
Liquid Phase Sintering of (Ti,Zr)C with WC-Co

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Procedures
2.1. Sintering Trials
2.2. Characterization of Sintered Samples
3. Results and Discussion
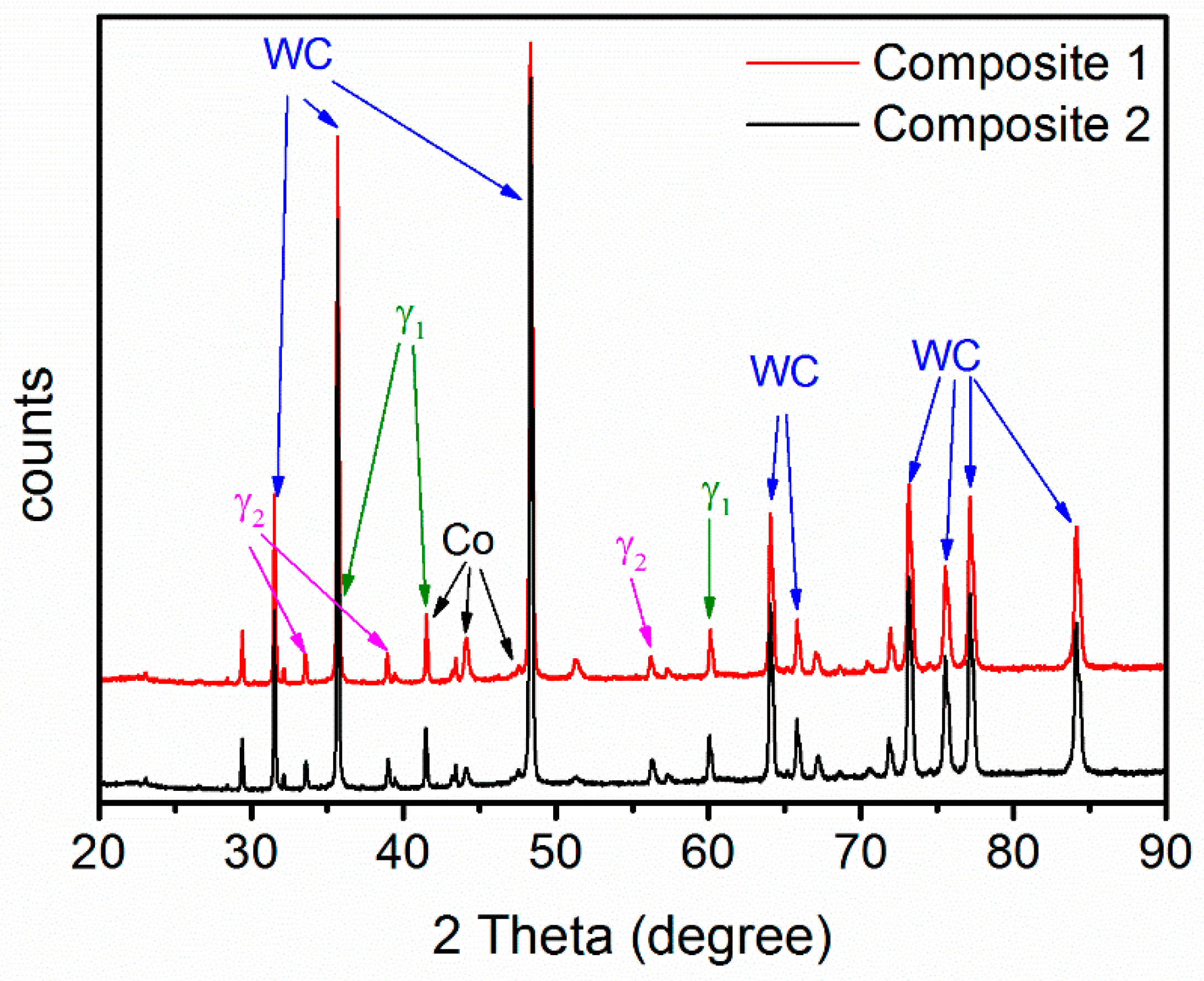
3.1. Final Microstructure and Properties
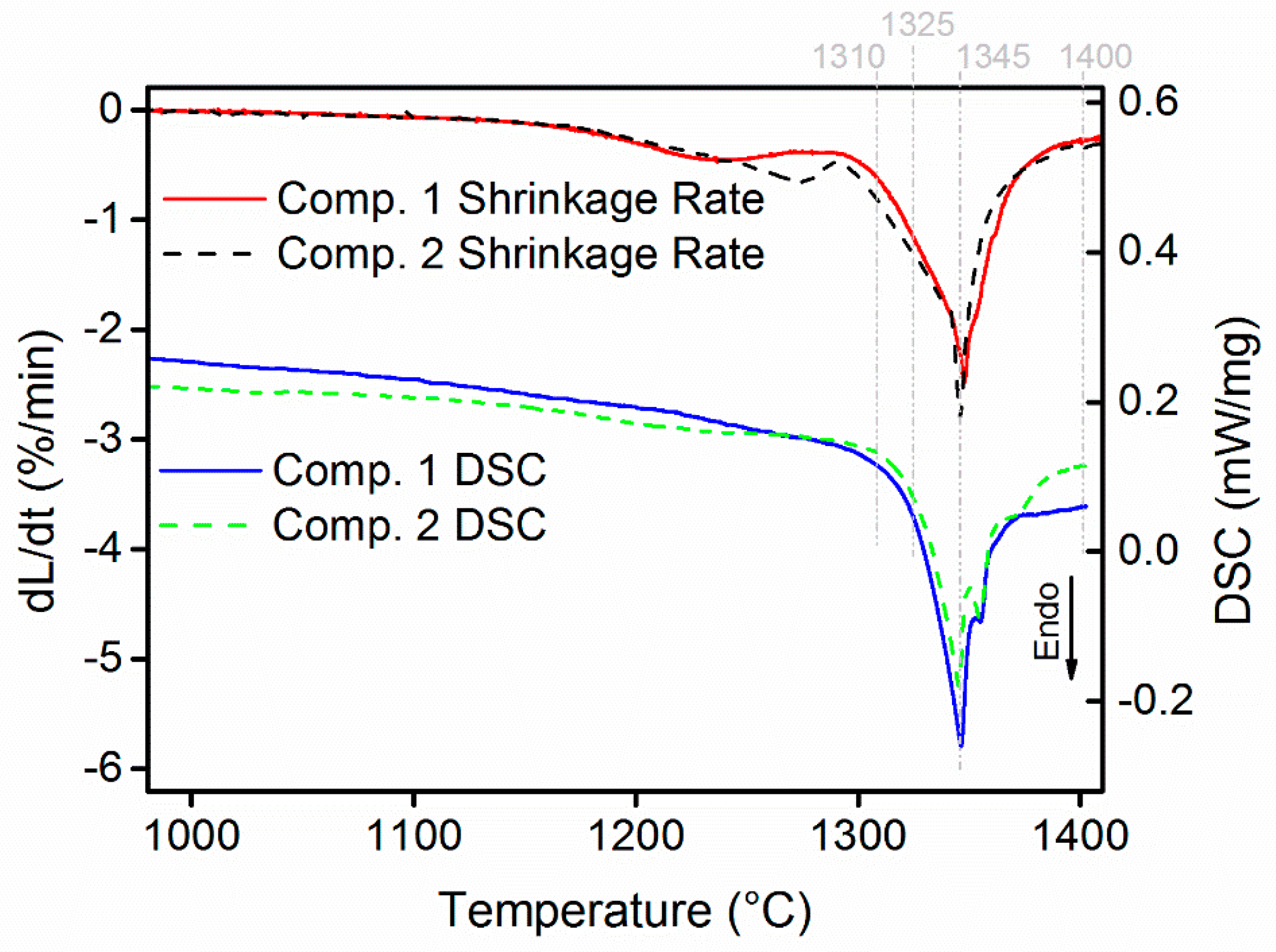
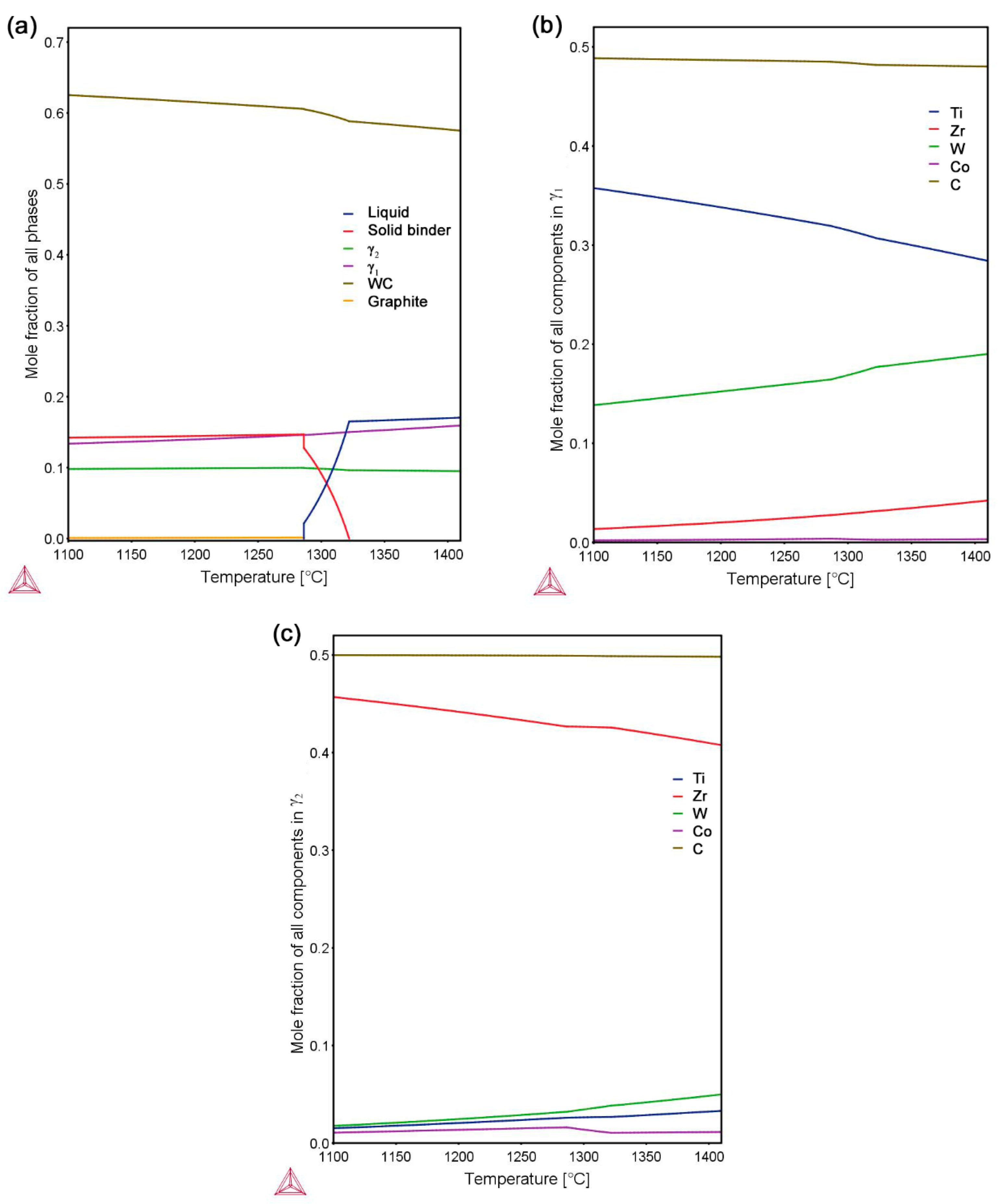
3.2. Sintering Behavior
3.3. Microstructural Evolution during Sintering
3.3.1. γ Phase in Composite 1
3.3.2. γ Phase in Composite 2
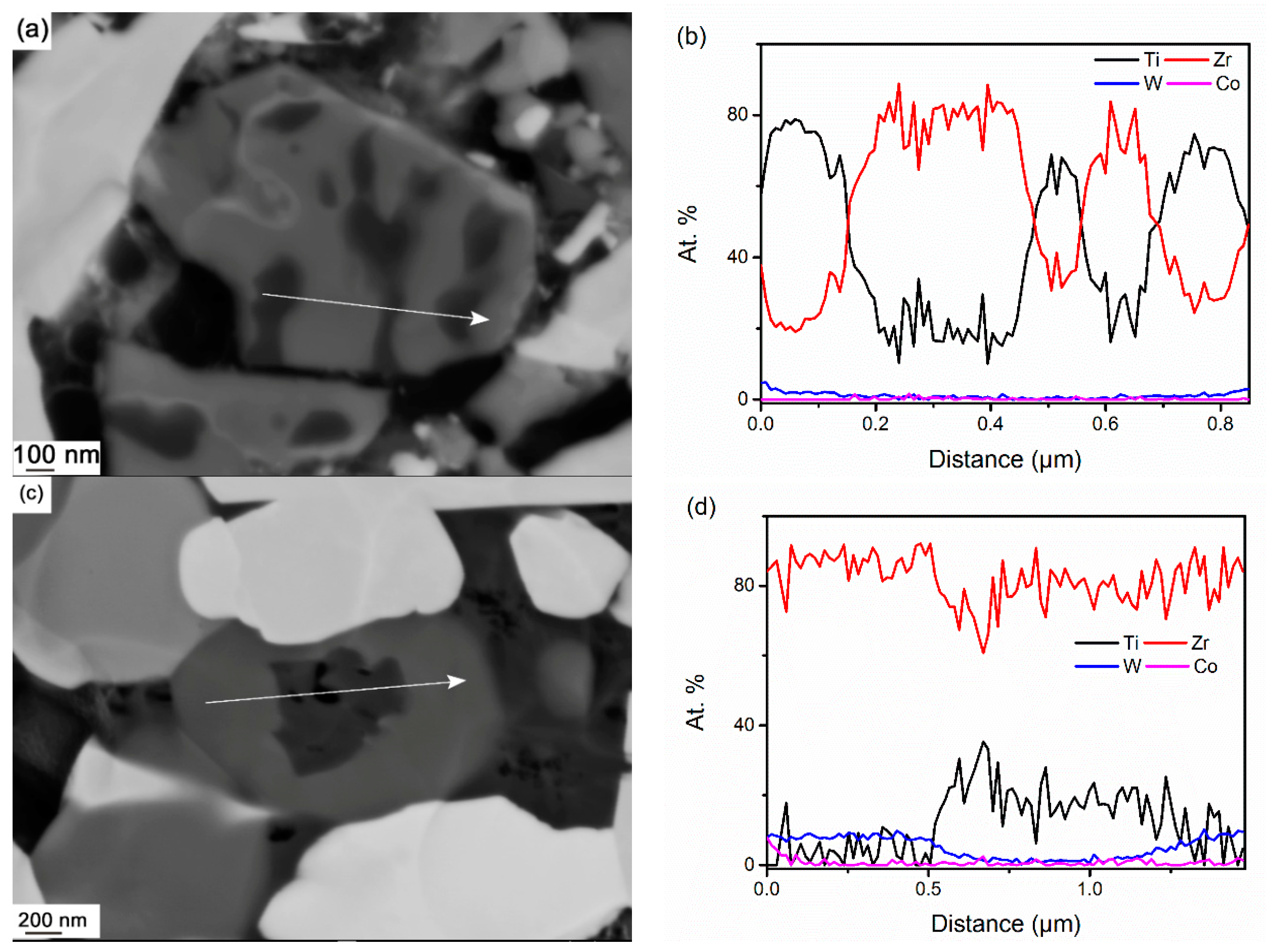
3.3.3. Phase Separation of (Ti,Zr)C
4. Conclusions
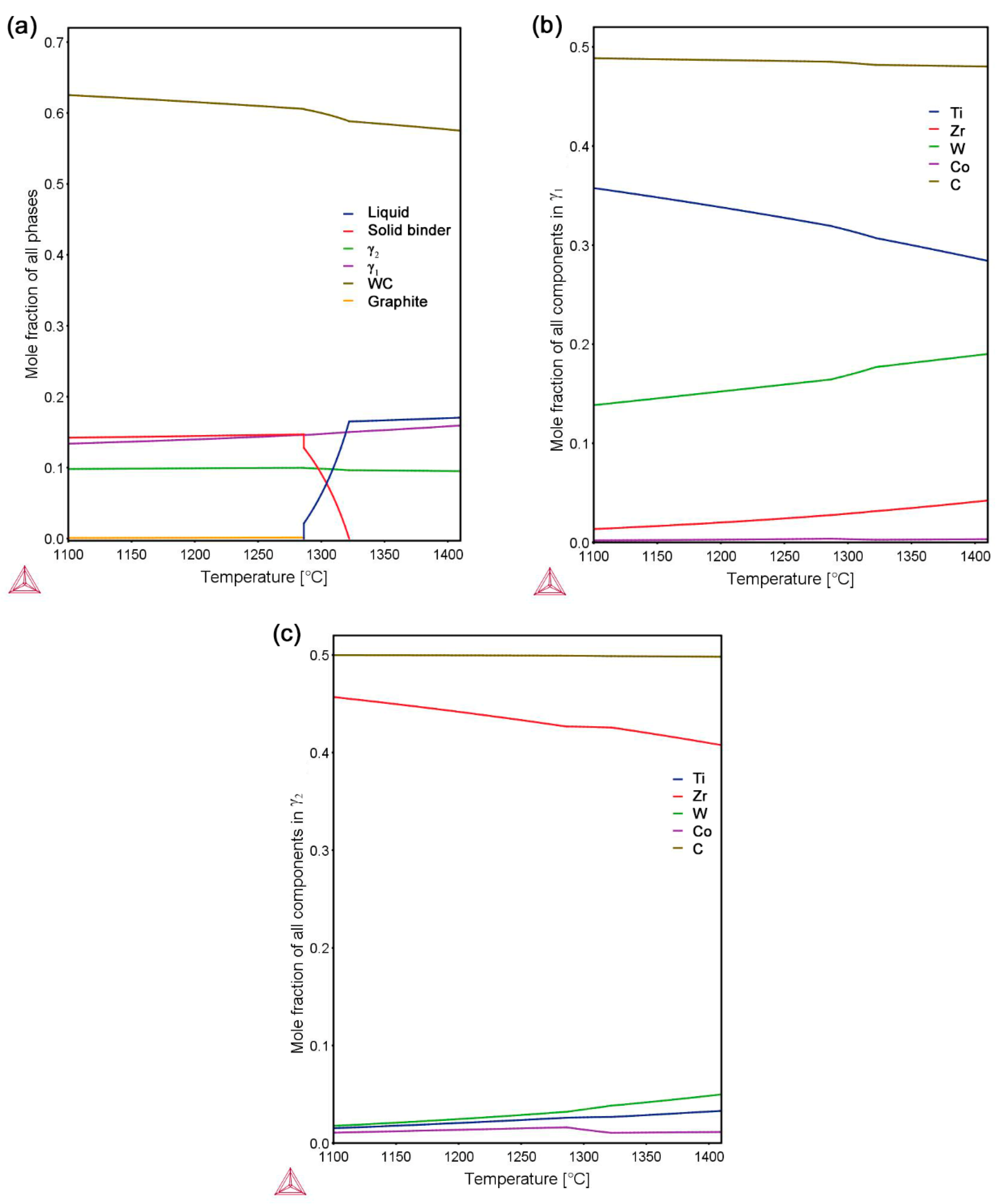
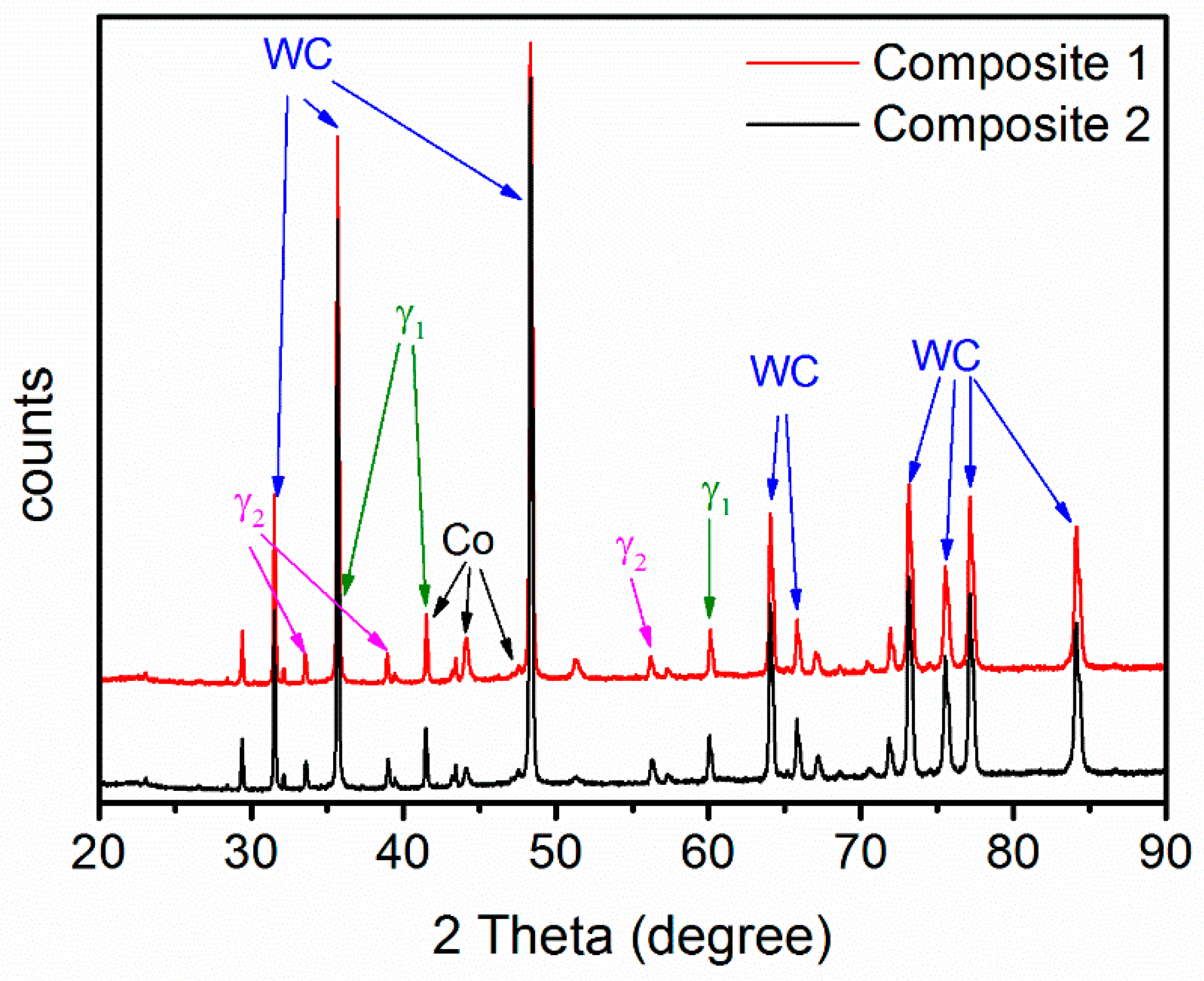
- WC-(Ti,Zr)C-Co and WC-TiC-ZrC-Co have been liquid phase sintered at 1410 °C in the present work. Both composites present similar shrinkage behavior, and WC-(Ti,Zr)C-Co has a lower total shrinkage, of 18.2%; both composites have a similar melting temperature of about 1322 °C; after sintering, both composites contain two γ phases—(Ti,W,Zr)C and (Zr,W,Ti)C cubic carbides.
- After full sintering the grain size of both WC and cubic carbides in WC-(Ti,Zr)C-Co is around 10% smaller than that in WC-TiC-ZrC-Co. The ternary carbide powder thus has a grain growth inhibiting effect, but only a small difference has been found in the measured hardness.
- During interrupted sintering, γ1 re-precipitates before binder melting, while γ2 re-precipitates directly after binder melting in both composites. The composition of γ phases measured by EDS is in good agreement with the thermodynamic calculations.
- In WC-TiC-ZrC-Co, γ1 and γ2 phases mainly re-precipitate on original TiC and ZrC grains respectively; in WC-(Ti,Zr)C-Co, except on the original (Ti,Zr)C grains, considerable amounts of γ1 and γ2 phases also re-precipitate as individual grains in the interparticle spaces.
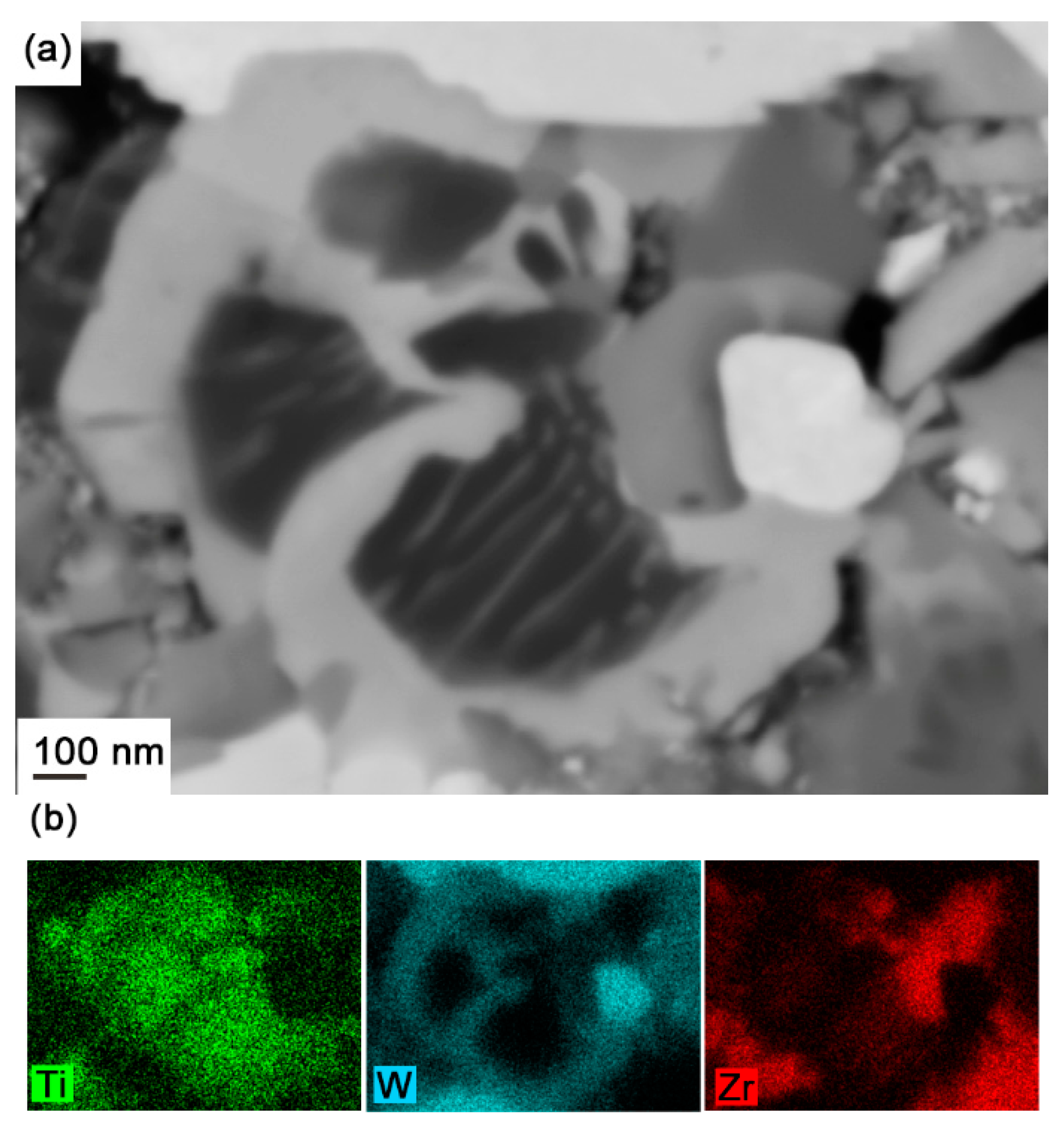
- In WC-(Ti,Zr)C-Co, (Ti,Zr)C phase decomposes into Ti-rich and Zr-rich nano-scale lamellae at a rapid rate during solid-state sintering; Ti-rich lamellae dissolve at a much faster rate than Zr-rich lamellae in liquid binder phase, and a small amount of Zr-rich (Ti,Zr)C phase is retained as darker cores in γ2 particles after full sintering.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Exner, H.E.; Gurland, J. A review of parameters influencing some mechanical properties of tungsten carbide-cobalt alloys. Powder Metall. 1970, 13, 13–31. [Google Scholar] [CrossRef]
- Brookes, K.J. World Dictionary and Handbook of Hard Metals and Hard Materials; International Carbide Data: London, UK, 1996. [Google Scholar]
- Norgren, S.; Kusoffsky, A. On the solubility of chromium in the cubic carbide and M7C 3 carbide in the WC-Co-Cr-Me systems (Me = Ti, Nb, Ta and Zr). Int. J. Refract. Met. Hard Mater. 2013, 40, 24–26. [Google Scholar] [CrossRef]
- Allibert, C.H. Sintering features of cemented carbides WC–Co processed from fine powders. Int. J. Refract. Met. Hard Mater. 2001, 19, 53–61. [Google Scholar] [CrossRef]
- Lee, H.R.; Kim, D.J.; Hwang, N.M.; Kim, D.-Y. Role of Vanadium Carbide Additive during Sintering of WC-Co: Mechanism of Grain Growth Inhibition. J. Am. Ceram. Soc. 2003, 86, 152–154. [Google Scholar] [CrossRef]
- Schwarzkopf, M.; Exner, H.E.; Fischmeister, H.F.; Schintlmeister, W. Kinetics of compositional modification of (W,Ti)C-WC-Co alloy surfaces. Mater. Sci. Eng. A 1988, 105–106, 225–231. [Google Scholar] [CrossRef]
- Frykholm, R.; Andrén, H.-O. Development of the microstructure during gradient sintering of a cemented carbide. Mater. Chem. Phys. 2001, 67, 203–208. [Google Scholar] [CrossRef]
- Andrén, H.-O. Microstructure development during sintering and heat-treatment of cemented carbides and cermets. Mater. Chem. Phys. 2001, 67, 209–213. [Google Scholar] [CrossRef]
- Shibata, D. Cutting Tool. U.S. Patent 6756110 B2, 29 June 2004. [Google Scholar]
- Holleck, H. Material selection for hard coatings. J. Vac. Sci. Technol. A 1986, 4, 2661. [Google Scholar] [CrossRef]
- Holleck, H. Ternäre Carbidsysteme der hochschmelzenden Übergangsmetalle- Teil I. Metall. Technol. 1981, 35, 999–1004. [Google Scholar]
- Borgh, I.; Hedström, P.; Blomqvist, A.; Århammar, C.; Ågren, J.; Odqvist, J. A study of the decomposition of the mixed (Ti,Zr)C phase. In Proceedings of the 18th International Plansee Seminar, Reutte, Austria, 3–7 June 2013.
- Borgh, I.; Hedström, P.; Blomqvist, A.; Ågren, J.; Odqvist, J. Synthesis and phase separation of (Ti,Zr)C. Acta Mater. 2014, 66, 209–218. [Google Scholar] [CrossRef]
- Ma, T.; Hedström, P.; Ström, V.; Masood, A.; Borgh, I.; Blomqvist, A.; Odqvist, J. Self-organizing nanostructured lamellar (Ti,Zr)C—A superhard mixed carbide. Int. J. Refract. Met. Hard Mater. 2015, 51, 25–28. [Google Scholar] [CrossRef]
- Markström, A.; Frisk, K. Experimental and thermodynamic evaluation of the miscibility gaps in MC carbides for the C-Co-Ti-V-W-Zr system. Calphad 2009, 33, 530–538. [Google Scholar] [CrossRef]
- Borgh, I.; Norgren, S.; Borgenstam, A.; Ågren, J. Influence of nitrogen gas pressure on the miscibility gap in the Ti-Zr carbonitride system. Int. J. Refract. Met. Hard Mater. 2012, 32, 11–15. [Google Scholar] [CrossRef]
- Weidow, J.; Zackrisson, J.; Jansson, B.; Andrén, H.-O. Characterisation of WC-Co with cubic carbide additions. Int. J. Refract. Met. Hard Mater. 2009, 27, 244–248. [Google Scholar] [CrossRef]
- Xu, R.; Huang, P.Y.H. Investigation on spinodal decomposition of WC-TiC-ZrC-Co cemented carbide. In Proceedings of the International Powder Metallurgy Conference, Orlando, FL, USA, 5–10 June 1988; pp. 15–21.
- Hall, F.W.; Retelsdorf, H.J. Sintered Hardmetals. U.S. Patent 4451292 A, 29 May 1984. [Google Scholar]
- Hovington, P.; Drouin, D.; Gauvin, R. CASINO: A new monte carlo code in C language for electron beam interaction—Part I: Description of the program. Scanning 2006, 19, 1–14. [Google Scholar] [CrossRef]
- Drouin, D.; Hovington, P.; Gauvin, R. CASINO: A new monte carlo code in C language for electron beam interactions-part II: Tabulated values of the mott cross section. Scanning 2006, 19, 20–28. [Google Scholar] [CrossRef]
- TCFE8 Steels/Fe-Alloys Database, version 8; Thermo-Calc Software: Stockholm, Sweden, 2016.
- Petersson, A. Sintering shrinkage of WC-Co and WC-(Ti,W)C-Co materials with different carbon contents. Int. J. Refract. Met. Hard Mater. 2004, 22, 211–217. [Google Scholar] [CrossRef]
- Petersson, A.; Ågren, J. Modelling WC-Co sintering shrinkage—Effect of carbide grain size and cobalt content. Mater. Sci. Eng. A 2007, 452–453, 37–45. [Google Scholar] [CrossRef]
- Zackrisson, J.; Andrén, H.-O.; Rolander, U. Development of cermet microstructures during sintering. Metall. Mater. Trans. A 2001, 32, 85–94. [Google Scholar] [CrossRef]
- Ma, T.; Borrajo-Pelaez, R.; Hedström, P.; Borgh, I.; Blomqvist, A.; Norgren, S.; Odqvist, J. Microstructure evolution during phase separation in Ti-Zr-C. Int. J. Refract. Met. Hard Mater. 2016, 61, 238–248. [Google Scholar] [CrossRef]
- Missiaen, J.-M. Solid-state spreading and sintering of multiphase materials. Mater. Sci. Eng. A 2008, 475, 2–11. [Google Scholar] [CrossRef]


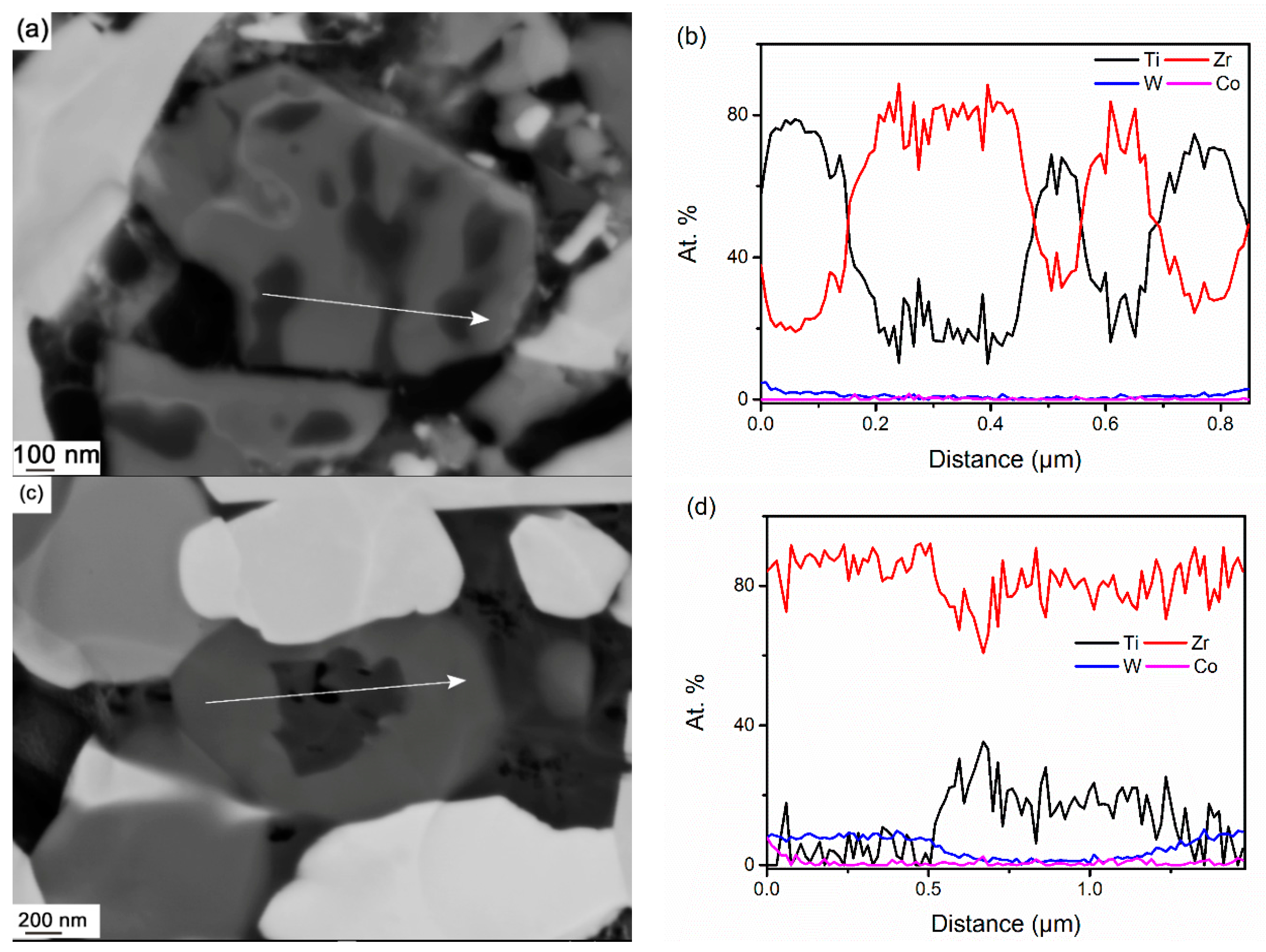

{kind=link}
{kind=link}
{kind=link}
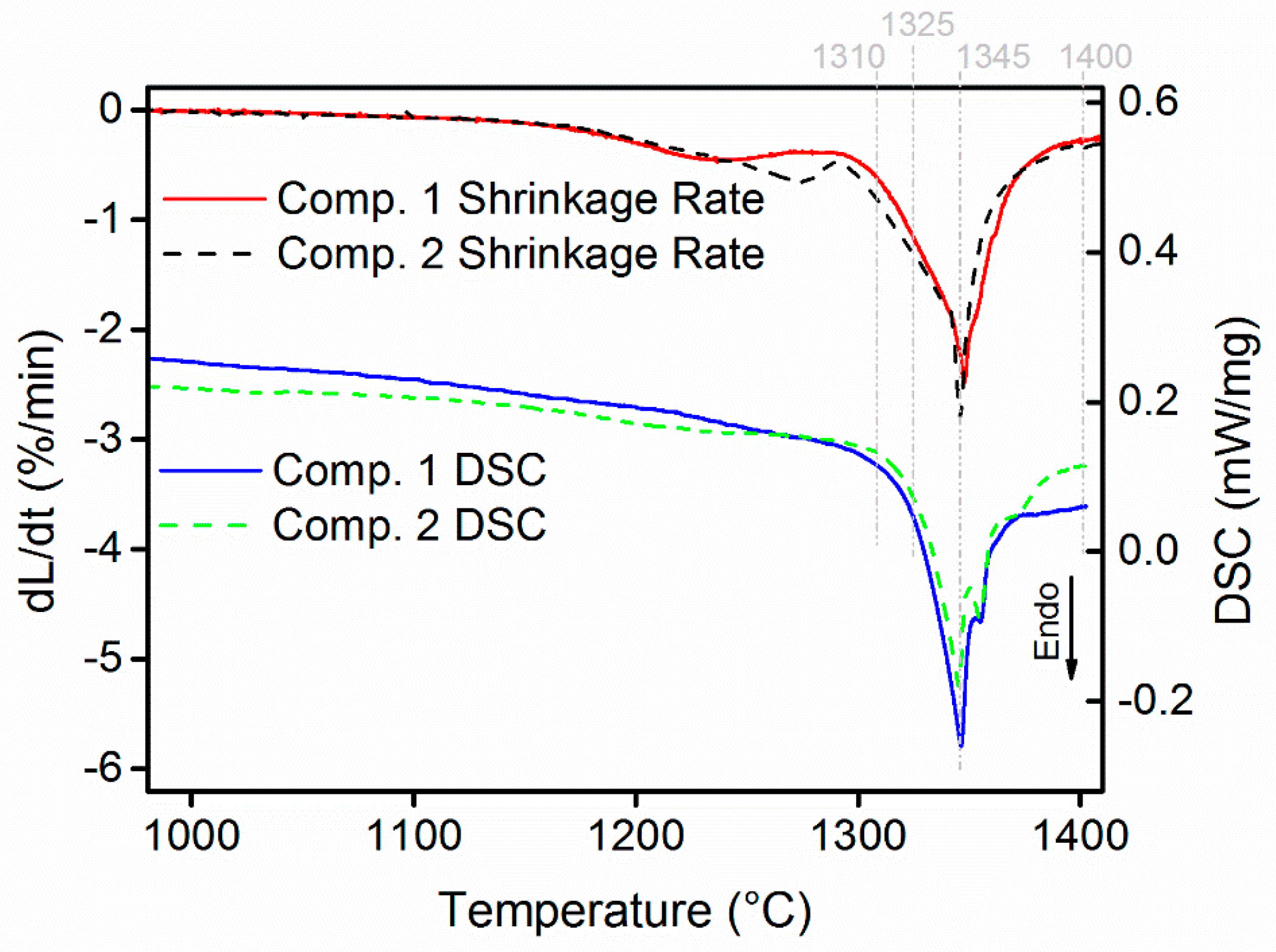{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composite | dWC (µm) | dγ1 (µm) | dγ2 (µm) | Hc (kA/m) | RMS | Hardness (HV3) | Porosity | Mass Loss (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.11 | 1.04 | 0.89 | 11.64 | 0.96 | 1448 | A00B00C00 | 2.10 |
| 2 | 1.21 | 1.19 | 1.03 | 10.73 | 0.97 | 1443 | A04B00C00 | 2.29 |
| Status | Comp. | Undissolved Cubic Carbides | Re-Precipitated Cubic Carbides | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ti-Rich | Zr-Rich | γ1 | γ2 | ||||||||||
| Ti | Zr | W | Ti | Zr | W | Ti | Zr | W | Ti | Zr | W | ||
| 1310 °C IS | 1 | 80 | 19 | 1 | 18 | 80 | 2 | 73 | 10 | 17 | - | ||
| 2 | 100 | 0 | 0 | 0 | 100 | 0 | 76 | 3 | 21 | - | |||
| 1325 °C IS | 1 | 89 | 9 | 2 | 23 | 74 | 3 | 63 | 13 | 24 | 15 | 73 | 12 |
| 2 | 99 | 0 | 1 | 0 | 99 | 1 | 67 | 6 | 27 | 5 | 85 | 10 | |
| 1345 °C IS | 1 | 82 | 15 | 3 | 15 | 78 | 7 | 64 | 7 | 29 | 9 | 80 | 11 |
| 2 | 100 | 0 | 0 | 1 | 97 | 2 | 65 | 6 | 29 | 7 | 80 | 13 | |
| 1400 °C IS | 1 | 89 | 5 | 6 | 13 | 80 | 7 | 61 | 8 | 31 | 6 | 85 | 9 |
| 2 | 100 | 0 | 0 | - | 61 | 8 | 31 | 6 | 83 | 11 | |||
| 1410 °C FS | 1 | - | 23 | 74 | 3 | 62 | 6 | 32 | 6 | 85 | 9 | ||
| 2 | - | - | 60 | 7 | 33 | 6 | 83 | 11 | |||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, T.; Borrajo-Pelaez, R.; Hedström, P.; Blomqvist, A.; Borgh, I.; Norgren, S.; Odqvist, J. Liquid Phase Sintering of (Ti,Zr)C with WC-Co. Materials 2017, 10, 57. https://doi.org/10.3390/ma10010057
Ma T, Borrajo-Pelaez R, Hedström P, Blomqvist A, Borgh I, Norgren S, Odqvist J. Liquid Phase Sintering of (Ti,Zr)C with WC-Co. Materials. 2017; 10(1):57. https://doi.org/10.3390/ma10010057
Chicago/Turabian StyleMa, Taoran, Rafael Borrajo-Pelaez, Peter Hedström, Andreas Blomqvist, Ida Borgh, Susanne Norgren, and Joakim Odqvist. 2017. "Liquid Phase Sintering of (Ti,Zr)C with WC-Co" Materials 10, no. 1: 57. https://doi.org/10.3390/ma10010057