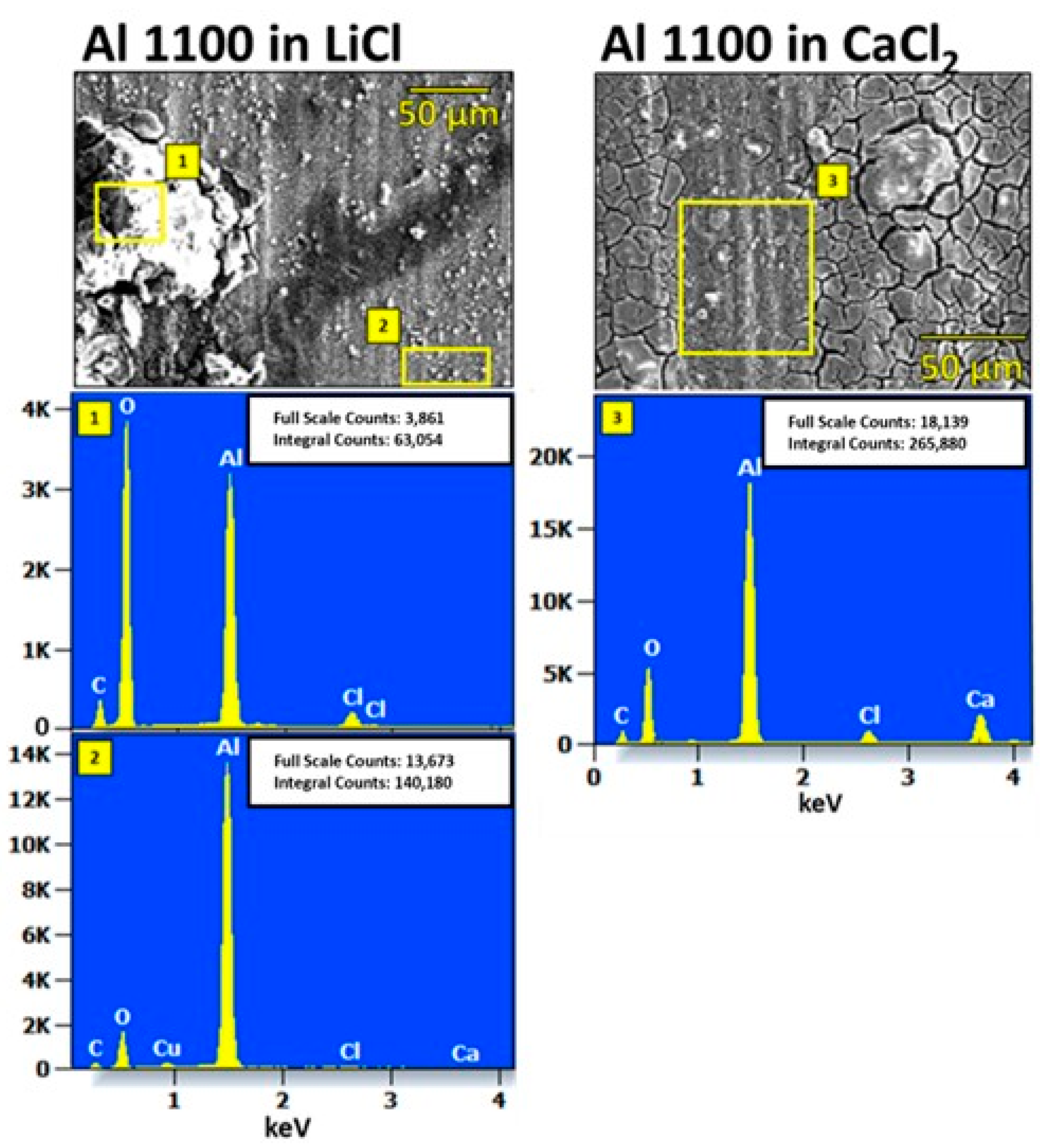
4.2. Characterization of Corroded Alloy Samples and Liquid Desiccant Solutions
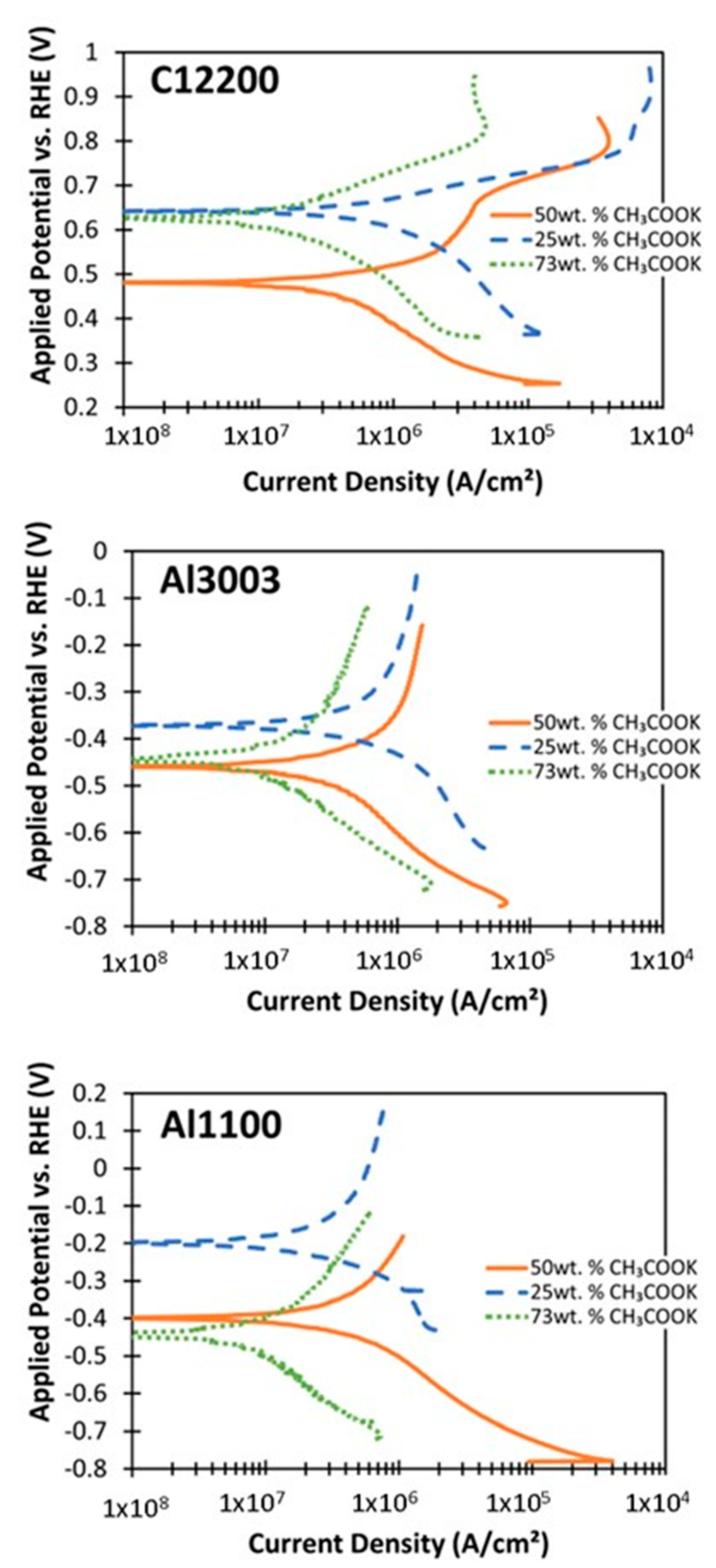
4.3. Corrosion in CH3COOK under Varied Conditions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Odukomaiya, A.; Woods, J.; James, N.; Kaur, S.; Gluesenkamp, K.R.; Kumar, N.; Mumme, S.; Jackson, R.; Prasher, R. Addressing energy storage needs at lower cost via on-site thermal energy storage in buildings. Energy Environ. Sci. 2021, 14, 5315–5329. [Google Scholar] [CrossRef]
- International Energy Agency. The Future of Cooling; International Energy Agency: Paris, France, 2018.
- Lowenstein, A.; Slayzak, S.; Ryan, J.; Pesaran, A. Advanced Commercial Liquid-Desiccant Technology Development Study; National Renewable Energy Lab. (NREL): Golden, CO, USA, 1998.
- Woods, J.; Kozubal, E. Combining liquid desiccant dehumidification with a dew-point evaporative cooler: A design analysis. HVAC&R Res. 2013, 19, 663–675. [Google Scholar]
- Eric Kozubal, J.W.; Judkoff, R. Development and Analysis of Desiccant Enhanced Evaporative Air Conditioner Prototype; National Renewable Energy Lab. (NREL): Golden, CO, USA, 2012.
- Kian Jon, C.; Islam, M.R.; Kim Choon, N.; Shahzad, M.W. Future of Air Conditioning. In Advances in Air Conditioning Technologies: Improving Energy Efficiency; Kian Jon, C., Islam, M.R., Kim Choon, N., Shahzad, M.W., Eds.; Springer: Singapore, 2021; pp. 17–52. [Google Scholar]
- Cheng, Q.; Zhang, X. Review of solar regeneration methods for liquid desiccant air-conditioning system. Energy Build. 2013, 67, 426–433. [Google Scholar] [CrossRef]
- Koronaki, I.P.; Christodoulaki, R.I.; Papaefthimiou, V.D.; Rogdakis, E.D. Thermodynamic analysis of a counter flow adiabatic dehumidifier with different liquid desiccant materials. Appl. Therm. Eng. 2013, 50, 361–373. [Google Scholar] [CrossRef]
- Mujahid Rafique, M.; Gandhidasan, P.; Ibrahim, N.I.; Bahaidarah, H.M. Recent Developments in Liquid Desiccant-Based Cooling Systems. In Encyclopedia of Sustainable Technologies; Abraham, M.A., Ed.; Elsevier: Oxford, UK, 2017; pp. 441–453. [Google Scholar]
- Liu, X.; Qu, M.; Liu, X.; Wang, L. Membrane-based liquid desiccant air dehumidification: A comprehensive review on materials, components, systems and performances. Renew. Sustain. Energy Rev. 2019, 110, 444–466. [Google Scholar] [CrossRef]
- Chen, Y.; Yin, Y.; Zhang, X. Performance analysis of a hybrid air-conditioning system dehumidified by liquid desiccant with low temperature and low concentration. Energy Build. 2014, 77, 91–102. [Google Scholar] [CrossRef]
- de Lucas, A.; Donate, M.; Rodríguez, J.F. Vapour pressures, densities, and viscosities of the (water+lithium bromide+potassium acetate) system and (water+lithium bromide + sodium lactate) system. J. Chem. Thermodyn. 2006, 38, 123–129. [Google Scholar] [CrossRef]
- Donate, M.; Rodriguez, L.; Lucas, A.D.; Rodríguez, J.F. Thermodynamic evaluation of new absorbent mixtures of lithium bromide and organic salts for absorption refrigeration machines. Int. J. Refrig. 2006, 29, 30–35. [Google Scholar] [CrossRef]
- Salikandi, M.; Ranjbar, B.; Shirkhan, E.; Shanmuga Priya, S.; Thirunavukkarasu, I.; Sudhakar, K. Recent trends in liquid desiccant materials and cooling systems: Application, performance and regeneration characteristics. J. Build. Eng. 2021, 33, 101579. [Google Scholar] [CrossRef]
- Watanabe, H.; Komura, T.; Matsumoto, R.; Ito, K.; Nakayama, H.; Nokami, T.; Itoh, T. Design of ionic liquids as liquid desiccant for an air conditioning system. Green Energy Environ. 2019, 4, 139–145. [Google Scholar] [CrossRef]
- Wen, T.; Lu, L.; Li, M.; Zhong, H. Comparative study of the regeneration characteristics of LiCl and a new mixed liquid desiccant solution. Energy 2018, 163, 992–1005. [Google Scholar] [CrossRef]
- Wen, T.; Lu, L.; Nie, Y.; Zhong, H. Development and investigation on the dehumidification and corrosion resistance performance of a new mixed liquid desiccant. Int. J. Heat Mass Transf. 2019, 130, 72–82. [Google Scholar] [CrossRef]
- Bouzenada, S.; Kaabi, A.N.; Frainkin, L.; Salmon, T.; Léonard, A. Experimental Comparative Study on Lithium Chloride and Calcium Chloride Desiccants. Procedia Comput. Sci. 2016, 83, 718–725. [Google Scholar] [CrossRef] [Green Version]
- Lowenstein, A.; Miller, J.A.; Hermans, T. LDDX: A High Efficiency Air Conditioner for DOD Buildings; AIL Research, Inc.: Hopewell, NJ, USA, 2017. [Google Scholar]
- Gomez-Vidal, J.C.; Tirawat, R. Corrosion of alloys in a chloride molten salt (NaCl-LiCl) for solar thermal technologies. Sol. Energy Mater. Sol. Cells 2016, 157, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCafferty, E. Validation of corrosion rates measured by the Tafel extrapolation method. Corros. Sci. 2005, 47, 3202–3215. [Google Scholar] [CrossRef]
- Perez, T.E. Corrosion in the Oil and Gas Industry: An Increasing Challenge for Materials. JOM 2013, 65, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Finšgar, M.; Jackson, J. Application of corrosion inhibitors for steels in acidic media for the oil and gas industry: A review. Corros. Sci. 2014, 86, 17–41. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Medina, M.A.; Carbajal-De la Torre, G.; Liu, H.B.; Martínez-Villafañe, A.; González-Rodriguez, J.G. Hot corrosion behaviour of Fe–Al based intermetallic in molten NaVO3 salt. Corros. Sci. 2009, 51, 1420–1427. [Google Scholar] [CrossRef]
- Rapp, R.A.; Zhang, Y.-S. Hot corrosion of materials: Fundamental studies. JOM 1994, 46, 47–55. [Google Scholar] [CrossRef]
- Gamry. Getting Started with Electrochemical, Corrosion Measurements; Gamry: Warminster, PA, USA, 2011. [Google Scholar]
- Tabrizi, M.R.; Lyon, S.B.; Thompson, G.E.; Ferguson, J.M. The long-term corrosion of aluminium in alkaline media. Corros. Sci. 1991, 32, 733–742. [Google Scholar] [CrossRef]
- Adams, F.V.; Akinwamide, S.O.; Obadele, B.; Olubambi, P.A. Comparison study on the corrosion behavior of aluminum alloys in different acidic media. Mater. Today Proc. 2021, 38, 1040–1043. [Google Scholar] [CrossRef]
- Paz Martínez-Viademonte, M.; Abrahami, S.T.; Hack, T.; Burchardt, M.; Terryn, H. A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection. Coatings 2020, 10, 1106. [Google Scholar] [CrossRef]
- Xhanari, K.; Finšgar, M.; Knez Hrnčič, M.; Maver, U.; Knez, Ž.; Seiti, B. Green corrosion inhibitors for aluminium and its alloys: A review. RSC Adv. 2017, 7, 27299–27330. [Google Scholar] [CrossRef]
- Xhanari, K.; Finšgar, M. Organic corrosion inhibitors for aluminum and its alloys in chloride and alkaline solutions: A review. Arab. J. Chem. 2019, 12, 4646–4663. [Google Scholar] [CrossRef]
- Frankel, G.S. Pitting Corrosion of Metals A Review of the Critical Factors. J. Electrochem. Soc. 1998, 145, 2186–2198. [Google Scholar] [CrossRef]
- Akpanyung, K.; Loto, R. Pitting corrosion evaluation: A review. J. Phys. Conf. Ser. 2019, 1378, 022088. [Google Scholar] [CrossRef]
- Chernova, G.P.; Kornienko, L.P. Effect of inhibiting additives on the electrochemical behavior of AMg-6 alloy in aqueous potassium acetate solutions at elevated temperatures. Prot. Met. Phys. Chem. Surf. 2010, 46, 354–358. [Google Scholar] [CrossRef]
- Ahangar, M.; Izadi, M.; Shahrabi, T.; Mohammadi, I. The synergistic effect of zinc acetate on the protective behavior of sodium lignosulfonate for corrosion prevention of mild steel in 3.5 wt% NaCl electrolyte: Surface and electrochemical studies. J. Mol. Liq. 2020, 314, 113617. [Google Scholar] [CrossRef]
- Cano, E.; Bastidas, J.M.; Polo, J.L.; Mora, N. Study of the Effect of Acetic Acid Vapor on Copper Corrosion at 40 and 80% Relative Humidity. J. Electrochem. Soc. 2001, 148, B431. [Google Scholar] [CrossRef]
- Occupational Safety and Health Administration. Hazard Classification Guidance for Manufacturers, Importers, and Employers; U.S. Department of Labor: Washington, DC, USA, 2016.
- Liu, X.; Li, H.; Zhao, X.; Chen, Y.; Wang, S. Comparison of the corrosion behavior of copper tubes in formic acid and acetic acid environment. Mater. Corros. 2021, 72, 1919–1927. [Google Scholar] [CrossRef]
- Wysocka, J.; Cieslik, M.; Krakowiak, S.; Ryl, J. Carboxylic acids as efficient corrosion inhibitors of aluminium alloys in alkaline media. Electrochim. Acta 2018, 289, 175–192. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal/Alloy | C12200 | Al3003 | Al1100 |
|---|---|---|---|
| Density (g/cm3) | 8.96 | 2.73 | 2.71 |
| Equivalent Weight (g) | 31.78 | 9.45 | 9.14 |
| Typical Composition (wt. %) | 99.9 Cu 0.015–0.040 P | 98.6 Al 0.05–0.2 Cu 1.0–1.5 Mn 0.7 Fe 0.6 Si 0.10 Zn 0.15 other | 99 Al 0.02–0.05 Cu 0.05 Mn 0.95 Si + Fe 0.10 Zn 0.15 other |
| Sample | Conditions | OCP vs. RHE (mV) | Ecorr vs. RHE (mV) | jcorr (µA/cm2) | CR (µm/Year) |
|---|---|---|---|---|---|
| LiCl | |||||
| C12200 | 23 °C, 30 wt. % | 237 ± 14 | 197 ± 8 | 1.56 ± 0.03 | 18.1 ± 0.4 |
| C12200 | 80 °C, 30 wt. % | −8.00 ± 0.48 | −28.6 ± 1.1 | 5.31 ± 0.11 | 61.7 ± 1.3 |
| C12200 | 120 °C, 30 wt. % | −18.0 ± 1.1 | −43.1 ± 1.7 | 12.2 ± 0.3 | 141 ± 3 |
| Al3003 | 23 °C, 30 wt. % | −542 ± 87 | −704 ± 113 | 3.93 ± 2.33 | 45.6 ± 27.1 |
| Al3003 | 80 °C, 30 wt. % | −446 ± 72 | −440 ± 71 | 12.1 ± 7.2 | 141 ± 84 |
| Al3003 | 120 °C, 30 wt. % | −516 ± 83 | −509 ± 82 | 59.9 ± 35.6 | 696 ± 413 |
| Al1100 | 23 °C, 30 wt. % | −515 ± 148 | −672 ± 236 | 0.785 ± 0.132 | 9.11 ± 1.53 |
| Al1100 | 80 °C, 30 wt. % | −434 ± 125 | −416 ± 146 | 13.3 ± 2.2 | 155 ± 26 |
| Al1100 | 120 °C, 30 wt. % | −453 ± 130 | −407 ± 143 | 67.5 ± 11.4 | 783 ± 132 |
| CaCl2 | |||||
| C12200 | 23 °C, 40 wt. % | 248 ± 15 | 209 ± 8 | 5.89 ± 0.12 | 68.4 ± 1.4 |
| C12200 | 80 °C, 40 wt. % | 8.00 ± 0.48 | −33.9 ± 1.3 | 1.56 ± 0.03 | 18.1 ± 0.4 |
| C12200 | 120 °C, 40 wt. % | −25.0 ± 1.5 | −34.7 ± 1.4 | 12.7 ± 0.3 | 147 ± 3 |
| Al3003 | 23 °C, 40 wt. % | −289 ± 46 | −287 ± 46 | 2.26 ± 1.34 | 26.3 ± 15.6 |
| Al3003 | 80 °C, 40 wt. % | −406 ± 65 | −425 ± 68 | 1.41 ± 0.84 | 16.4 ± 9.7 |
| Al3003 | 120 °C, 40 wt. % | −484 ± 78 | -480 ± 77 | 56.7 ± 33.7 | 658 ± 391 |
| Al1100 | 23 °C, 40 wt. % | −415 ± 119 | −354 ± 125 | 0.646 ± 0.109 | 7.50 ± 1.26 |
| Al1100 | 80 °C, 40 wt. % | −405 ± 116 | −460 ± 162 | 6.00 ± 1.01 | 69.6 ± 11.7 |
| Al1100 | 120 °C, 40 wt. % | −518 ± 149 | −515 ± 181 | 34.4 ± 5.8 | 399 ± 67 |
| CH3COOK | |||||
| C12200 | 23 °C, 50 wt. % | 551 ± 33 | 481 ± 19 | 0.329 ± 0.007 | 3.82 ± 0.08 |
| C12200 | 80 °C, 50 wt. % | 671 ± 40 | 645 ± 25 | 0.669 ± 0.014 | 7.77 ± 0.16 |
| C12200 | 120 °C, 50 wt. % | 721 ± 43 | 566 ± 22 | 3.01 ± 0.06 | 34.9 ± 0.7 |
| Al3003 | 23 °C, 50 wt. % | −458 ± 74 | −457 ± 73 | 0.272 ± 0.161 | 3.15 ± 1.87 |
| Al3003 | 80 °C, 50 wt. % | −373 ± 60 | −394 ± 63 | 6.97 ± 4.14 | 80.9 ± 48.0 |
| Al3003 | 120 °C, 50 wt. % | −332 ± 53 | −337 ± 54 | 17.7 ± 10.5 | 205 ± 122 |
| Al1100 | 23 °C, 50 wt. % | −482 ± 138 | −396 ± 139 | 0.321 ± 0.054 | 3.73 ± 0.63 |
| Al1100 | 80 °C, 50 wt. % | −176 ± 51 | −191 ± 67 | 0.516 ± 0.087 | 5.99 ± 1.01 |
| Al1100 | 120 °C, 50 wt. % | −281 ± 81 | −311 ± 110 | 29.4 ± 5.0 | 342 ± 58 |
| Sample | Conditions | OCP vs. RHE (mV) | Ecorr vs. RHE (mV) | jcorr (µA/cm2) | CR (mm/year) |
|---|---|---|---|---|---|
| CH3COOK, 23 °C | |||||
| C12200 | 73 wt. % | 657 ± 40 | 624 ± 25 | 0.104 ± 0.002 | 1.20 ± 0.03 |
| Al3003 | 73 wt. % | −423 ± 68 | −446 ± 72 | 0.0878 ± 0.0521 | 1.02 ± 0.61 |
| Al1100 | 73 wt. % | −420 ± 121 | −446 ± 157 | 0.0642 ± 0.0108 | 0.745 ± 0.126 |
| C12200 | 25 wt. % | 663 ± 40 | 643 ± 25 | 0.387 ± 0.008 | 4.49 ± 0.09 |
| Al3003 | 25 wt. % | −334 ± 54 | −369 ± 59 | 0.619 ± 0.368 | 7.19 ± 4.27 |
| Al1100 | 25 wt. % | −132 ± 38 | −201 ± 70 | 0.176 ± 0.030 | 2.04 ± 0.34 |
| CH3COOK, 50 wt. %, 23 °C | |||||
| C12200 | pH 11.3 | 705 ± 42 | 679 ± 27 | 0.802 ± 0.017 | 9.31 ± 0.20 |
| Al3003 | pH 11.3 | −539 ± 87 | −559 ± 90 | 1.20 ± 0.71 | 13.9 ± 8.3 |
| Al1100 | pH 11.3 | −425 ± 122 | −483 ± 170 | 0.731 ± 0.123 | 8.48 ± 1.43 |
| C12200 | pH 6.6 | 442 ± 27 | 417 ± 16 | 1.36 ± 0.03 | 15.8 ± 0.3 |
| Al3003 | pH 6.6 | 19.0 ± 3.1 | 19.9 ± 3.2 | 0.0802 ± 0.0476 | 0.931 ± 0.553 |
| Al1100 | pH 6.6 | −21.0 ± 6.0 | 9.58 ± 3.37 | 0.436 ± 0.074 | 5.06 ± 0.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rippy, K.C.; Volk, E.; Beers, R.; Kozubal, E.; Gauderman, K.; Vidal, J. Corrosion of Metal Alloys in Potassium Acetate Solutions for Liquid Desiccant Dehumidification and Air Conditioning. Energies 2022, 15, 4421. https://doi.org/10.3390/en15124421
Rippy KC, Volk E, Beers R, Kozubal E, Gauderman K, Vidal J. Corrosion of Metal Alloys in Potassium Acetate Solutions for Liquid Desiccant Dehumidification and Air Conditioning. Energies. 2022; 15(12):4421. https://doi.org/10.3390/en15124421
Chicago/Turabian StyleRippy, Kerry C., Emily Volk, Reagan Beers, Eric Kozubal, Kristin Gauderman, and Judith Vidal. 2022. "Corrosion of Metal Alloys in Potassium Acetate Solutions for Liquid Desiccant Dehumidification and Air Conditioning" Energies 15, no. 12: 4421. https://doi.org/10.3390/en15124421