Recent Advances in Enzymatic Fuel Cells: Experiments and Modeling

Abstract
:1. Introduction
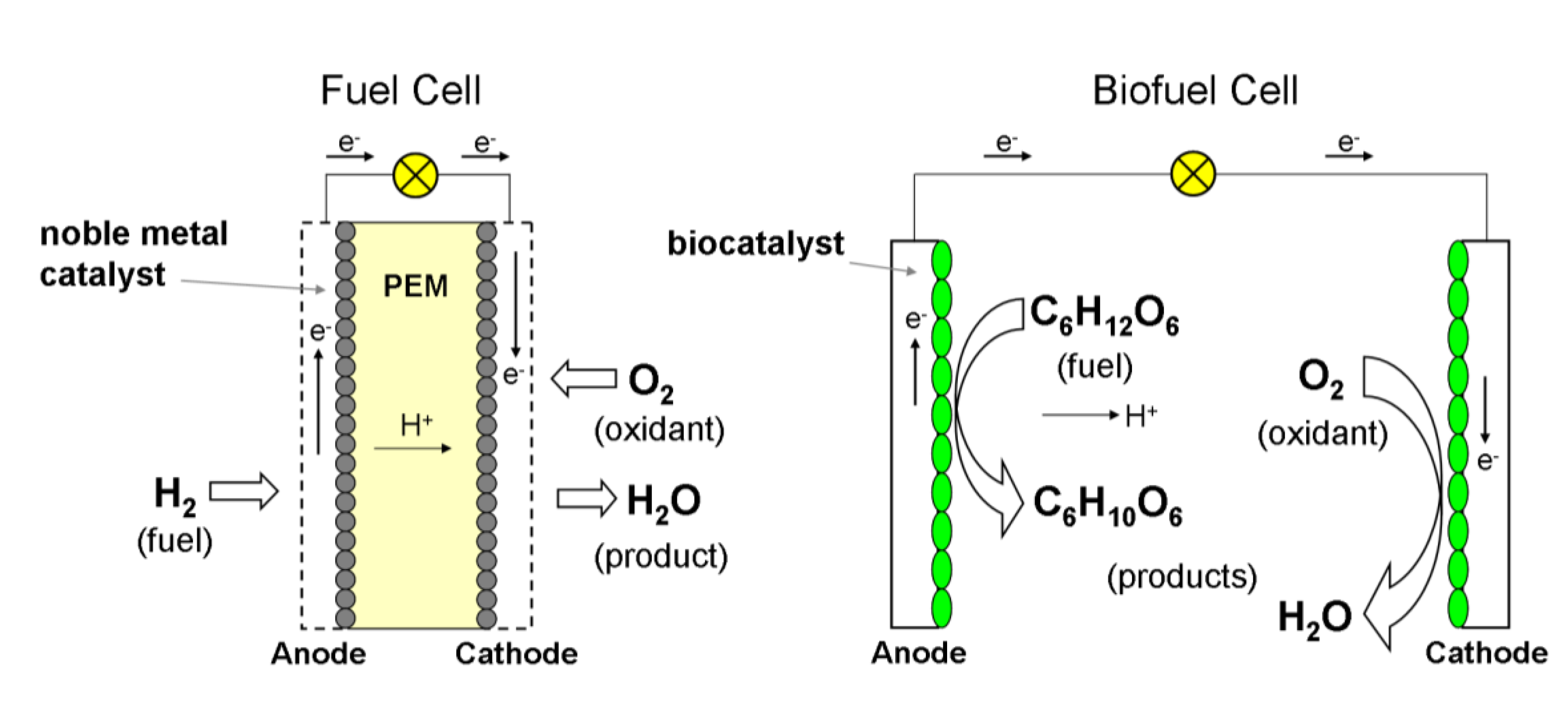
2. Enzymatic Fuel Cells in the Context of Biofuel Cells
3. Future Applications of Biofuel Cells
4. Fuels, Oxidants and Biocatalysts Used in Enzymatic Fuel Cells
4.1. Fuels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fuel | Enzyme | Co-factor | Half-Cell Reaction | Natural Acceptor |
|---|---|---|---|---|
| glucose | glucose oxidase, EC 1.1.3.4 | FAD | glucose → glucono-1,5-lactone + 2H+ + 2e− | O2 |
| glucose dehydrogenase, EC 1.1.1.47 | NAD | see above | NAD | |
| glucose dehydrogenase, EC 1.1.5.2 | PQQ | see above | quinone | |
| cellobiose dehydrogenase, EC 1.1.99.18 | FAD, heme | see above | acceptor | |
| fructose | fructose dehydrogenase, EC 1.1.99.11 | FAD, heme | fructose → 5-dehydrofructose + 2H+ + 2e− | acceptor |
| cellobiose | cellobiose dehydrogenase, EC 1.1.99.18 | FAD, heme | cellobiose → cellobiono-1,5-lactone + 2H+ + 2e− | acceptor |
| lactose | cellobiose dehydrogenase, EC 1.1.99.18 | FAD, heme | lactose → 4-O-(galactopyranosyl)-glucono-1,5-lactone + 2H+ + 2e− | acceptor |
| methanol | alcohol dehydrogenase*, EC 1.1.1.1 | NAD | alcohol → aldehyde + 2H+ + 2e− | NAD |
| aldehyde dehydrogenase*, EC 1.2.1.5 | NAD | aldehyde + H2O → acid + 2H+ + 2e− | NAD | |
| formate dehydrogenase*, EC 1.2.1.2 | NAD | formate → CO2 + 2H+ + 2e− | NAD | |
| alcohol dehydrogenase, EC 1.1.99.8 | PQQ, heme | alcohol → aldehyde + 2H+ + 2e− | acceptor | |
| ethanol | alcohol dehydrogenase*, EC 1.1.1.1 | NAD | see above | see above |
| aldehyde dehydrogenase*, EC 1.2.1.5 | NAD | see above | see above | |
| alcohol dehydrogenase, EC 1.1.99.8 | PQQ, heme | see above | see above | |
| glycerol | alcohol dehydrogenase*, - | PQQ, heme | alcohol → aldehyde + 2H+ + 2e− | - |
| aldehyde dehydrogenase*, - | PQQ, heme | aldehyde + H2O → acid + + 2H+ + 2e− | - | |
| oxalate oxidase*, EC 1.2.3.4 | FAD, Mn | oxalate → 2CO2 + 2H+ + 2e− | O2 | |
| pyruvate | pyruvate dehydrogenase*, EC 1.2.4.1 | NAD | pyruvate + CoA→ acetylCoA + 2H+ + 2e− | NAD |
| hydrogen | membrane-bound hydrogenase, - | - | H2 → 2H+ + 2e− | - |
4.2. Oxidants
| Oxidant | Enzyme | Metal/ Co-factor | Half-cell Reaction |
|---|---|---|---|
| oxygen | laccase, EC 1.10.3.2 | Cu | O2 + 4H+ + 4e− → 2H2O |
| bilirubin oxidase, EC 1.3.3.5 | Cu | see above | |
| cytochrome oxidase, EC 1.9.3.1 | Cu, Fe / heme | see above | |
| cytochrome c, - | Fe / heme | - | |
| hydrogen peroxide | microperoxidase-11, - | Fe / heme | H2O2 + 2H+ + 2e− → 2H2O |
| horseradish peroxidase, EC 1.11.1.7 | Fe / heme | see above | |
| (glucose, GOx) microperoxidase-8, - | Fe / heme | see above | |
| cumene peroxide | microperoxidase-11, - | Fe / heme | C9H12O2 + 2H+ + 2e− → C9H12O + H2O |
4.3. Enzymes
4.3.1. Enzymes for the anodic reaction.
4.3.2. Enzymes for the cathodic reaction.
5. Typical Enzymatic Electrode Assemblies
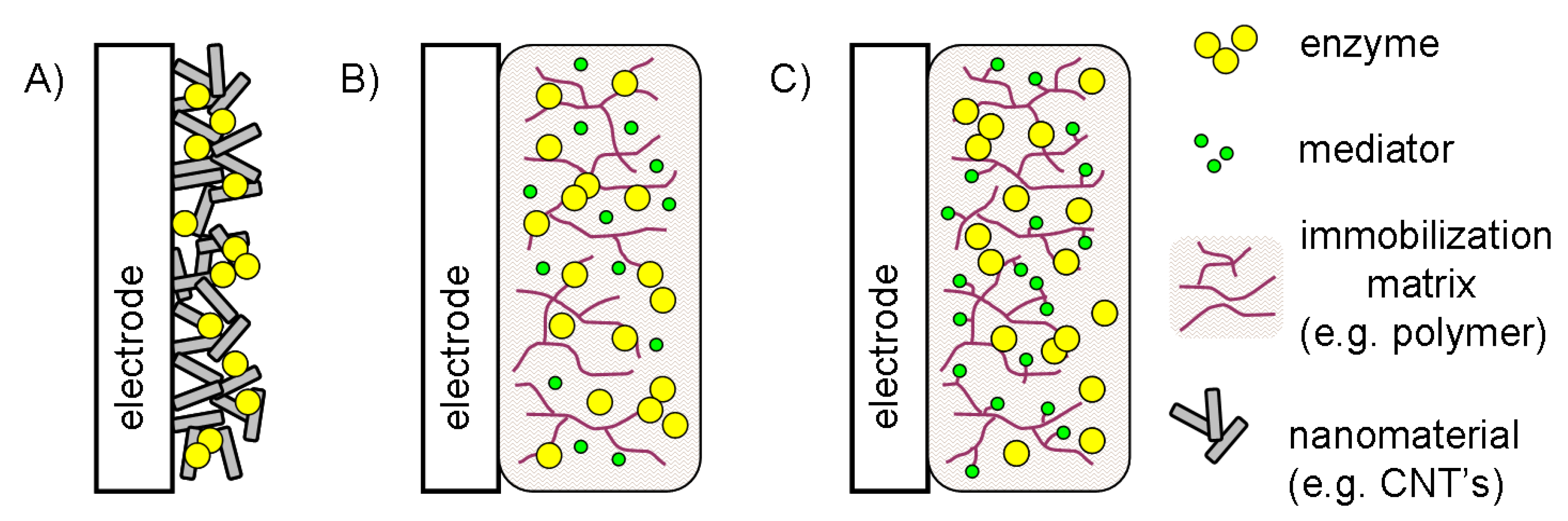
5.1. Electron transfer as a factor determining the electrode architecture
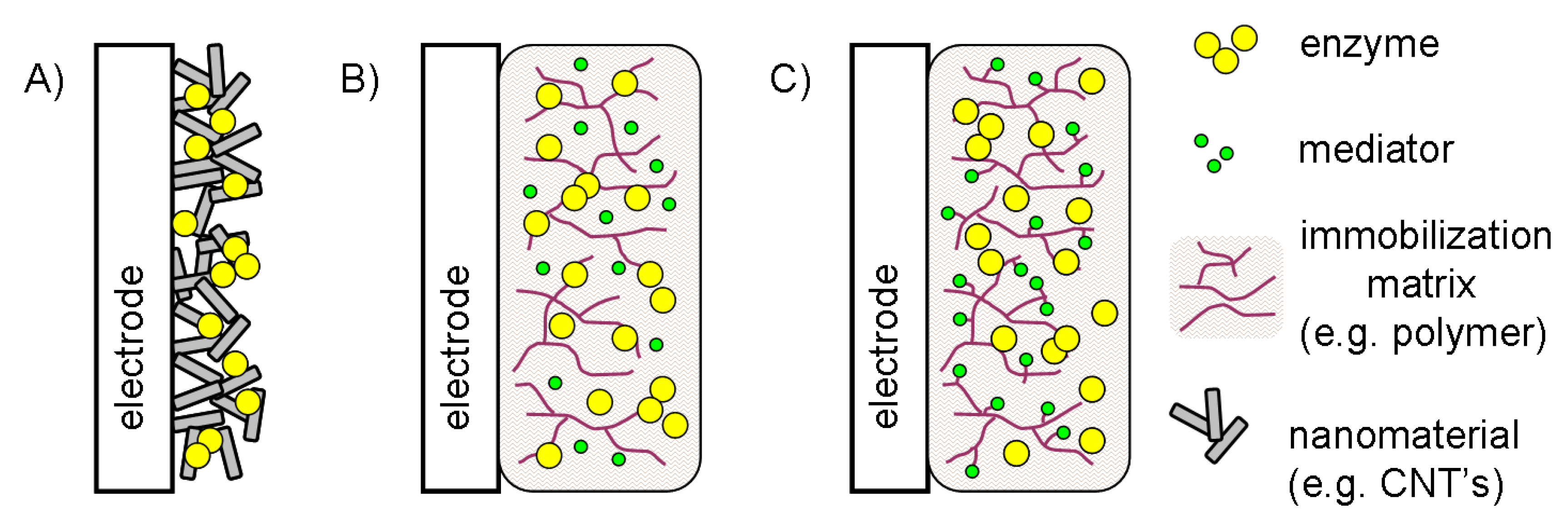
5.2. Configurations without electron transfer mediator
5.3. Configurations based on diffusional and immobilized mediators
5.3.1. Typical mediators employed in enzymatic fuel cells.
| Mediator | Redox Potential, V | Recalculated Redox Potential, V vs. SHE | pH | Reference |
|---|---|---|---|---|
| ferrocene monocarboxylic acid | ~0.34 Ag/AgCl | ~0.54 | 7 | [58] |
| p-benzoquinone | ~0.02 SCE | ~0.26 | 7 | [22] |
| phenazine methosufate | 0.08 SCE | 0.32 | 6 | [63] |
| pyrroloquinoline quinone | –0.13 SCE | 0.11 | 7 | [37] |
| 8-hydroxyquinoline-5-sulfonic acid | 0.065 SCE | 0.305 | 5 | [64] |
| tetrathiafulvalene | 0.222 Ag/AgCl | 0.419 | 7 | [65] |
| Mediator | Redox Potential, V | Recalculated Redox Potential, V vs. SHE | pH | Reference |
|---|---|---|---|---|
| poly(methylene blue) | –0.10 Ag/AgCl | 0.1 | 6 | [66] |
| poly(brilliant cresyl blue) | –0.11 Ag/AgCl | 0.09 | 7 | [62] |
| methylene green* | –0.20/–0.05 Ag/AgCl | 0/0.15 | 6 | [56] |
| Meldola blue | - | - | - | [67] |
| Nile blue | –0.35 Ag/AgCl | –0.15 | 7 | [68] |
| Thionine | - | - | - | [57] |
| Fuel | Mediator | Redox Potential, V | Recalculated Redox Potential, V vs. SHE | pH | Reference |
|---|---|---|---|---|---|
| methanol | benzylviologen | –0.55 SCE | –0.31 | 7.5 | [30] |
| tetramethyl-p-phenylenediamine | –0.055/0.037 SCE | 0.185/0.277 | 7/10 | [24] | |
| ethanol | Nile blue | –0.35 Ag/AgCl | –0.15 | 7 | [68] |
| poly(methylene green) | - | - | - | [44] | |
| glycerol | poly(methylene green) | - | - | - | [23] |
| pyruvate | poly(methylene green) | - | - | - | [45] |
5.3.2. Architectures with diffusional mediators.
5.3.3. Architectures with immobilized mediators.
5.4. Mediators attached to a polymer backbone
| Fuel/Oxidant | Mediator | Redox Potential, V | Recalculated Redox Potential, V vs. SHE | pH | Reference | |
|---|---|---|---|---|---|---|
| glucose | Os polymer | –0.19 Ag/AgCl | 0.01 | 5 | [80] | |
| –0.19 Ag/AgCl | 0.01 | 7 | [86] | |||
| 0.095 Ag/AgCl | 0.292 | 5 | [81] | |||
| –0.16 Ag/AgCl | 0.04 | 7 | [85] | |||
| –0.11 Ag/AgCl | 0.09 | 5 | [87] | |||
| poly(vinylferrocene) | 0.30 Ag/AgCl | 0.50 | 7 | [25] | ||
| 2-methyl-1,4-naphtoquinone (vitamin K3) | on PLL | –0.27 Ag/AgCl | –0.07 | 7 | [20] | |
| on PAAm | –0.25 Ag/AgCl | –0.05 | 7 | [28] | ||
| lactose | Os polymer | 0.15 Ag/AgCl | 0.35 | - | [88] | |
| oxygen | Os polymer | 0.36 Ag/AgCl | 0.56 | 7 | [86] | |
| 0.55 Ag/AgCl | 0.75 | 5 | [79] | |||

6. Kinetics of Bioelectrochemical Reactions on Biofuel Cell Electrodes
6.1. Anode reaction
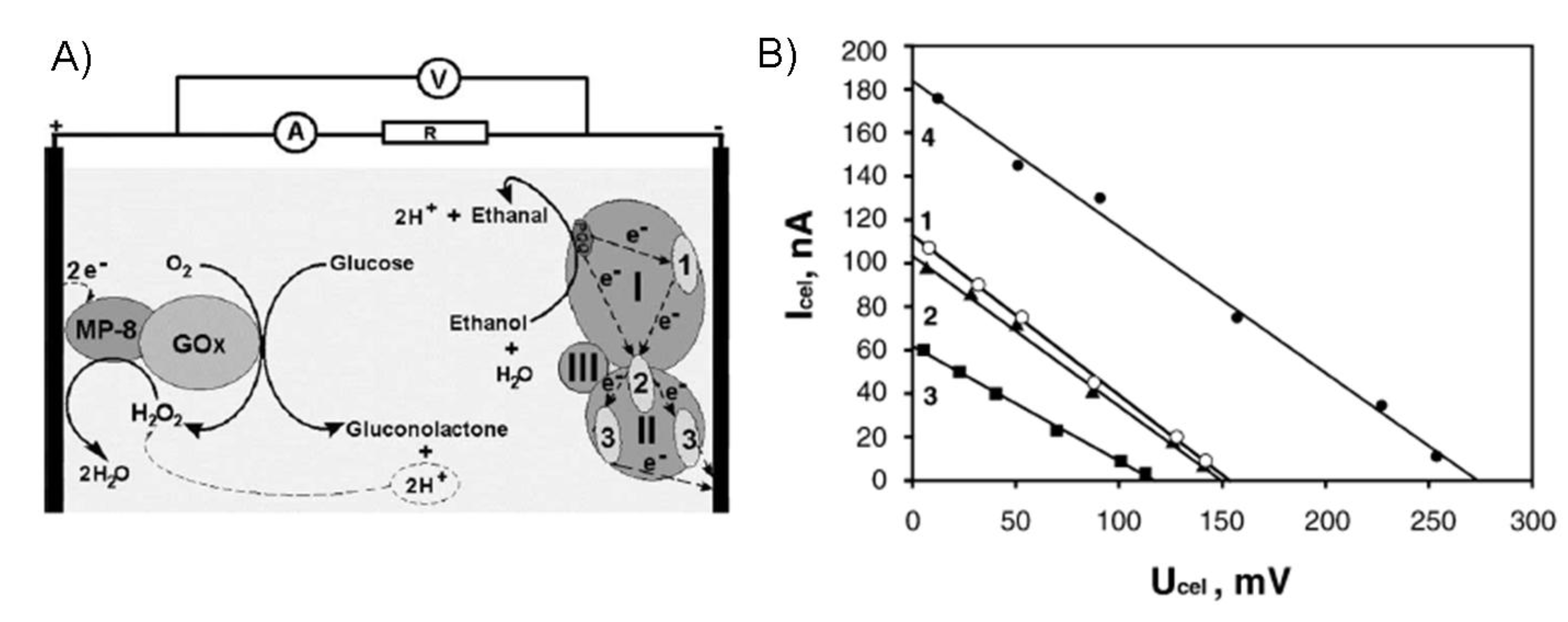
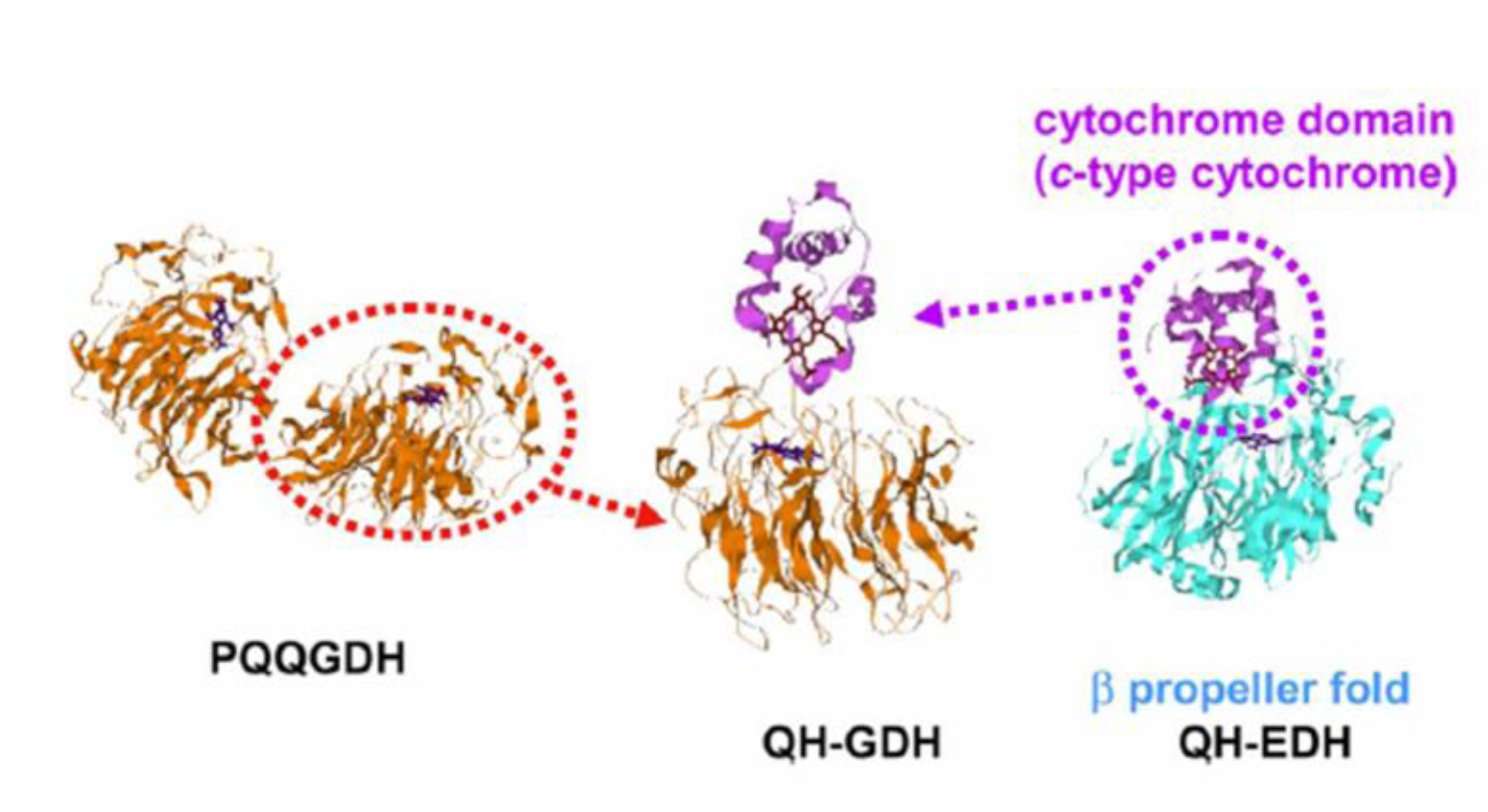
6.1.1. Bioelectrochemical oxidation of glucose.
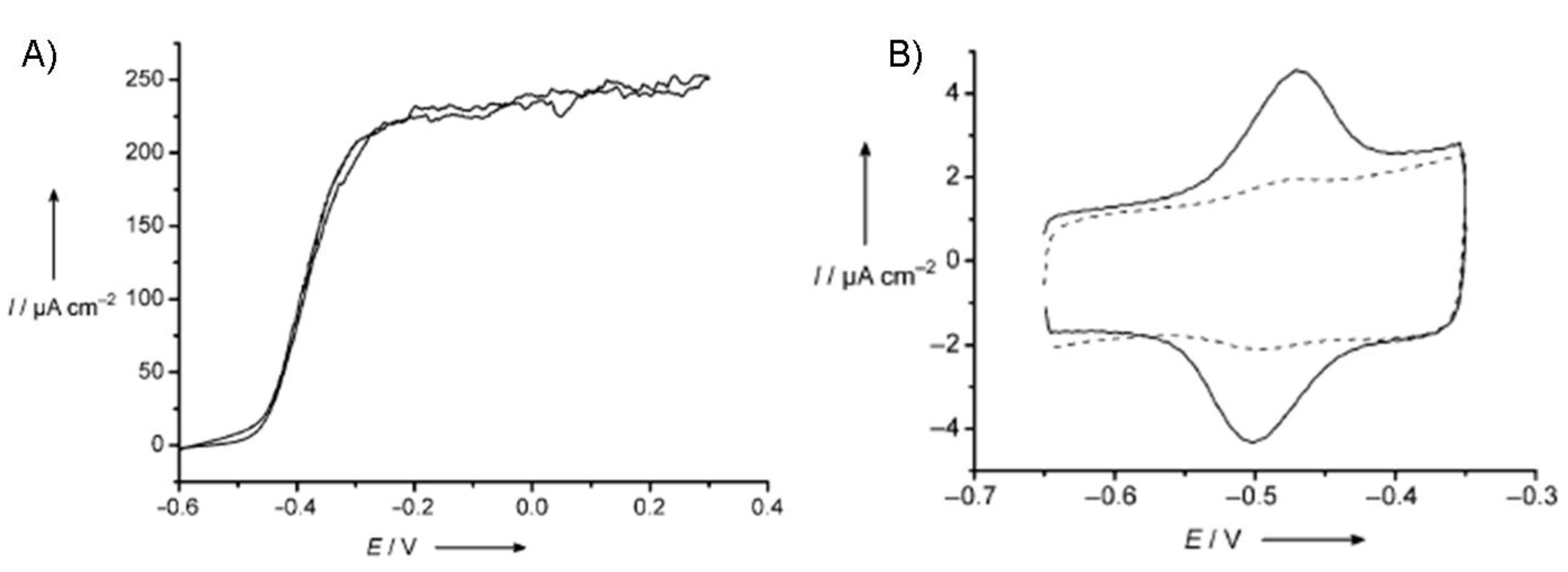
6.1.2. Bioelectrochemical oxidation of other fuels.
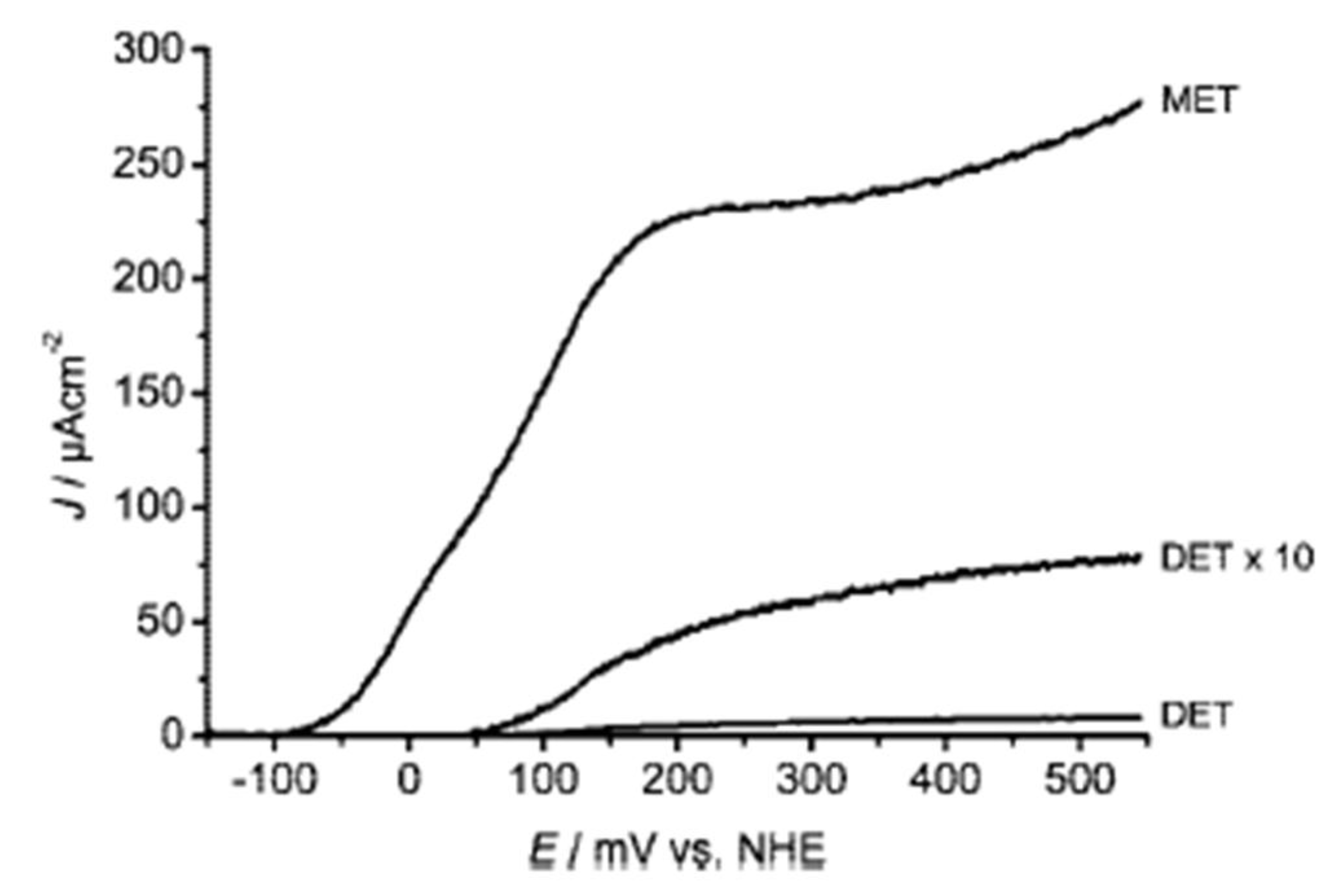
6.2. Cathode reaction
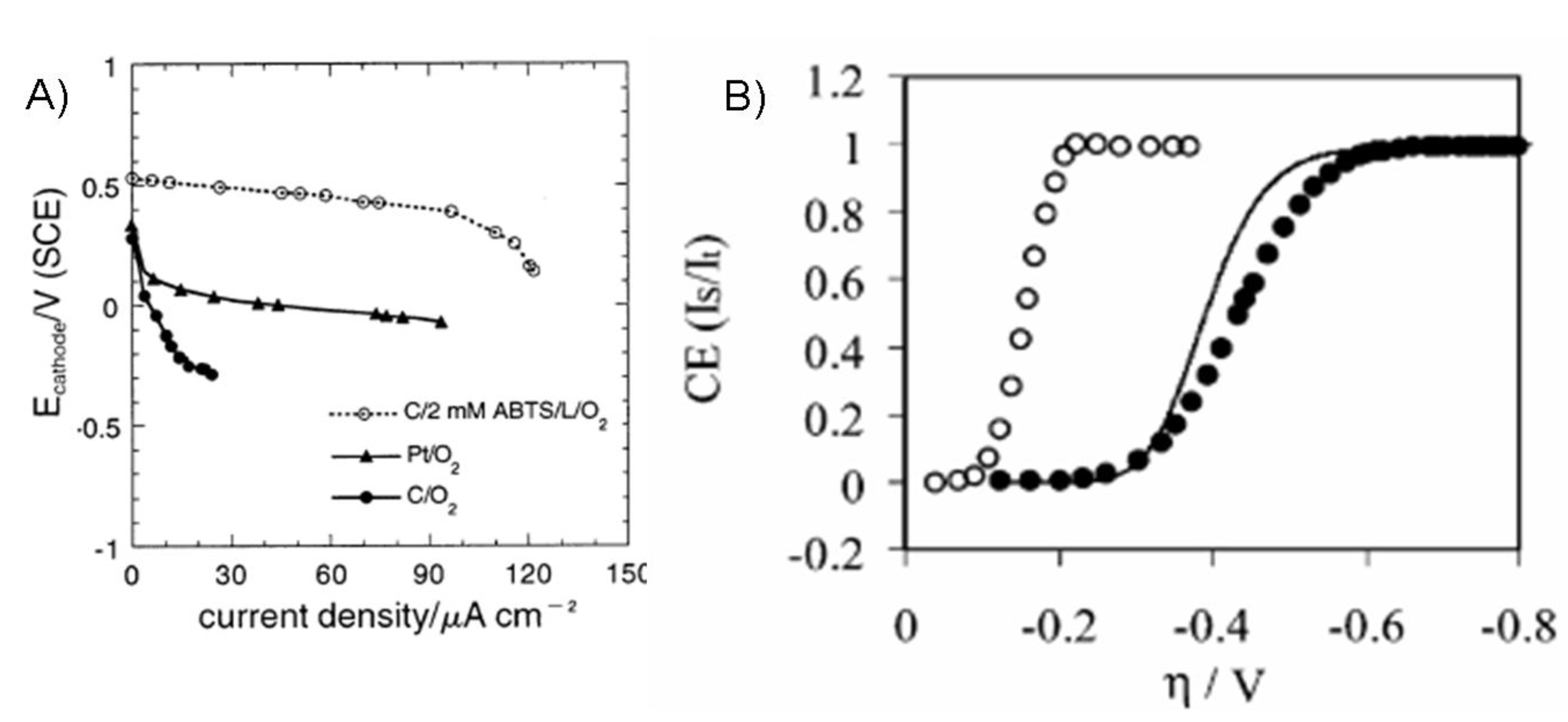
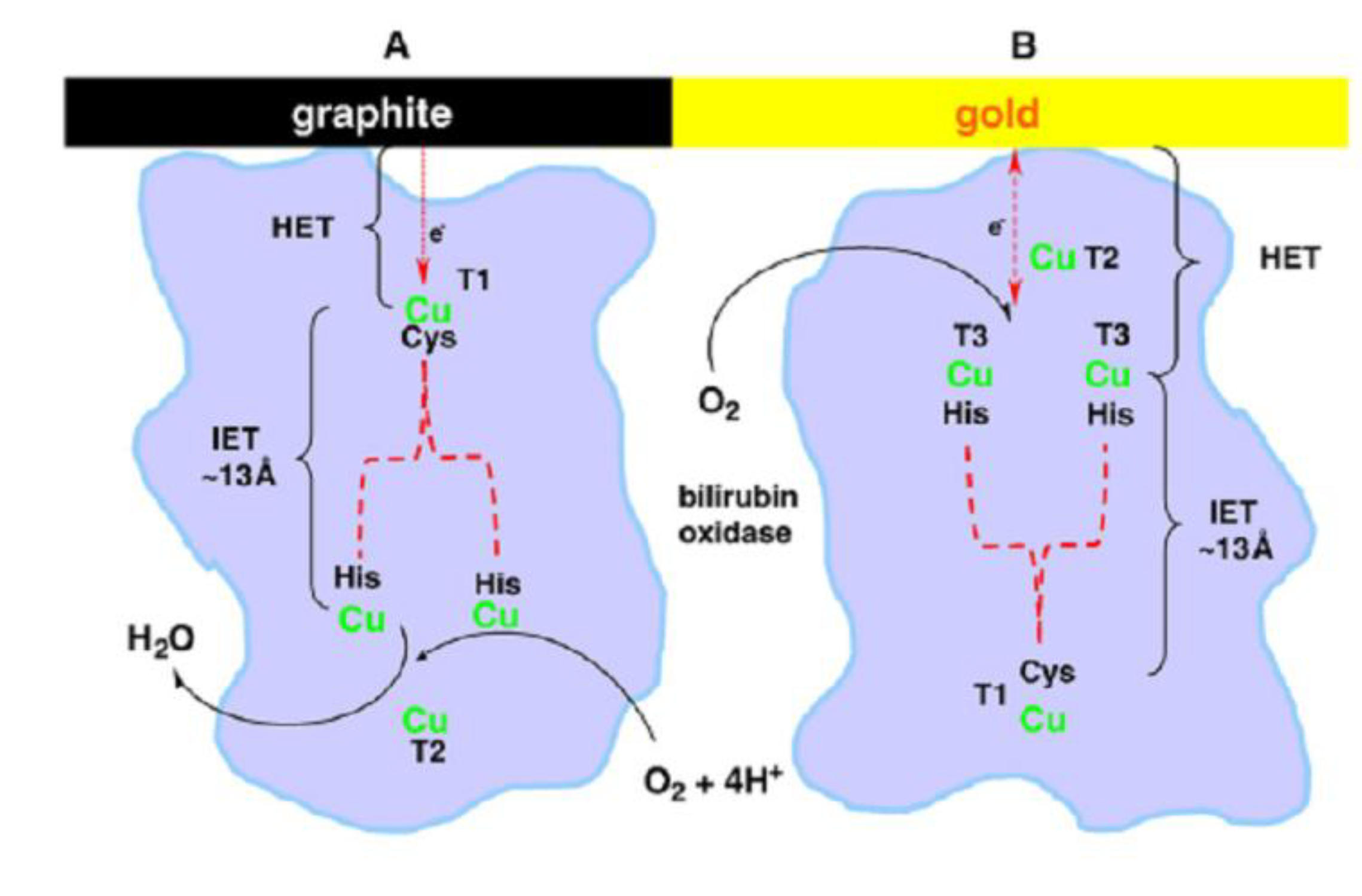
7. Fuel Cell Configurations and Performance
7.1. Biofuel cells based on glucose as a fuel and GOx as the biocatalyst
7.1.1. Mediators entrapped in different matrices.
| Fuel/Oxidant | Enzymes Anode/Cathode | Mediators Anode/Cathode | Power Density, μW cm−2 | Fuel Concentration, mM | Reference |
|---|---|---|---|---|---|
| glucose/O2 | GOx/COx, cytochrome c | PQQ/- | 5 | 1 | [49] |
| glucose/O2 | GOx/laccase | ferrocene/- | 15.8 | 10 | [54] |
| glucose/O2 | GOx/BOD | HQS/ABTS | 42 | 10 | [76] |
| glucose/O2 | GOx/laccase | TTF/ABTS | 7 | 15 | [65] |
| glucose/O2 | GOx/laccase | Os polymer/Os polymer | 137-350 | 15 | [79-81] |
| glucose/O2 | GOx/BOD | Os polymer/Os polymer | 50-480 | 15 | [82-85] |
| glucose/H2O2 | GOx/MP-11 | PQQ/- | 160 | 1 | [37] |
| glucose/H2O2 | GOx/HRP | ferrocene/ferrocene | 0.15 | 1 | [50] |
| glucose/O2 | GDH/laccase | azine dyes/- | 58/38.7 | 45/60 | [56,67] |
| glucose/O2 | GDH/BOD | azine dyes/- | 52/53.9 | 40 | [59,62] |
| glucose/O2 | GDH/BOD | VK3/ferricyanide | 1450 | 400 | [32] |
| glucose/O2 | CDH/- | Os polymer/- | 157 | 100 | [18] |
| lactose/O2 | CDH/laccase | Os polymer/Os polymer | 1.9 | 34 | [88] |
| fructose/O2 | FDH/laccase | -/- | 850 | 200 | [43] |
| fructose/O2 | FDH/BOD | -/- | 126 | 200 | [42] |
| methanol/O2 | ADH/- | benzylviologen/- | 670 | 100 | [30] |
| ethanol/(H2O2) | -/- | 1.5 | 25 | [46] | |
| ethanol/O2 | poly(methylene green)/Ru(bpy)3 | 460 | 1 | [69] | |
| glycerol/O2 | poly(methylene green)/- | 1320 | 100 | [47] | |
| pyruvate/O2 | poly(methylene green)/- | 930 | 100 | [45] |
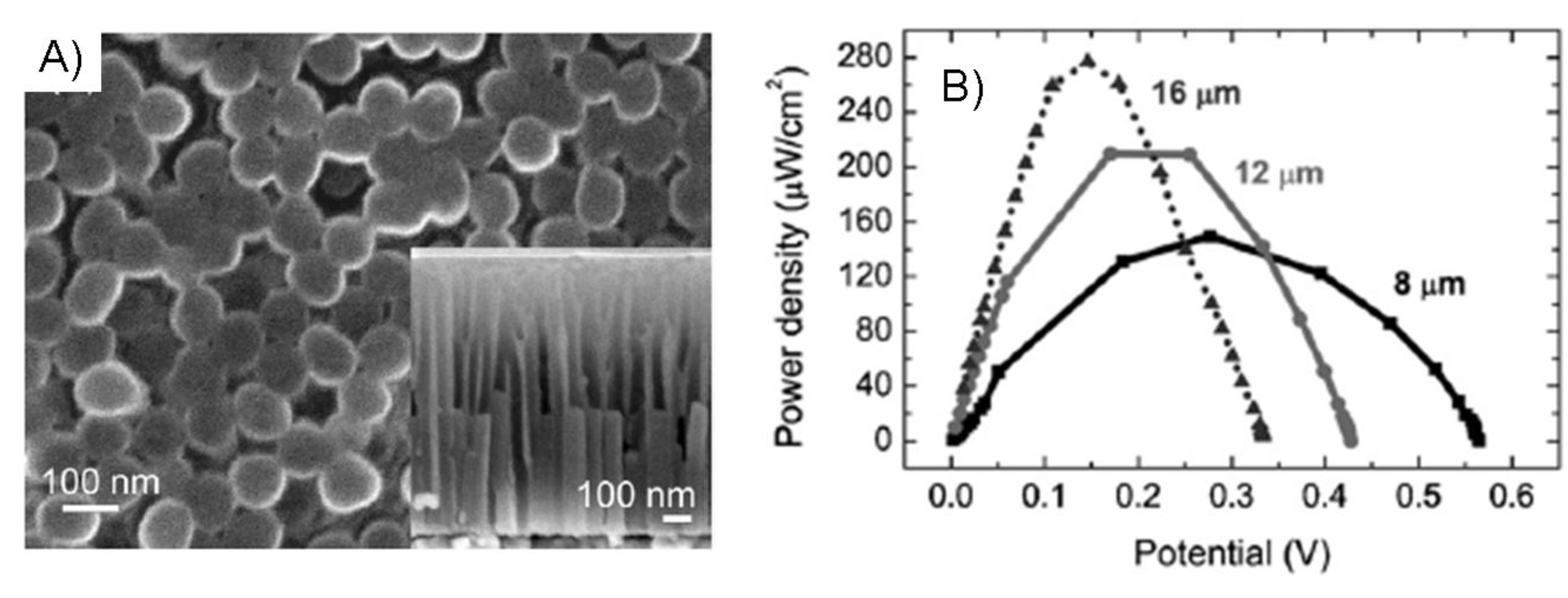
7.1.2. Mediators attached to a polymer backbone
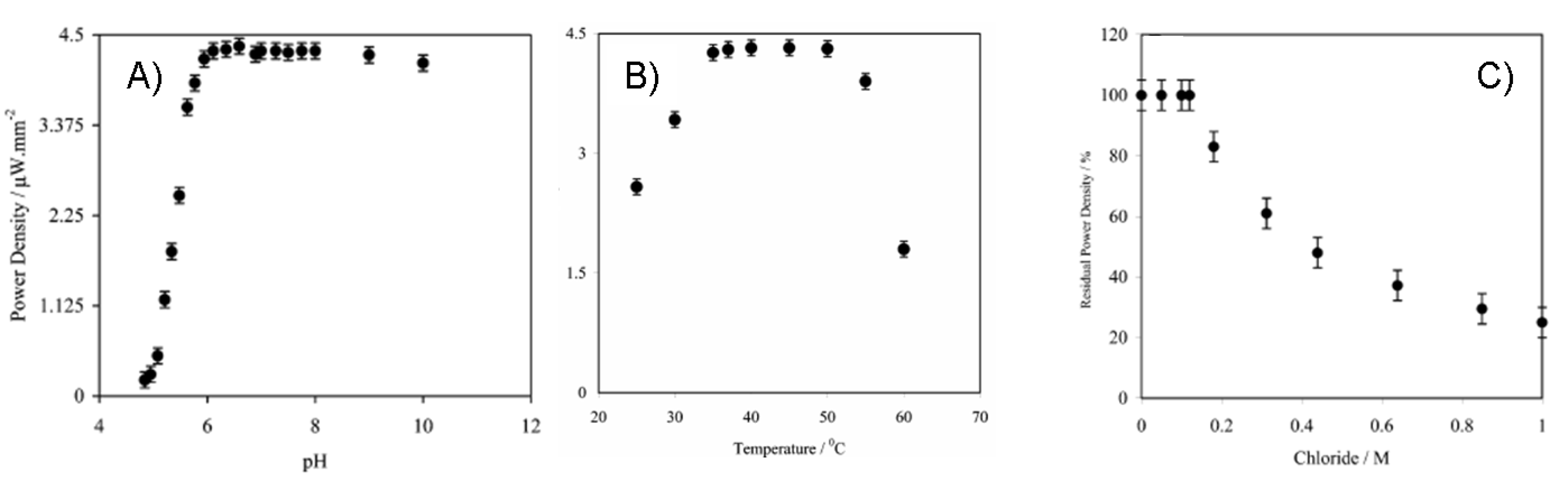
7.2. Biofuel cells based on glucose as a fuel and GDH and CDH as biocatalysts
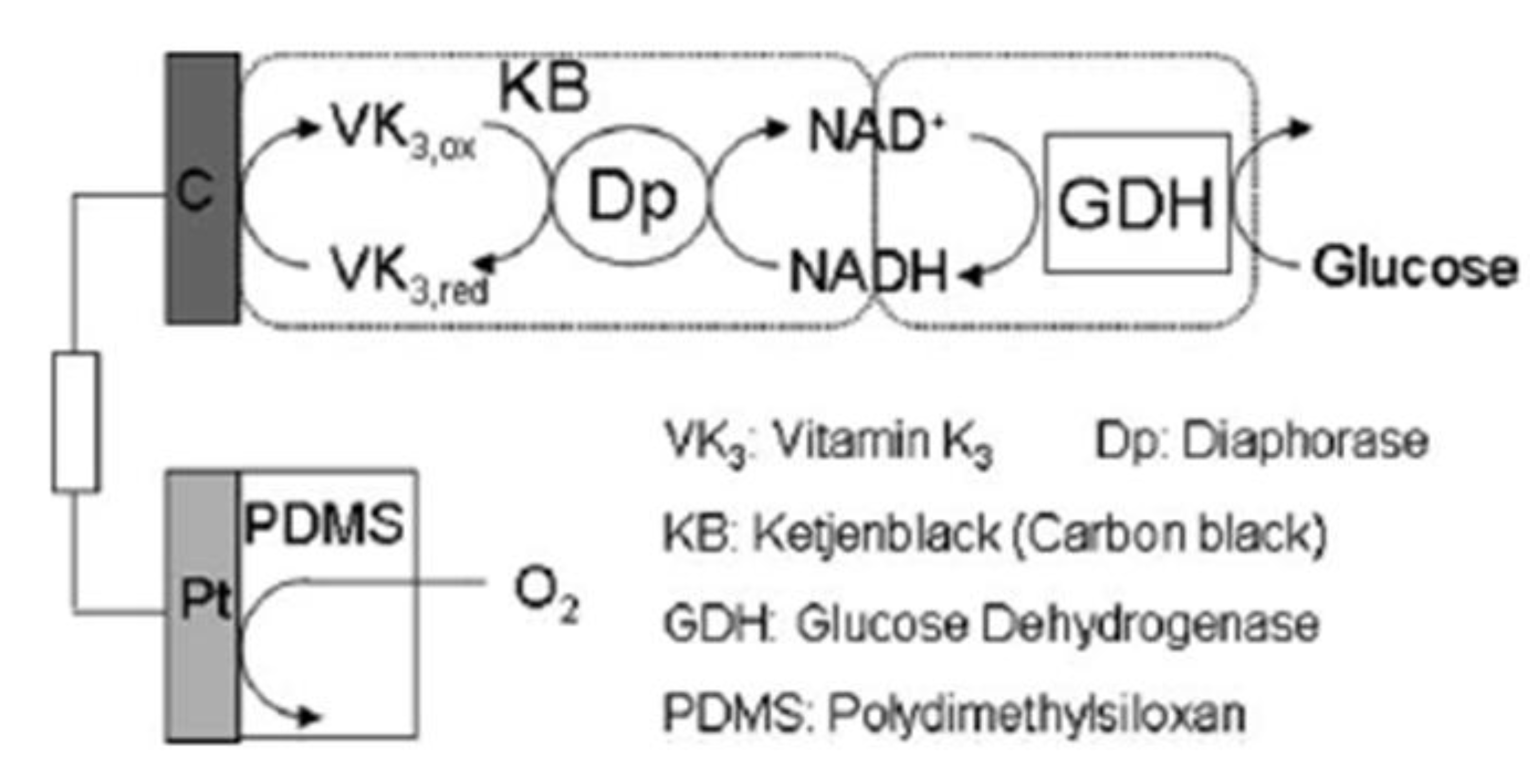
7.3. Biofuel cells based on fuels other than glucose
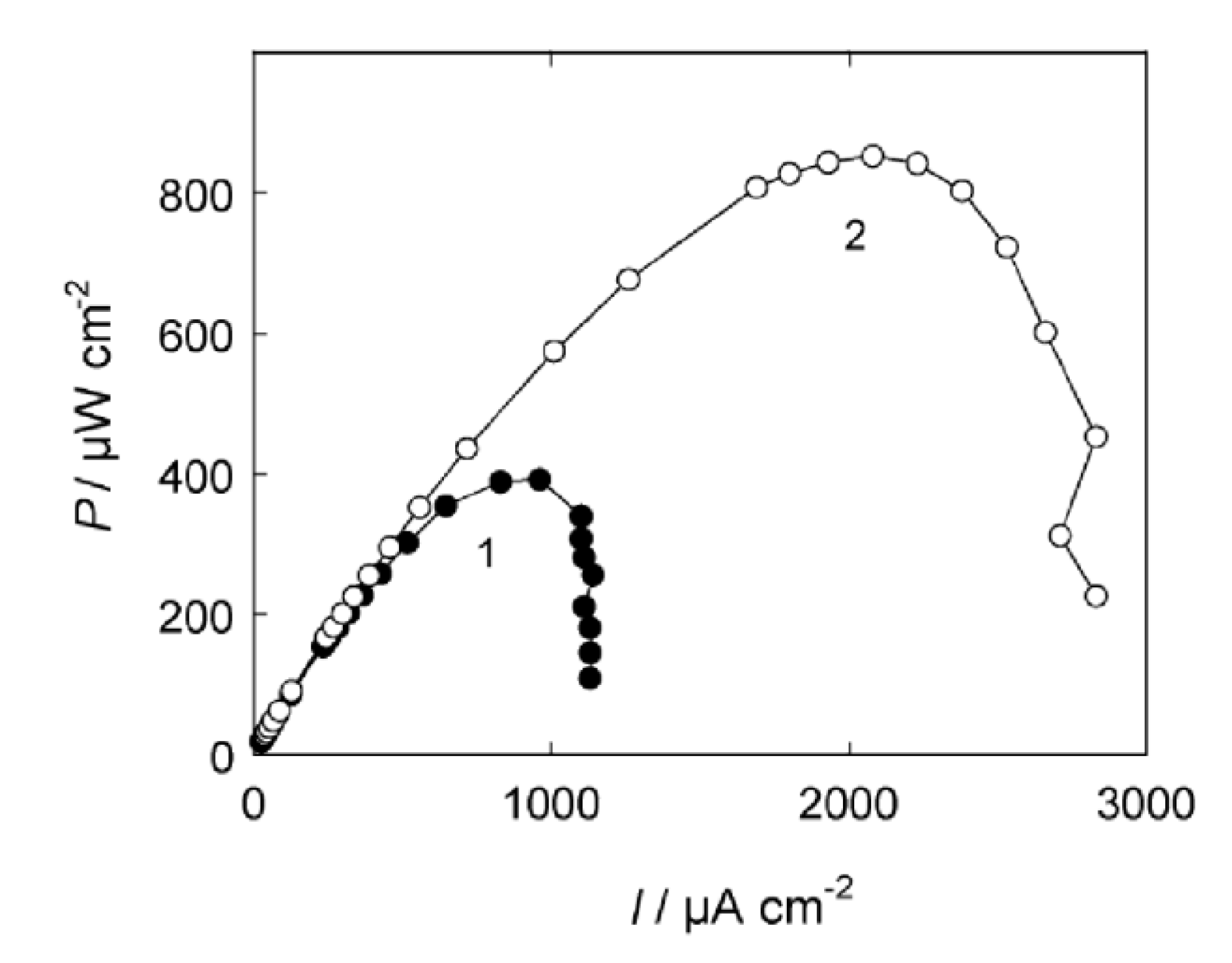
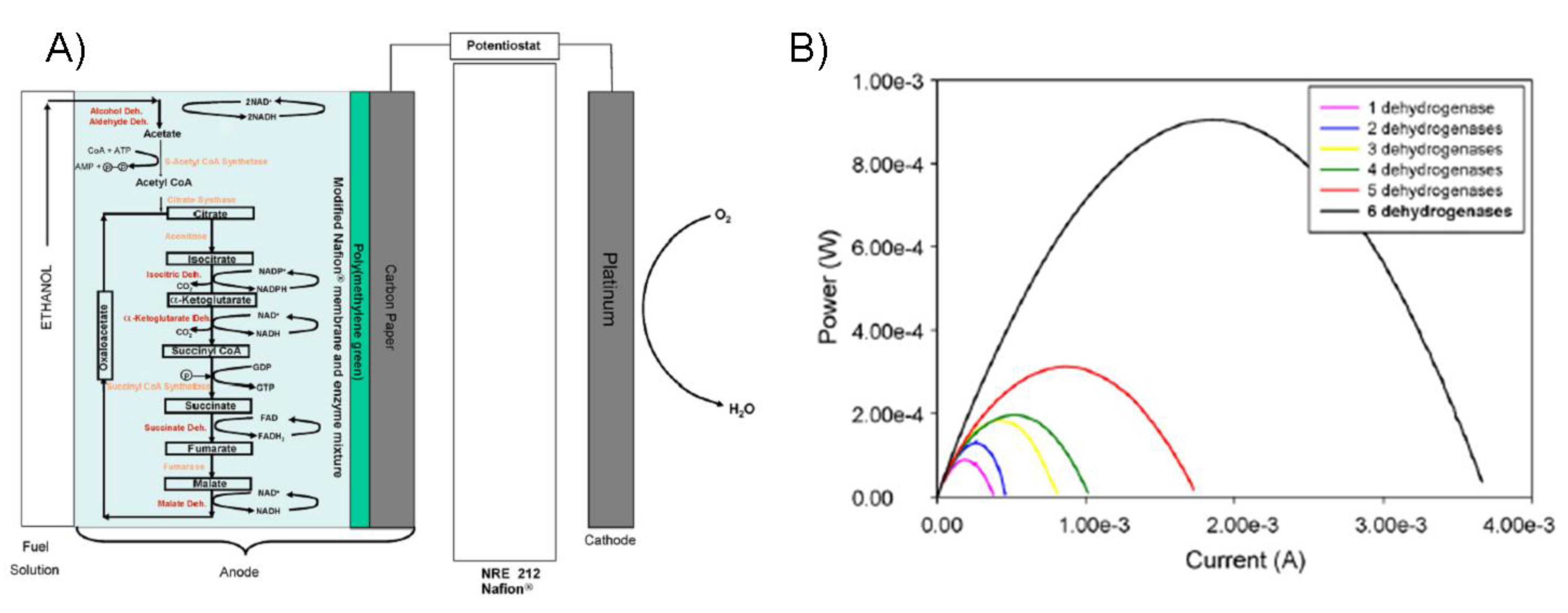
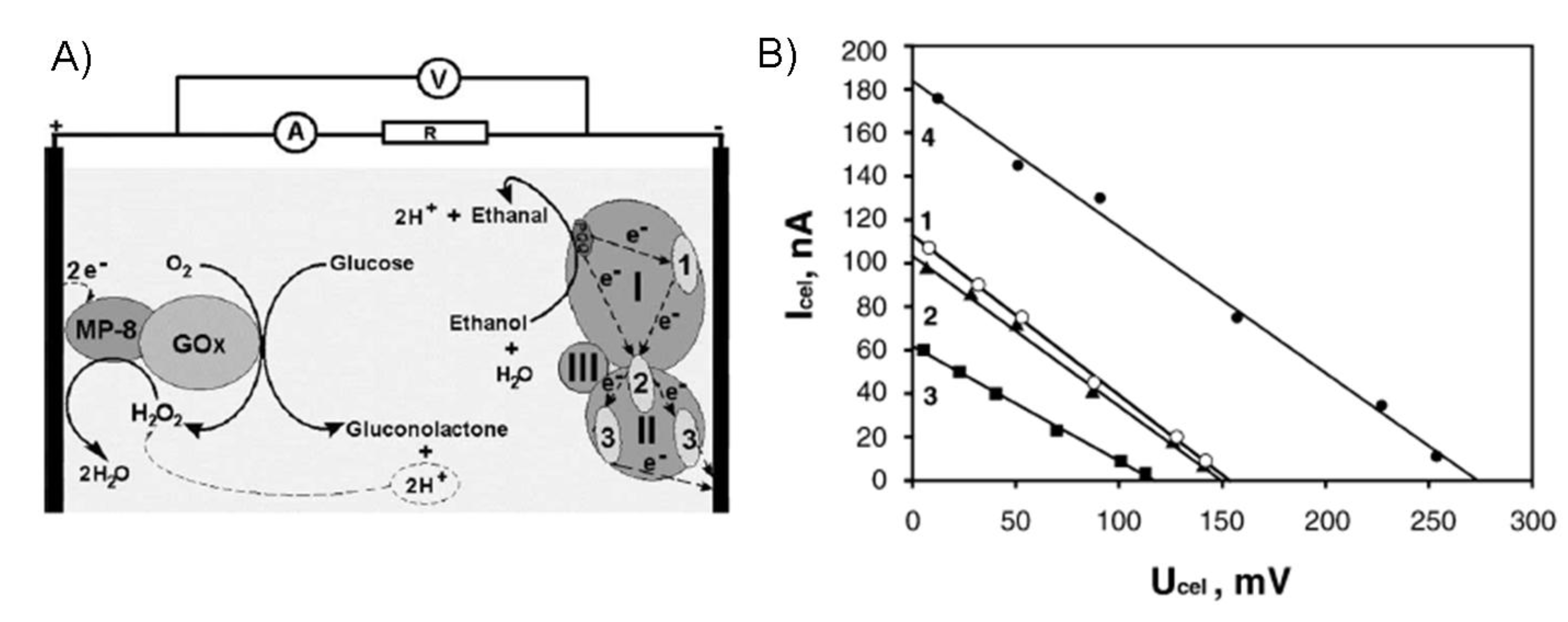
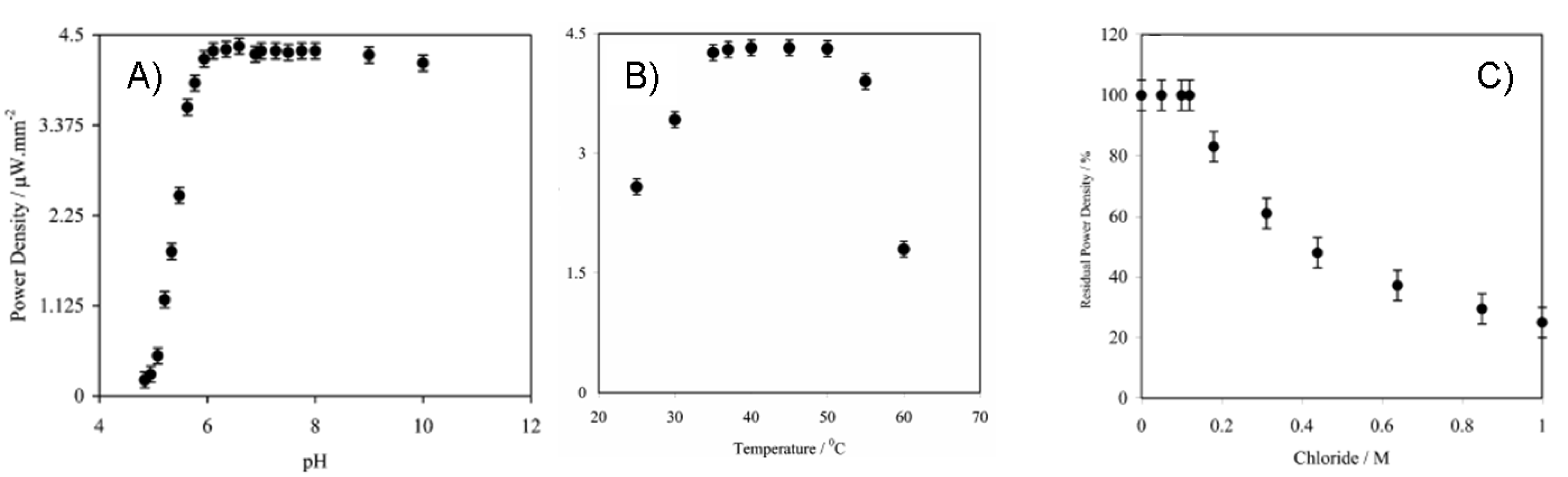
7.4. Long-term stability testing
8. Typical Designs of Enzymatic Fuel Cells
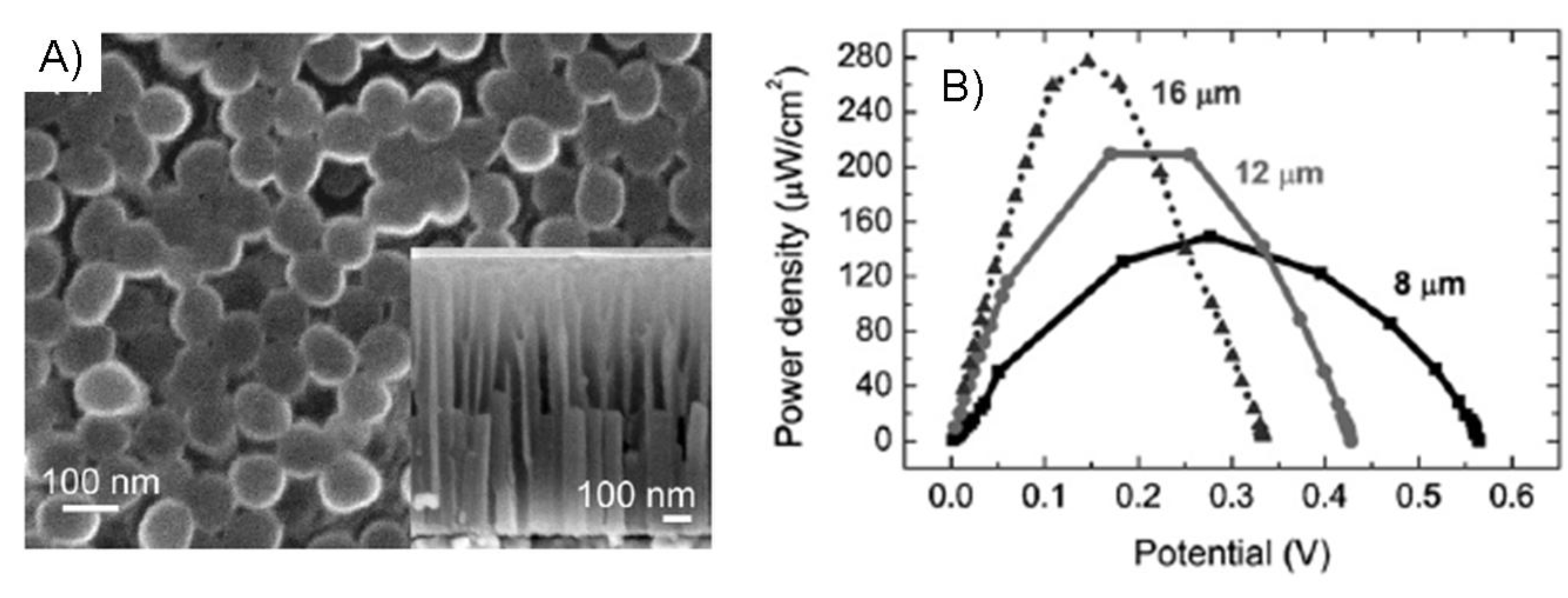
8.1. Microfluidic systems
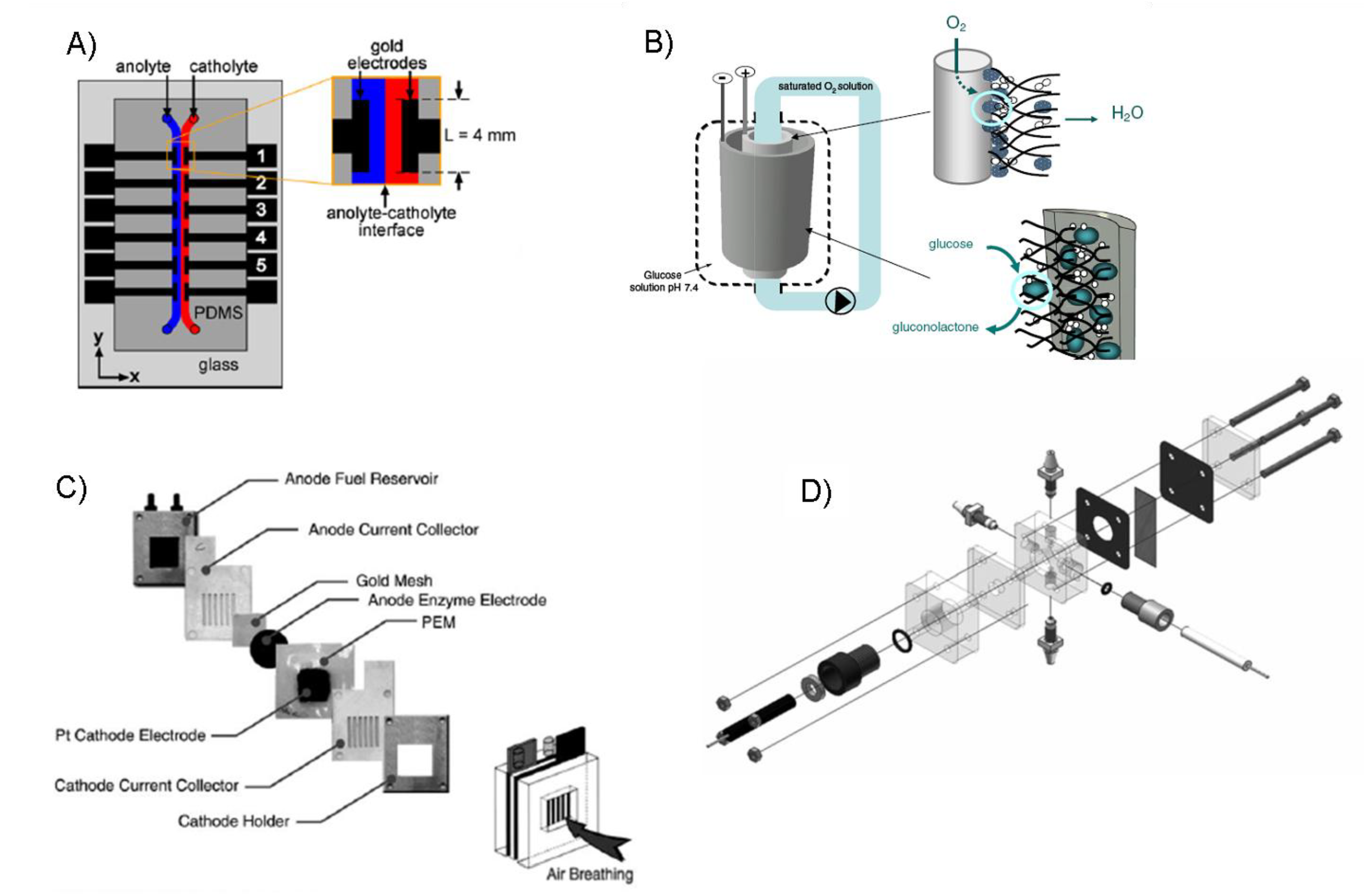
8.2. Concentric biofuel cell
8.3. Designs using MEA’s
8.4. Air-breathing cathodes
8.5. Modular stack cell
9. Modeling of Enzymatic Biofuel Cells
9.1. Complete fuel cell modeling
9.2. Modeling of enzymatic electrodes
10. Enzymatic Fuel Cells Optimization
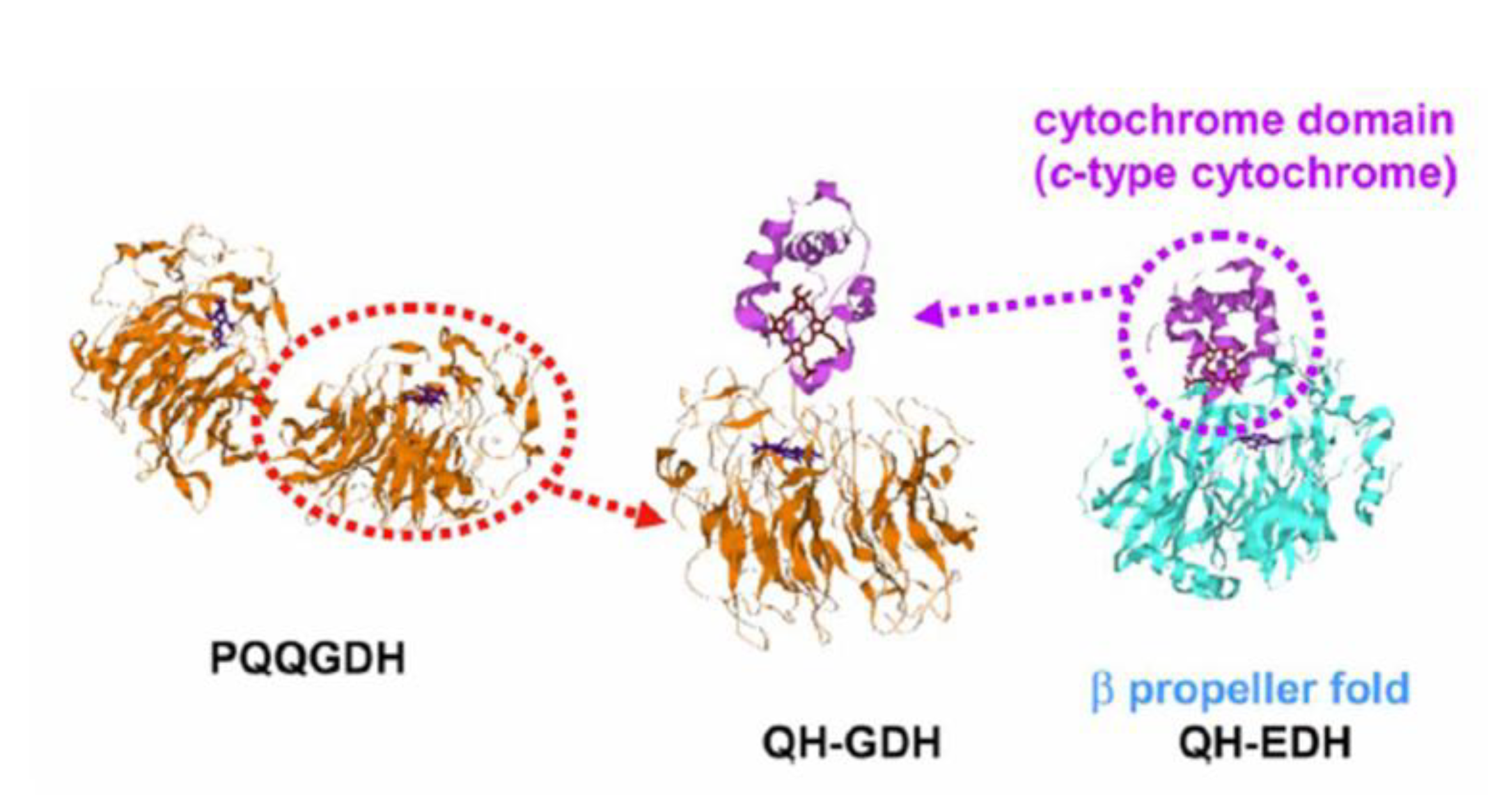
11. Summary
References:
- Cooney, M.J.; Svoboda, V.; Lau, C.; Martin, G.; Minteer, S.D. Enzyme catalysed biofuel cells. Energy Environ. Sci. 2008, 1, 320–337. [Google Scholar] [CrossRef]
- Minteer, S.D.; Liaw, B.Y.; Cooney, M.J. Enzyme-based biofuel cells. Curr. Opin. Biotechnol. 2007, 18, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.M.; Renugopalakrishnan, V.; Filipek, S.; Li, P.; Audette, G.F.; Munukutla, L. Bio-Batteries and Bio-Fuel Cells: Leveraging on Electronic Charge Transfer Proteins. In International Conference on Bionano Science (ICONBS 2007), Taipei, Taiwan, December 2007; pp. 1665–1678.
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef] [PubMed]
- Heller, A. Miniature biofuel cells. Phys. Chem. Chem. Phys. 2004, 6, 209–216. [Google Scholar] [CrossRef]
- Willner, I.; Yan, Y.M.; Willner, B.; Tel-Vered, R. Integrated enzyme-based biofuel cells—A review. Fuel Cells 2009, 9, 7–24. [Google Scholar] [CrossRef]
- Zayats, M.; Willner, B.; Willner, I. Design of amperometric biosensors and biofuel cells by the reconstitution of electrically contacted enzyme electrodes. Electroanalysis 2008, 20, 583–601. [Google Scholar] [CrossRef]
- Barton, S.C.; Gallaway, J.; Atanassov, P. Enzymatic biofuel cells for Implantable and microscale devices. Chem. Rev. 2004, 104, 4867–4886. [Google Scholar] [CrossRef] [PubMed]
- Bullen, R.A.; Arnot, T.C.; Lakeman, J.B.; Walsh, F.C. Biofuel cells and their development. Biosens. Bioelectron. 2006, 21, 2015–2045. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Vonderheit, A.; Wei, X.; Kuttel, C.; Stemmer, A. A novel biofuel cell harvesting energy from activated human macrophages. Biosens. Bioelectron. 2009, 25, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Habrioux, A.; Servat, K.; Tingry, S.; Kokoh, K.B. Enhancement of the performances of a single concentric glucose/O-2 biofuel cell by combination of bilirubin oxidase/nafion cathode and Au-Pt anode. Electrochem. Commun. 2009, 11, 111–113. [Google Scholar] [CrossRef]
- Zheng, W.; Zho, H.M.; Zheng, Y.F.; Wang, N. A comparative study on electrochemistry of laccase at two kinds of carbon nanotubes and its application for biofuel cell. Chem. Phys. Lett. 2008, 457, 381–385. [Google Scholar] [CrossRef]
- Smolander, M.; Boer, H.; Valkiainen, M.; Roozeman, R.; Bergelin, M.; Eriksson, J.E.; Zhang, X.C.; Koivula, A.; Viikari, L. Development of a printable laccase-based biocathode for fuel cell applications. In Proceedings of the 10th International Congress on Biotechnology in Pulp and Paper, Madison, WI, USA, June 2007; pp. 93–102.
- Habrioux, A.; Sibert, E.; Servat, K.; Vogel, W.; Kokoh, K.B.; Alonso-Vante, N. Activity of platinum-gold alloys for glucose electrooxidation in biofuel cells. J. Phys. Chem. B 2007, 111, 10329–10333. [Google Scholar] [CrossRef] [PubMed]
- Colmati, F.; Yoshioka, S.A.; Silva, V.; Varela, H.; Gonzalez, E.R. Enzymatic based biocathode in a polymer electrolyte membrane fuel cell. Int. J. Electrochem. Sci. 2007, 2, 195–202. [Google Scholar]
- Hudak, N.S.; Barton, S.C. Mediated biocatalytic cathode for direct methanol membrane-electrode assemblies. J. Electrochem. Soc. 2005, 152, A876–A881. [Google Scholar] [CrossRef]
- Palmore, G.T.R.; Kim, H.H. Electro-enzymatic reduction of dioxygen to water in the cathode compartment of a biofuel cell. J. Electroanal. Chem. 1999, 464, 110–117. [Google Scholar] [CrossRef]
- Tasca, F.; Gorton, L.; Harreither, W.; Haltrich, D.; Ludwig, R.; Noll, G. Highly efficient and versatile anodes for biofuel cells based on cellobiose dehydrogenase from Myriococcum thermophilum. J. Phys. Chem. C 2008, 112, 13668–13673. [Google Scholar] [CrossRef]
- Muguruma, H. Biofuel cell based on a complex between glucose oxidase and a plasma-polymerized film containing a redox site. IEICE Trans. Electron. 2008, E91C, 1811–1815. [Google Scholar] [CrossRef]
- Togo, M.; Takamura, A.; Asai, T.; Kaji, H.; Nishizawa, M. An enzyme-based microfluidic biofuel cell using vitamin K-3-mediated glucose oxidation. Electrochim. Acta 2007, 52, 4669–4674. [Google Scholar] [CrossRef]
- Okuda, J.; Yamazaki, T.; Fukasawa, M.; Kakehi, N.; Sode, K. The application of engineered glucose dehydrogenase to a direct electron-transfer-type continuous glucose monitoring system and a compartmentless biofuel cell. Anal. Lett. 2007, 40, 431–440. [Google Scholar] [CrossRef]
- Kuwahara, T.; Oshima, K.; Shimomura, M.; Miyauchi, S. Properties of the enzyme electrode fabricated with a film of polythiophene derivative and its application to a glucose fuel cell. J. Appl. Polym. Sci. 2007, 104, 2947–2953. [Google Scholar] [CrossRef]
- Arechederra, R.L.; Treu, B.L.; Minteer, S.D. Development of glycerol/O-2 biofuel cell. J. Power Sources 2007, 173, 156–161. [Google Scholar] [CrossRef]
- Zhang, X.C.; Ranta, A.; Halme, A. Direct methanol biocatalytic fuel cell—Considerations of restraints on electron transfer. Biosens. Bioelectron. 2006, 21, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, T.; Yamaguchi, T. High-surface-area three-dimensional biofuel cell electrode using redox-polymer-grafted carbon. Ind. Eng. Chem. Res. 2006, 45, 3050–3058. [Google Scholar] [CrossRef]
- Kakehi, N.; Yamazaki, T.; Tsugawa, W.; Sode, K. A novel wireless glucose sensor employing direct electron transfer principle based enzyme fuel cell. In 9th World Congress on Biosensors, Toronto, Canada, May, 2006; pp. 2250–2255.
- Fischback, M.B.; Youn, J.K.; Zhao, X.Y.; Wang, P.; Park, H.G.; Chang, H.N.; Kim, J.; Ha, S. Miniature biofuel cells with improved stability under continuous operation. Electroanalysis 2006, 18, 2016–2022. [Google Scholar] [CrossRef]
- Sato, F.; Togo, M.; Islam, M.K.; Matsue, T.; Kosuge, J.; Fukasaku, N.; Kurosawa, S.; Nishizawa, M. Enzyme-based glucose fuel cell using Vitamin K-3-immobilized polymer as an electron mediator. Electrochem. Commun. 2005, 7, 643–647. [Google Scholar] [CrossRef]
- Akers, N.L.; Moore, C.M.; Minteer, S.D. Development of alcohol/O-2 biofuel cells using salt-extracted tetrabutylammonium bromide/Nafion membranes to immobilize dehydrogenase enzymes. Electrochim. Acta 2005, 50, 2521–2525. [Google Scholar] [CrossRef]
- Palmore, G.T.R.; Bertschy, H.; Bergens, S.H.; Whitesides, G.M. A methanol/dioxygen biofuel cell that uses NAD(+)-dependent dehydrogenases as catalysts: application of an electro-enzymatic method to regenerate nicotinamide adenine dinucleotide at low overpotentials. J. Electroanal. Chem. 1998, 443, 155–161. [Google Scholar] [CrossRef]
- Choi, Y.K.; Wang, G.; Nayfeh, M.H.; Yau, S.T. A hybrid biofuel cell based on electrooxidation of glucose using ultra-small silicon nanoparticles. Biosens. Bioelectron. 2009, 24, 3103–3107. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Nakagawa, T.; Tokita, Y.; Hatazawa, T.; Ikeda, T.; Tsujimura, S.; Kano, K. A high-power glucose/oxygen biofuel cell operating under quiescent conditions. Energy Environ. Sci. 2009, 2, 133–138. [Google Scholar] [CrossRef]
- Heller, A. Potentially implantable miniature batteries. In Proceedings of the Annual Meeting of the Gesellschaft-Deutscher-Chemiker, Regensburg, Germany, February 2005; pp. 469–473.
- Heller, A. Integrated medical feedback systems for drug delivery. In Meeting on Lessening the Pain and Worry of Diabetic People held at the AICHE Annual Meeting, Austin, TX, USA, November 2004; pp. 1054–1066.
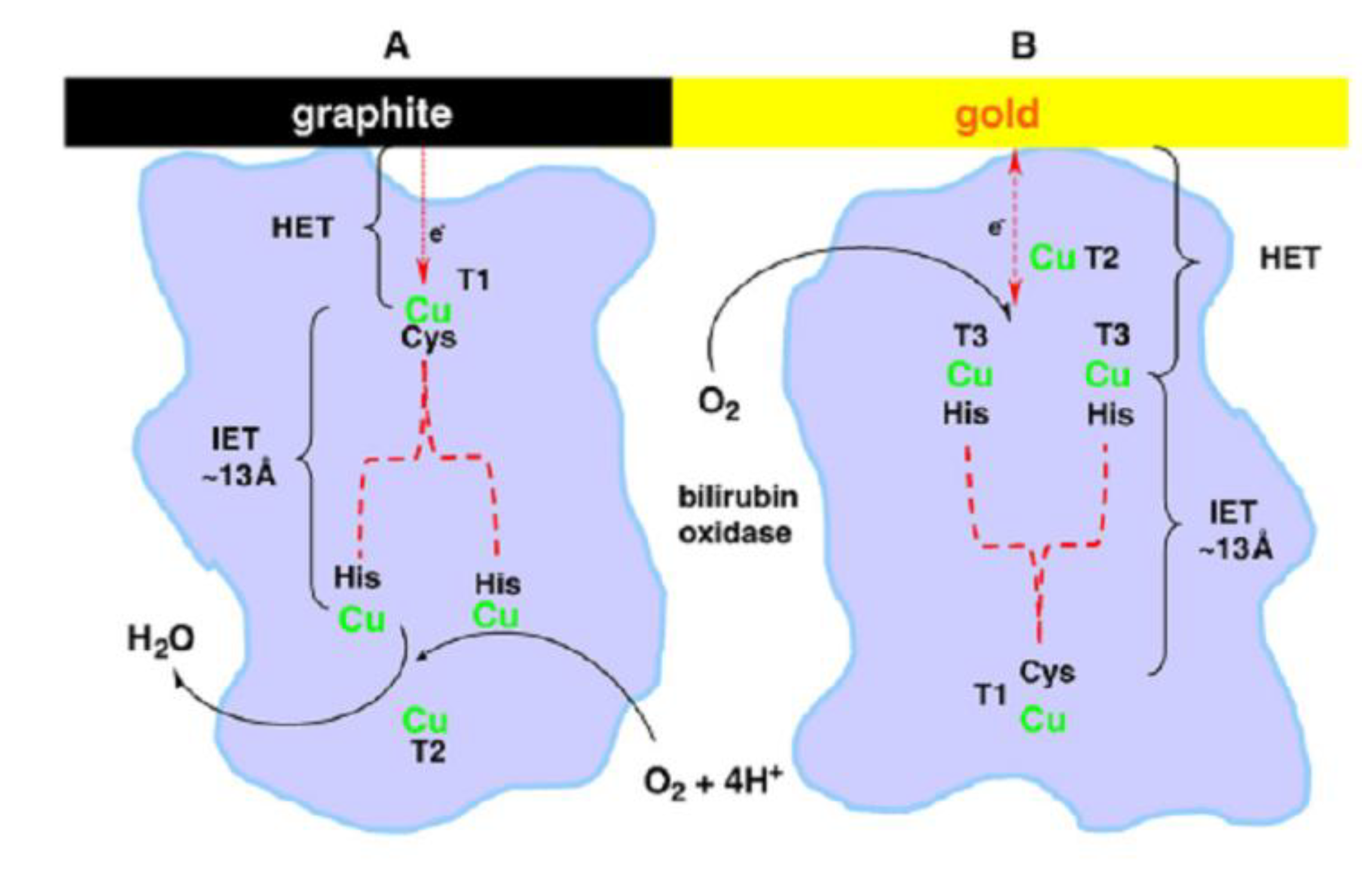
- Ramirez, P.; Mano, N.; Andreu, R.; Ruzgas, T.; Heller, A.; Gorton, L.; Shleev, S. Direct electron transfer from graphite and functionalized gold electrodes to T1 and T2/T3 copper centers of bilirubin oxidase. Biochim. Biophys. Acta, Bioenerg. 2008, 1777, 1364–1369. [Google Scholar] [CrossRef]
- Shleev, S.; Jarosz-Wilkolazka, A.; Khalunina, A.; Morozova, O.; Yaropolov, A.; Ruzgas, T.; Gorton, L. Direct electron transfer reactions of laccases from different origins on carbon electrodes. Bioelectrochemistry 2005, 67, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.; Katz, E.; Patolsky, F.; Buckmann, A.F. Biofuel cell based on glucose oxidase and microperoxidase-11 monolayer-fundionalized electrodes. J. Chem. Soc., Perkin Trans. 1998, 2, 1817–1822. [Google Scholar] [CrossRef]
- Katz, E.; Filanovsky, B.; Willner, I. A biofuel cell based on two immiscible solvents and glucose oxidase and microperoxidase-11 monolayer-functionalized electrodes. New J. Chem. 1999, 23, 481–487. [Google Scholar] [CrossRef]
- Wilson, R.; Turner, A.P.F. Glucose-oxidase - An ideal enzyme. Biosens. Bioelectron. 1992, 7, 165–185. [Google Scholar] [CrossRef]
- Coman, V.; Vaz-Dominguez, C.; Ludwig, R.; Herreither, W.; Haltrich, D.; De Lacey, A.L.; Ruzgas, T.; Gorton, L.; Shleev, S. A membrane-, mediator-, cofactor-less glucose/oxygen biofuel cell. Phys. Chem. Chem. Phys. 2008, 10, 6093–6096. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Kano, K.; Ikeda, T. Glucose/O-2 biofuel cell operating at physiological conditions. Electrochemistry 2002, 70, 940–942. [Google Scholar]
- Wu, X.; Zhao, F.; Varcoe, J.R.; Thumser, A.E.; Avignone-Rossa, C.; Slade, R.C.T. A one-compartment fructose/air biological fuel cell based on direct electron transfer. Biosens. Bioelectron. 2009, 25, 326–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamitaka, Y.; Tsujimura, S.; Setoyama, N.; Kajino, T.; Kano, K. Fructose/dioxygen biofuel cell based on direct electron transfer-type bioelectrocatalysis. Phys. Chem. Chem. Phys. 2007, 9, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Sokic-Lazic, D.; Minteer, S.D. Citric acid cycle biomimic on a carbon electrode. Biosens. Bioelectron. 2008, 24, 939–944. [Google Scholar] [CrossRef]
- Sokic-Lazic, D.; Minteer, S.D. Pyruvate/air enzymatic biofuel cell capable of complete oxidation. Electrochem. Solid-State Lett. 2009, 12, F26–F28. [Google Scholar] [CrossRef]
- Ramanavicius, A.; Kausaite, A.; Ramanaviciene, A. Enzymatic biofuel cell based on anode and cathode powered by ethanol. Biosens. Bioelectron. 2008, 24, 761–766. [Google Scholar] [CrossRef]
- Arechederra, R.L.; Minteer, S.D. Complete oxidation of glycerol in an enzymatic biofuel cell. Fuel Cells 2009, 9, 63–69. [Google Scholar] [CrossRef]
- Vincent, K.A.; Cracknell, J.A.; Lenz, O.; Zebger, I.; Friedrich, B.; Armstrong, F.A. Electrocatalytic hydrogen oxidation by an enzyme at high carbon monoxide or oxygen levels. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16951–16954. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Willner, I.; Kotlyar, A.B. A non-compartmentalized glucose vertical bar O-2 biofuel cell by bioengineered electrode surfaces. J. Electroanal. Chem. 1999, 479, 64–68. [Google Scholar] [CrossRef]
- Pizzariello, A.; Stred'ansky, M.; Miertus, S. A glucose/hydrogen peroxide biofuel cell that uses oxidase and peroxidase as catalysts by composite bulk-modified bioelectrodes based on a solid binding matrix. In Proceedings of the 16th International Symposium on Bioelectrochemistry and Bioenergetics, Bratislava, Slovakia, Jun 2001; pp. 99–105.
- Ramanavicius, A.; Kausaite, A.; Ramanaviciene, A. Biofuel cell based on direct bioelectrocatalysis. In Proceedings of the 8th World Congress on Biosensors, Granada, Spain, May 2004; pp. 1962–1967.
- Ramanavicius, A.; Ramanaviciene, A. Hemoproteins in design of biofuel cells. Fuel Cells 2009, 9, 25–36. [Google Scholar] [CrossRef]
- Habermuller, L.; Mosbach, M.; Schuhmann, W. Electron-transfer mechanisms in amperometric biosensors. Fresenius' J. Anal. Chem. 2000, 366, 560–568. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Zhou, H.M.; Zhang, J.X.; Zheng, W.; Zheng, Y.F. Carbon nanotube-hydroxyapatite nanocomposite: A novel platform for glucose/O-2 biofuel cell. Biosens. Bioelectron. 2009, 25, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Yang, F.; Silva, M.; Zarow, A.; Wang, Y.B.; Iqbal, Z. Membrane-less and mediator-free enzymatic biofuel cell using carbon nanotube/porous silicon electrodes. Electrochem. Commun. 2009, 11, 34–37. [Google Scholar] [CrossRef]
- Li, X.C.; Zhou, H.J.; Yu, P.; Su, L.; Ohsaka, T.; Mao, L.Q. A miniature glucose/O-2 biofuel cell with single-walled carbon nanotubes-modified carbon fiber microelectrodes as the substrate. Electrochem. Commun. 2008, 10, 851–854. [Google Scholar] [CrossRef]
- Deng, L.; Shang, L.; Wang, Y.Z.; Wang, T.; Chen, H.J.; Dong, S.J. Multilayer structured carbon nanotubes/poly-L-lysine/laccase composite cathode for glucose/O-2 biofuel cell. Electrochem. Commun. 2008, 10, 1012–1015. [Google Scholar] [CrossRef]
- Yan, Y.M.; Su, L.; Mao, L.Q. Multi-walled carbon nanotube-based glucose/O-2 biofuel cell with glucose oxidase and laccase as biocatalysts. J. Nanosci. Nanotechnol. 2007, 7, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Su, L.; Ohsaka, T.; Mao, L. A miniature glucose/O-2 biofuel cell with a high tolerance against ascorbic acid. Fuel Cells 2009, 9, 85–91. [Google Scholar] [CrossRef]
- Togo, M.; Takamura, A.; Asai, T.; Kaji, H.; Nishizawa, M. Structural studies of enzyme-based microfluidic biofuel cells. J. Power Sources 2008, 178, 53–58. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, S.J. A biofuel cell harvesting energy from glucose-air and fruit juice-air. Biosens. Bioelectron. 2007, 23, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yan, Y.M.; Su, L.; Wang, L.; Mao, L.Q. An enzymatic glucose/O-2 biofuel cell: Preparation, characterization and performance in serum. Electrochem. Commun. 2007, 9, 989–996. [Google Scholar] [CrossRef]
- Bedekar, A.S.; Feng, J.J.; Krishnamoorthy, S.; Lim, K.G.; Palmore, G.T.R.; Sundaram, S. Oxygen limitation in microfluidic biofuel cells. Chem. Eng. Commun. 2008, 195, 256–266. [Google Scholar] [CrossRef]
- Brunel, L.; Denele, J.; Servat, K.; Kokoh, K.B.; Jolivalt, C.; Innocent, C.; Cretin, M.; Rolland, M.; Tingry, S. Oxygen transport through laccase biocathodes for a membrane-less glucose/O-2 biofuel cell. Electrochem. Commun. 2007, 9, 331–336. [Google Scholar] [CrossRef]
- Nazaruk, E.; Smolinski, S.; Swatko-Ossor, M.; Ginalska, G.; Fiedurek, J.; Rogalski, J.; Bilewicz, R. Enzymatic biofuel cell based on electrodes modified with lipid liquid-crystalline cubic phases. J. Power Sources 2008, 183, 533–538. [Google Scholar] [CrossRef]
- Yan, Y.M.; Zheng, W.; Su, L.; Mao, L.Q. Carbon-nanotube-based glucose/O-2 biofuel cells. Adv. Mater. 2006, 18, 2639. [Google Scholar] [CrossRef]
- Zhou, M.; Deng, L.; Wen, D.; Shang, L.; Jin, L.H.; Dong, S.J. Highly ordered mesoporous carbons-based glucose/O-2 biofuel cell. Biosens. Bioelectron. 2009, 24, 2904–2908. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.M.; Yehezkeli, O.; Willner, I. Integrated, electrically contacted NAD(P)(+)-dependent enzyme - carbon nanotube electrodes for biosensors and biofuel cell applications. Chem.- Eur. J. 2007, 13, 10168–10175. [Google Scholar] [CrossRef] [PubMed]
- Topcagic, S.; Minteer, S.D. Development of a membraneless ethanol/oxygen biofuel cell. In Proceedings of the International Conference on Electrode Processes, Szczyrk, Poland, September 2004; pp. 2168–2172.
- Liu, Y.; Wang, M.K.; Zhao, F.; Liu, B.F.; Dong, S.J. A low-cost biofuel cell with pH-dependent power output based on porous carbon as matrix. Chem. Eur. J. 2005, 11, 4970–4974. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, S.J. A biofuel cell with enhanced power output by grape juice. Electrochem. Commun. 2007, 9, 1423–1427. [Google Scholar] [CrossRef]
- Tan, Y.M.; Deng, W.F.; Ge, B.; Xie, Q.J.; Huang, J.H.; Yao, S.Z. Biofuel cell and phenolic biosensor based on acid-resistant laccase-glutaraldehyde functionalized chitosan-multiwalled carbon nanotubes nanocomposite film. Biosens. Bioelectron. 2009, 24, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Merle, G.; Habrioux, A.; Servat, K.; Rolland, M.; Innocent, C.; Kokoh, K.B.; Tingry, S. Long-term activity of covalent grafted biocatalysts during intermittent use of a glucose/O-2 biofuel cell. Electrochim. Acta 2009, 54, 2998–3003. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.I.; Yoo, K.H. Polypyrrole nanowire-based enzymatic biofuel cells. Biosens. Bioelectron. 2009, 25, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Komaba, S.; Mitsuhashi, T.; Shraishi, S. Optimization of enzyme anode and cathode with polyion complex for the application to biofuel cells. Electrochemistry 2008, 76, 619–624. [Google Scholar] [CrossRef]
- Habrioux, A.; Merle, G.; Servat, K.; Kokoh, K.B.; Innocent, C.; Cretin, M.; Tingry, S. Concentric glucose/O-2 biofuel cell. J. Electroanal. Chem. 2008, 622, 97–102. [Google Scholar] [CrossRef]
- Ivanov, I.; Vidakovic, T.R.; Sundmacher, K. Glucose electrooxidation for biofuel cell applications. Chem. Biochem. Eng. 2009, 23, 77–86. [Google Scholar]
- Tan, Y.M.; Xie, Q.J.; Huang, J.H.; Duan, W.S.; Ma, M.; Yao, S.Z. Study on glucose biofuel cells using an electrochemical noise device. Electroanalysis 2008, 20, 1599–1606. [Google Scholar] [CrossRef]
- Soukharev, V.; Mano, N.; Heller, A. A four-electron O-2-electroreduction biocatalyst superior to platinum and a biofuel cell operating at 0.88 V. J. Am. Chem. Soc. 2004, 126, 8368–8369. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Mao, F.; Shin, W.; Chen, T.; Heller, A. A miniature biofuel cell operating at 0.78 V. Chem. Commun. 2003, 518–519. [Google Scholar] [CrossRef]
- Chen, T.; Barton, S.C.; Binyamin, G.; Gao, Z.Q.; Zhang, Y.C.; Kim, H.H.; Heller, A. A miniature biofuel cell. J. Am. Chem. Soc. 2001, 123, 8630–8631. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Mao, F.; Heller, A. A miniature membrane-less biofuel cell operating at +0.60 V under physiological conditions. ChemBioChem 2004, 5, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Mao, F.; Heller, A. Characteristics of a miniature compartment-less glucose-O-2 biofuel cell and its operation in a living plant. J. Am. Chem. Soc. 2003, 125, 6588–6594. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Heller, A. A miniature membraneless biofuel cell operating at 0.36 V under physiological conditions. J. Electrochem. Soc. 2003, 150, A1136–A1138. [Google Scholar] [CrossRef]
- Kim, H.H.; Mano, N.; Zhang, X.C.; Heller, A. A miniature membrane-less biofuel cell operating under physiological conditions at 0.5 V. J. Electrochem. Soc. 2003, 150, A209–A213. [Google Scholar] [CrossRef]
- Mano, N.; Mao, F.; Heller, A. A miniature biofuel cell operating in a physiological buffer. J. Am. Chem. Soc. 2002, 124, 12962–12963. [Google Scholar] [CrossRef] [PubMed]
- Barriere, F.; Kavanagh, P.; Leech, D. A laccase-glucose oxidase biofuel cell prototype operating in a physiological buffer. In Proceedings of 18th International Symposium on Bioelectrochemistry and Bioenergetics/3rd Spring Meeting of the International-Society-of-Electrochemistry, Coimbra, Portugal, Jun 2005; pp. 5187–5192.
- Stoica, L.; Dimcheva, N.; Ackermann, Y.; Karnicka, K.; Guschin, D.A.; Kulesza, P.J.; Rogalski, J.; Haltrich, D.; Ludwig, R.; Gorton, L.; Schuhmann, W. Membrane-less biofuel cell based on cellobiose dehydrogenase (anode)/laccase (cathode) wired via specific os-redox polymers. Fuel Cells 2009, 9, 53–62. [Google Scholar] [CrossRef]
- Barriere, F.; Ferry, Y.; Rochefort, D.; Leech, D. Targetting redox polymers as mediators for laccase oxygen reduction in a membrane-less biofuel cell. Electrochem. Commun. 2004, 6, 237–241. [Google Scholar] [CrossRef]
- Zafar, M.N.; Tasca, F.; Gorton, L.; Patridge, E.V.; Ferry, J.G.; Noll, G. Tryptophan Repressor-Binding Proteins from Escherichia coli and Archaeoglobus fulgidus as New Catalysts for 1,4-Dihydronicotinamide Adenine Dinucleotide-Dependent Amperometric Biosensors and Biofuel Cells. Anal. Chem. 2009, 81, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- Heller, A. Electron-conducting redox hydrogels: design, characteristics and synthesis. Curr. Opin. Chem. Biol. 2006, 10, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Courjean, O.; Mano, N. An improved glucose/O-2 membrane-less biofuel cell through glucose oxidase purification. Biosens. Bioelectron. 2009, 25, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Mano, N. A 280 μW cm–2 biofuel cell operating at low glucose concentration. Chem. Commun. 2008, 2221–2223. [Google Scholar] [CrossRef]
- Courjean, O.; Gao, F.; Mano, N. Deglycosylation of glucose oxidase for direct and efficient glucose electrooxidation on a glassy carbon electrode. Angew. Chem., Int. Ed. 2009, 48, 5897–5899. [Google Scholar] [CrossRef]
- Kim, J.; Parkey, J.; Rhodes, C.; Gonzalez-Martin, A. Development of a biofuel cell using glucose-oxidase- and bilirubin-oxidase-based electrodes. In Proceedings of the 213th Electrochemistry-Society Meeting 2008, Phoenix, AZ, USA, May 18, 2008; pp. 1043–1050.
- Mano, N.; Fernandez, J.L.; Kim, Y.; Shin, W.; Bard, A.J.; Heller, A. Oxygen is electroreduced to water on a "wired" enzyme electrode at a lesser overpotential than on platinum. J. Am. Chem. Soc. 2003, 125, 15290–15291. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Vidaković, T.; Sundmacher, K. A flow-through glucose-oxygen fuel cell based on enzymatic anode and Pt cathode. In Proceedings of the 60th Annual Meeting of the ISE, Beijing, China, August 2009.
- Arechederra, R.; Minteer, S.D. Organelle-based biofuel cells: Immobilized mitochondria on carbon paper electrodes. In Proceedings of 58th Annual Meeting of the ISE, Banff, Canada, September 2007; pp. 6698–6703.
- Tsujimura, S.; Fujita, M.; Tatsumi, H.; Kano, K.; Ikeda, T. Bioelectrocatalysis-based dihydrogen/dioxygen fuel cell operating at physiological pH. Phys. Chem. Chem. Phys. 2001, 3, 1331–1335. [Google Scholar] [CrossRef]
- Zebda, A.; Renaud, J.; Cretin, M.; Pichot, F.; Innocent, C.; Ferrigno, R.; Tingry, S. A microfluidic glucose biofuel cell to generate micropower from enzymes at ambient temperature. Electrochem. Commun. 2009, 11, 592–595. [Google Scholar] [CrossRef]
- Lim, K.G.; Palmore, G.T.R. Microfluidic biofuel cells: The influence of electrode diffusion layer on performance. Biosens. Bioelectron. 2007, 22, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Hudak, N.S.; Gallaway, J.W.; Barton, S.C. Mediated biocatalytic cathodes operating on gas-phase air and oxygen in fuel cells. J. Electrochem. Soc. 2009, 156, B9–B15. [Google Scholar] [CrossRef]
- Svoboda, V.; Cooney, M.; Liaw, B.Y.; Minteer, S.; Piles, E.; Lehnert, D.; Barton, S.C.; Rincon, R.; Atanassov, P. Standardized characterization of electrocatalytic electrodes. Electroanalysis 2008, 20, 1099–1109. [Google Scholar] [CrossRef]
- Zebda, A.; Renaud, L.; Cretin, M.; Innocent, C.; Pichot, F.; Ferrigno, R.; Tingry, S. Electrochemical performance of a glucose/oxygen microfluidic biofuel cell. J. Power Sources 2009, 193, 602–606. [Google Scholar] [CrossRef]
- Moore, C.M.; Minteer, S.D.; Martin, R.S. Microchip-based ethanol/oxygen biofuel cell. Lab. Chip 2005, 5, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, F.; Chen, H.J.; Shang, L.; Wang, L.; Wang, T.; Dong, S.J. A biofuel cell with enhanced performance by multilayer biocatalyst immobilized on highly ordered macroporous electrode. Biosens. Bioelectron. 2008, 24, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Glykys, D.J.; Banta, S. Metabolic control analysis of an enzymatic biofuel cell. Biotechnol. Bioeng. 2009, 102, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.W.; Lee, J.Y.; Lee, J.H.; Kang, S.W.; Park, C.H.; Kim, S.W. Optimization of cell conditions for enzymatic fuel cell using statistical analysis. J. Ind. Eng. Chem. 2008, 14, 338–343. [Google Scholar] [CrossRef]
- Kjeang, E.; Sinton, D.; Harrington, D.A. Strategic enzyme patterning for microfluidic biofuel cells. J. Power Sources 2006, 158, 1–12. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Toh, C.S.; Calvo, E.J.; Flexer, V. Modelling biosensor responses. In Bioelectrochemistry : Fundamentals, Experimental Techniques and Applications; Bartlett, P.N., Ed.; Wiley: Chichester, England, 2008; pp. 267–325. [Google Scholar]
- Weber, A.Z.; Newman, J. Modeling transport in polymer-electrolyte fuel cells. Chem. Rev. 2004, 104, 4679–4726. [Google Scholar] [CrossRef] [PubMed]
- Kano, K.; Ikeda, T. Fundamentals and practices of mediated bioelectrocatalysis. Anal. Sci. 2000, 16, 1013–1021. [Google Scholar] [CrossRef]
- Tamaki, T.; Ito, T.; Yamaguchi, T. Modelling of reaction and diffusion processes in a high-surface-area biofuel cell electrode made of redox polymer-grafted carbon. Fuel Cells 2009, 9, 37–43. [Google Scholar] [CrossRef]
- Gallaway, J.W.; Barton, S.A.C. Kinetics of redox polymer-mediated enzyme electrodes. J. Am. Chem. Soc. 2008, 130, 8527–8536. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.N.; Pratt, K.F.E. Theoretical treatment of diffusion and kinetics in amperometric immobilized enzyme electrodes. 1. Redox mediator entrapped within the film. J. Electroanal. Chem. 1995, 397, 61–78. [Google Scholar] [CrossRef]
- Matsumoto, R.; Kano, K.; Ikeda, T. Theory of steady-state catalytic current of mediated bioelectrocatalysis. J. Electroanal. Chem. 2002, 535, 37–40. [Google Scholar] [CrossRef]
- Wong, T.S.; Schwaneberg, U. Protein engineering in bioelectrocatalysis. Curr. Opin. Biotechnol. 2003, 14, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.W.; Momeu, C.; Zakhartsev, M.; Schwaneberg, U. Making glucose oxidase fit for biofuel cell applications by directed protein evolution. Biosens. Bioelectron. 2006, 21, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, C.M.; Simon, E.; Toh, C.S.; Cass, A.E.G.; Bartlett, P.N. The design of dehydrogenase enzymes for use in a biofuel cell: the role of genetically introduced peptide tags in enzyme immobilization on electrodes. In Proceedings of the 16th International Symposium on Bioelectrochemistry and Bioenergetics, Bratislava, Slovakia, June 2001; pp. 21–23.
- Spadiut, O.; Pisanelli, I.; Maischberger, T.; Peterbauer, C.; Gorton, L.; Chaiyen, P.; Haltrich, D. Engineering of pyranose 2-oxidase: Improvement for biofuel cell and food applications through semi-rational protein design. J. Biotechnol. 2009, 139, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, S.; Okuda, J.; Ikebukuro, K.; Sode, K. Molecular engineering of PQQGDH and its applications. Arch. Biochem. Biophys. 2004, 428, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Yuhashi, N.; Tomiyama, M.; Okuda, J.; Igarashi, S.; Ikebukuro, K.; Sode, K. Development of a novel glucose enzyme fuel cell system employing protein engineered PQQ glucose dehydrogenase. In Proceedings of the 8th World Congress on Biosensors, Granada, Spain, May 2004; pp. 2145–2150.
- Okuda-Shimazaki, J.; Kakehi, N.; Yamazaki, T.; Tomiyama, M.; Sode, K. Biofuel cell system employing thermostable glucose dehydrogenase. Biotechnol. Lett. 2008, 30, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Kamitaka, Y.; Tsujimura, S.; Kataoka, K.; Sakurai, T.; Ikeda, T.; Kano, K. Effects of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J. Electroanal. Chem. 2007, 601, 119–124. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ivanov, I.; Vidaković-Koch, T.; Sundmacher, K. Recent Advances in Enzymatic Fuel Cells: Experiments and Modeling. Energies 2010, 3, 803-846. https://doi.org/10.3390/en3040803
Ivanov I, Vidaković-Koch T, Sundmacher K. Recent Advances in Enzymatic Fuel Cells: Experiments and Modeling. Energies. 2010; 3(4):803-846. https://doi.org/10.3390/en3040803
Chicago/Turabian StyleIvanov, Ivan, Tanja Vidaković-Koch, and Kai Sundmacher. 2010. "Recent Advances in Enzymatic Fuel Cells: Experiments and Modeling" Energies 3, no. 4: 803-846. https://doi.org/10.3390/en3040803