Li-Decorated β12-Borophene as Potential Candidates for Hydrogen Storage: A First-Principle Study
by

Tingting Liu
1,2, 
Yuhong Chen
1,2,*,
Haifeng Wang
3,
Meiling Zhang
2,4,
Lihua Yuan
2 and
Cairong Zhang
1,2 1
State Key Laboratory of Advanced Processing and Recycling of No-ferrous Metals, Lanzhou University of Technology, Lanzhou 730050, China
2
School of Science, Lanzhou University of Technology, Lanzhou 730050, China
3
Department of Physics, College of Science, Shihezi University, Xinjiang 832003, China
4
The School of Nuclear Science and Technology, Lanzhou University, Lanzhou 730000, China
*
Author to whom correspondence should be addressed.
Materials 2017, 10(12), 1399; https://doi.org/10.3390/ma10121399
Submission received: 15 November 2017
/
Revised: 1 December 2017
/
Accepted: 4 December 2017
/
Published: 7 December 2017
Abstract
:The hydrogen storage properties of pristine β12-borophene and Li-decorated β12-borophene are systemically investigated by means of first-principles calculations based on density functional theory. The adsorption sites, adsorption energies, electronic structures, and hydrogen storage performance of pristine β12-borophene/H2 and Li-β12-borophene/H2 systems are discussed in detail. The results show that H2 is dissociated into Two H atoms that are then chemisorbed on β12-borophene via strong covalent bonds. Then, we use Li atom to improve the hydrogen storage performance and modify the hydrogen storage capacity of β12-borophene. Our numerical calculation shows that Li-β12-borophene system can adsorb up to 7 H2 molecules; while 2Li-β12-borophene system can adsorb up to 14 H2 molecules and the hydrogen storage capacity up to 10.85 wt %.
1. Introduction
As the gap between energy supply and demand has become increasingly prominent, sources of renewable energy has been investigated urgently. Hydrogen is an inexhaustible source of clean energy, making it important for society to develop and utilize this energy [1,2]. Hydrogen storage is one of the most critical technical problems in the development of hydrogen energy sources. The average adsorption energy of the ideal physical hydrogen storage method should be between chemical and physical adsorption energy (0.1~0.8 eV) [3,4]. The US Department of Energy (DOE) and the International Energy Agency (IEA) reported that the ideal hydrogen storage capacity should be greater than 5.5 wt % [5]. At present, one of the best types of hydrogen storage methods involves physical adsorption, which results in low adsorption heat, small activation energy, fast hydrogen adsorption and desorption, and reversible cyclization performance. Carbon nanomaterials have become a hotspot of physical hydrogen storage materials due to their characteristics of a large specific surface area, good adsorption kinetic properties and reversible hydrogen storage [6,7]. However, clean carbon nanomaterials adsorb H2 molecules with weak binding capacity, which means that they have low hydrogen storage capacity and are not ideal. Therefore, it is essential to find a suitable physical adsorbent.
Recently, 2D (two-dimensional) borophene created from Boron elements was artificially synthesized [8]. Although there are many theoretical studies about the possible 2D borophene structure [9], only three types of stable structures have been synthesized for borophene so far [8,10]. Borophene’s unique metal properties, mechanical properties, and optical properties have been extensively studied [11,12,13,14,15], but only a few studies have considered its hydrogen storage properties. Borophene and graphene [16] have a similar 2D planar structure with a large specific surface area. Moreover, the relative atomic mass of B atom is smaller than the relative atomic mass of C atom. Therefore, we suspect that borophene has better hydrogen storage properties than graphene (it exhibits a triangular lattice with different periodic arrangements and is flat without obvious vertical undulation). Feng et al. [10] reported that β12-borophene is more stable than the other two types of borophene. Chen et al. [17] used the first-principles method to study the hydrogen storage properties of Ca-β12-borophene and found that it has a larger adsorption energy compared to other types of borophene. Therefore, we selected β12-borophene as the research focus. In this work, we performed theoretical calculations for the hydrogen storage properties of pure β12-borophene and Li-β12-borophene based on the first-principle study. We found that H2 molecules were completely dissociated into two H atoms that were adsorbed on the B–B bridge sites to form H–B covalent bonds, thus making it difficult to dissociate. Comparison of the improvement in hydrogen storage properties of graphene found that the graphene surface was modified by alkali metal (Li, Na, K) [18], alkali-earth metal (Ca) [19], light metal (Al) [20] and transition metals (Cu, Pd, Y) [21,22,23,24], which can change the chemical activity of the graphene surface and could effectively change the hydrogen storagecapability. The quality of alkali metal (Li atoms) is very light, which helps to enhance the hydrogen storage density [25]. The transition metal atom-modified nanostructures are highly reactive and can easily cause the dissociation of H2 molecules, which is detrimental to the reversible storage of hydrogen [26]. Therefore, we selected the lightest Li atom to modify the β12-borophenen. H2 adsorbed on Li-β12-borophene by physical adsorption, which improved the reversible hydrogen storage performance and significantly increased the amount of hydrogen storage. It is expected that this work can provide theoretical support for β12-borophene being used as hydrogen materials.
2. Computational Methods
All density functional theory (DFT) calculations are carried out using the Cambridge Sequential Total Energy Package (CASTEP) [27], and the DFT evaluation is based on the plane-wave expansion. We use the Generalized Gradient Approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [28] to describe exchange and correlation effects. The van der waals forces of H2 adsorption on Li-β12-borophenen is modified by DFT-D methods. While the DFT-D perform poorly for energetics in layered materials [29], it is important to deal with the molecules adsorption system. We select the Ultrasoft Pseudopotential [30] to describe the interaction of electron-ion, and the electron wave functions are expanded by plane wave. The convergence tolerance energy, the force on each atoms and displacement convergence criterions are set to 5.0 × 10−6 eV/atom, 0.01 eV/Å and 0.001 Å, respectively. All atoms are relaxed in our calculations. In order to eliminate the interaction of the interlayer we select the vacuum thickness 20 Å. Considering the calculation accuracy and computational efficiency, all calculations are using a cutoff energy of 600 eV and 9 × 16 × 5 k-point mesh in the Brillouin zone.
The adsorption energy () and average adsorption energy ( of H2 adsorption on Li-β12-borophene are calculated by the following formulas [31]:
The average adsorption energy of Li atom on β12-borophenec [32] is defined as:
where , and are the total energy of the n Li-β12-borophene with i, i − 1 H2 molecules and β12-borophene with n Li atoms, respectively. , and are the total energy of the β12-borophene, free Li atom and an isolated H2, respectively. n is the number of adsorbed Li atoms.
3. Results and Discussion
3.1. H2 Adsorption on β12-Borophene
The optimized lattice parameters of the primitive cell of β12-borophenen are a = 5.069 Å, b = 2.929 Å, agree well with the experimental result (a = 5 Å and b = 3 Å) [10] and other theoretical calculation results [33,34,35]. In our follow calculations, we choose a 2 × 2 unit cell (see Figure 1) of the β12-borophenen containing 20 B atoms (in Figure 1) to investigate the hydrogen storage adsorbed on β12-borophene.
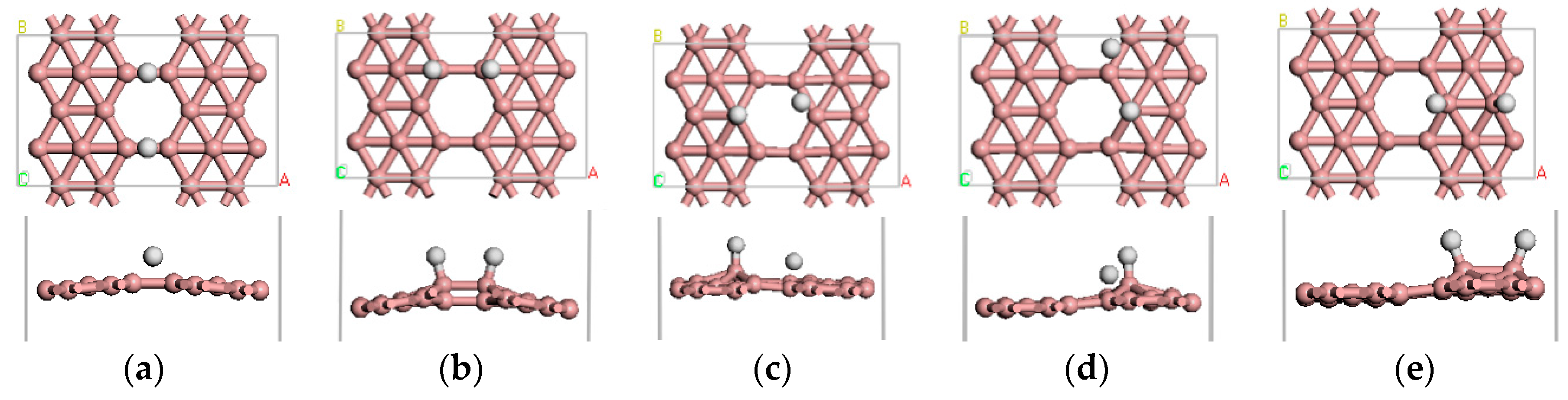
We first investigated the adsorption behavior of one H2 molecule on β12-borophene. The H2 molecule is initially placed in a parallel or vertical direction at different positions of the β12-borophene plane. We found that there are five stable adsorbed configurations in total, as illustrated in Figure 2. In all cases, the H2 molecule is dissociated into two separate H atoms after adsorption and the distance between the H atoms will change from 0.753 to 2.366 Å. Furthermore, the distance between H and its nearest B atom (rH-B) greatly increased from 1.217 to 1.358 Å. The most stable case among all the five adsorption configurations is shown in Figure 2a. In this case, the H2 molecule dissociated into two H atoms that are adsorbed on the B1–B3 and B2–B4 bridge sites with an value of −0.536 eV, which is related to chemical adsorption. Mulliken analysis demonstrates that there is 0.23 e− transferred from B to H, which occurs mainly in the H 1s orbital and B 2p orbital. The B–H bond population is 0.4, indicating it is a covalent bond, with difficult desorption of the β12-borophene/H2 system. In addition, we further studied the transition states of the stable adsorption configurations by combining linear synchronous and quadratic synchronous transits [21,36]. We found that the most stable adsorption configurations of the activation energy barrier from the reactant to transition state was 1.584 eV, which is smaller than the activation energy barrier of other adsorption methods, indicating it was difficult for the reaction of the H2 molecules adsorbed on the surface to take place.
3.2. H2 Adsorption on Li-β12-Borophene
3.2.1. The Adsorption Structure of Li-β12-Borophene
It is well known that doping alkali metal atoms to modify hydrogen storage materials may can greatly improve the hydrogen storage properties and increase the hydrogen storage capacity. Specially, lithium (Li) has been widely employed to functionalize 2D materials and improve the hydrogen storage ability. Therefore, in the following section, we chose to add Li atoms to modify the hydrogen storage properties of β12-borophene.
We examined the adsorption of Li atoms on pure β12-borophene. After optimization, we obtained three different stable adsorption structures, as shown in Figure 3a–c. Similar to Li-decorating graphene [37], the most favorable Li adsorption site on β12-borophene is the hollow center of B ring (Figure 3a).
Doping alkali metal atoms to modify hydrogen storage materials requires the average adsorption energy of the metal atoms on the substrate to be greater than the cohesive energy of the metal atoms in the solid form [38]. The average adsorption energy of Li atom on the β12-borophene is −3.088 eV, which is significantly greater than the cohesive energy of −1.795 eV of Li [39]. This indicates that Li atoms can be dispersed uniformly on β12-borophene, instead of forming metal clusters.
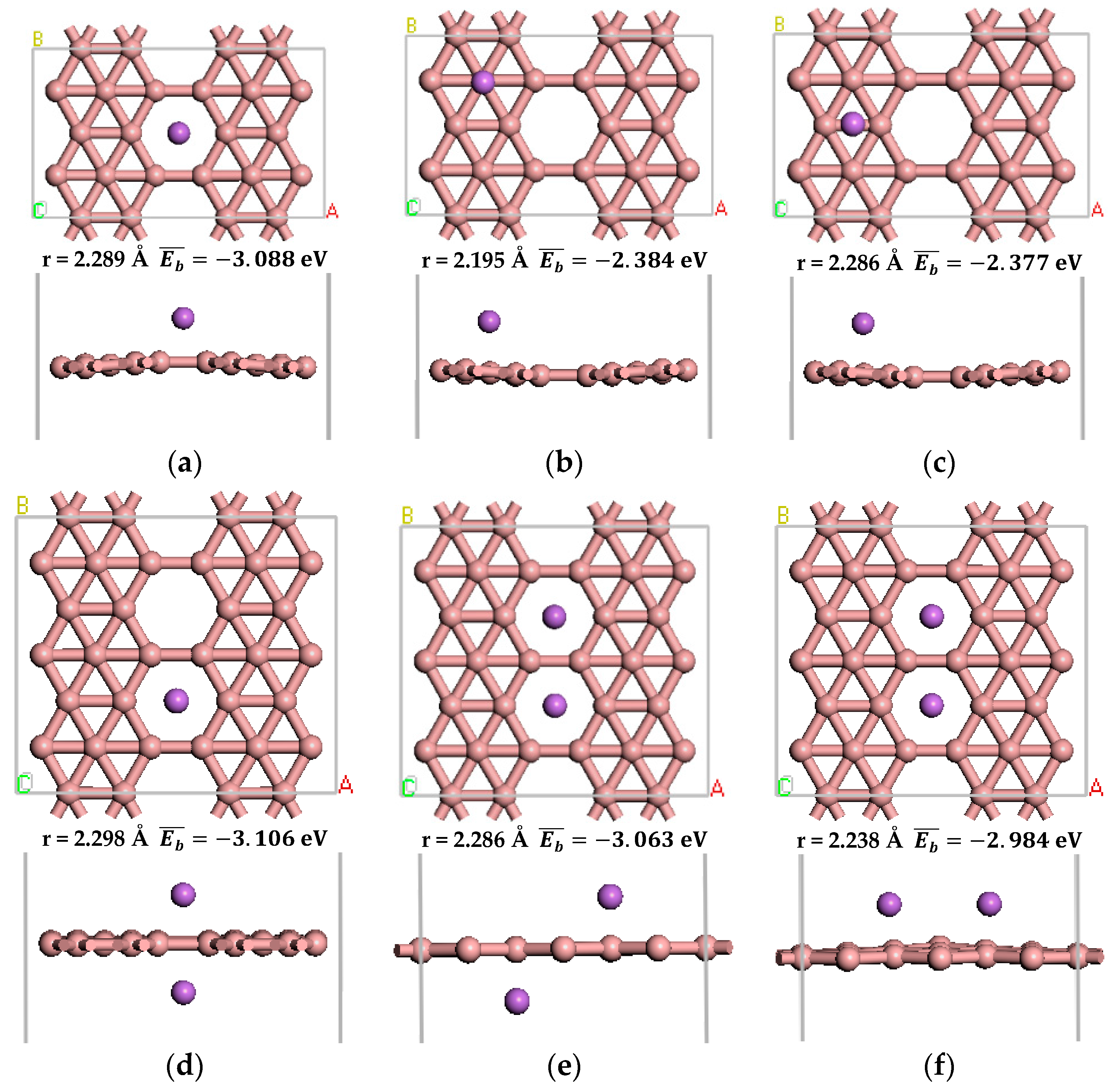
There are three stable adsorption structures of two Li atoms after adsorption on the β12-borophene as shown in Figure 3d–f, respectively. One of the most stable adsorption sites involves the two Li atoms being located on both sides of the same B ring. The distance between Li and the nearest B is 2.298 Å. The average adsorption energy is −3.106 eV, which is larger than the cohesive energy of Li atoms. After optimization, the relaxation of β12-borophene is very small. Each Li atom in the Li-β12-borophene system is an active adsorption site, allowing a large number of H2 molecules to be adsorbed around the Li atom in order to significantly increase the hydrogen storage capacity.
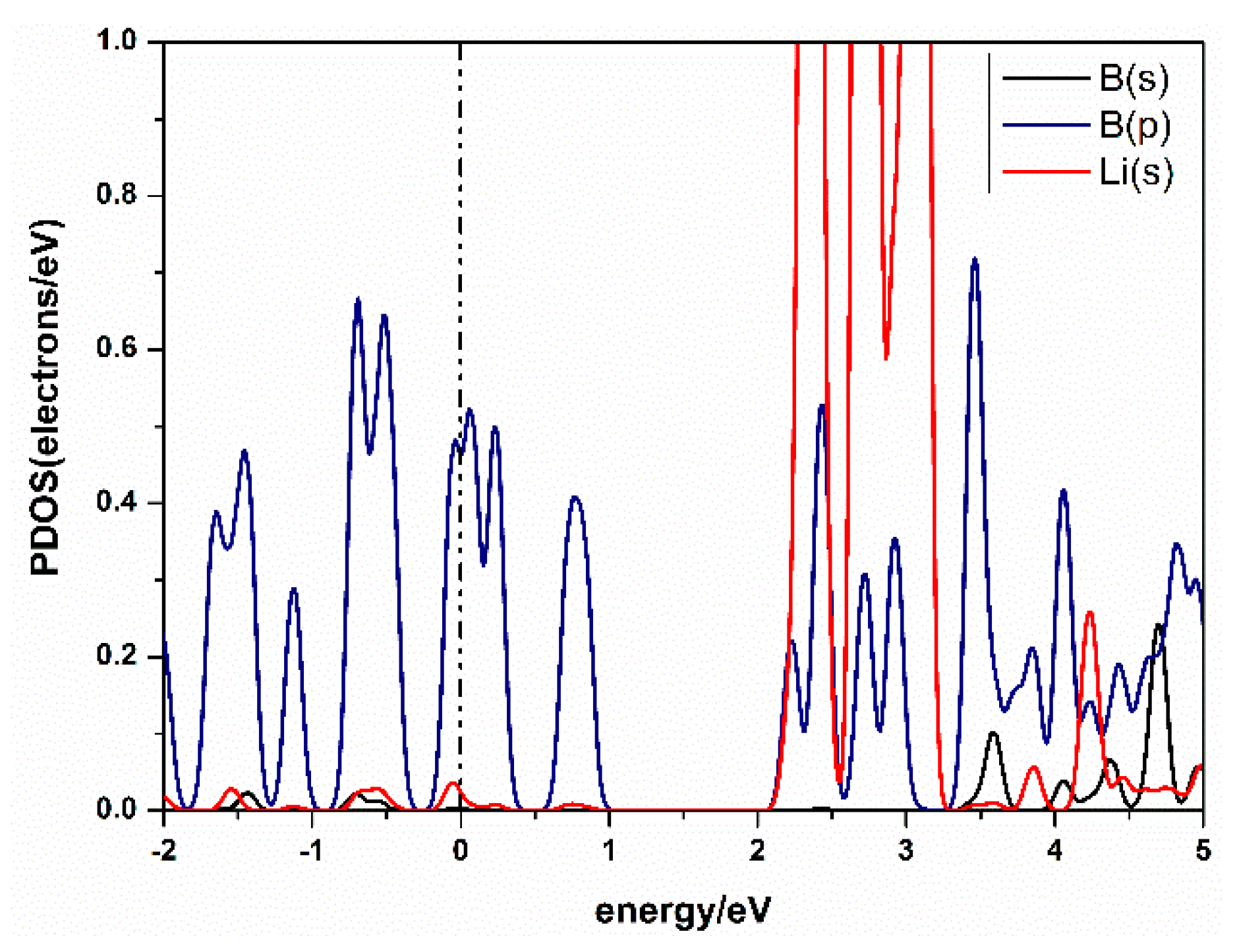
The charge transfer between atoms can be analyzed by Mulliken analysis [40], which shows that the charge was transferred from Li to B. From the Partial Densities of States (PDOS) of the Li-β12-borophene structure in Figure 4, we found the peak of B atom’s 2p orbital overlaps with the peak of the Li atom’s 1s orbital. This suggests a strong hybridization between B and Li atoms. A similar binding mechanism has also been confirmed in other metal-modified nanostructures [41]. In addition, it can be seen from the PDOS that the metal properties of β12-borophene did not change after modification of Li atom.
3.2.2. Adsorption of H2 Molecules on Li-β12-Borophene
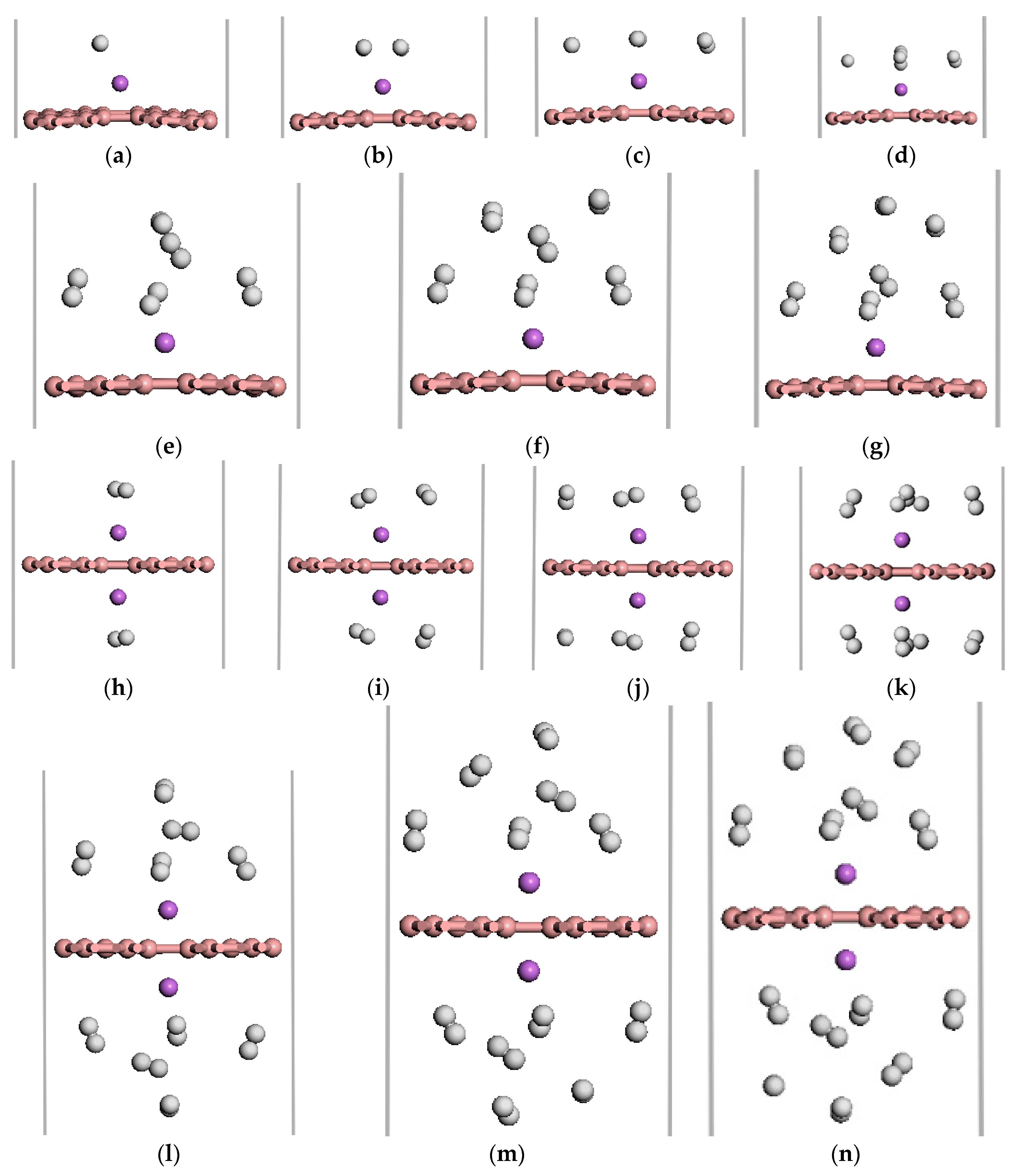
We investigated the adsorption properties of H2 molecules on Li-β12-borophene. Figure 5 shows the optimized geometries of 1–7 H2 molecules adsorbed on the Li-modified β12-borophene. Table 1 lists the adsorption energy and average adsorption energy calculated by the GGA PBE functional and DFT-D methods. First, we investigate the adsorption sites of H2 molecules on Li-β12-borophene. For the first adsorbed H2 molecules, many adsorption sites were considered in order to find the most stable site. The most stable site involves H2 being parallel to the β12-borophene plane, which is opposite to the H2 vertical adsorption on Ca-β12-borophene [17]. After adsorption, the corresponding rH-H of the adsorbed H2 is 0.756 Å, which is larger than the distance of free H2 (0.753 Å). To investigate the maximum storage capacity of single Li atom-modified β12-borophene, more H2 was added around Li gradually. The minimum distance between the H and Li atom are range of 2.164 to 6.368 Å. The first four H2 molecules were parallel to the β12-borophene and were around the Li atom at the same level. When the fifth H2 molecule was added to the system, two H2 molecules moved to an upper layer after relaxation. This may be due to the limited space around the Li atom and the repulsive interactions between the adsorbed H2. The average adsorption energy slowly reduced from −0.385 to −0.210 eV/H2 due to the strong steric interactions between the adsorbed H2. Interestingly, the adsorption energy suddenly rose to –0.388 eV after the second H2 molecule was added to the system. With an increase in the number of H2 molecules, the H2 molecules becomes further away from the Li atom and the adsorption weakens. The average adsorption energy was at its minimum (−0.210 eV/H2) when the seventh H2 molecule was adsorbed. At this time, the hydrogen storage capacity reached 5.90 wt %, which exceeded the ideal hydrogen storage capacity (over 5.5 wt %). In order to further increase the hydrogen storage capacity, we added two Li atoms to decorate the β12-borophene to adsorb H2 molecules. 2Li-β12-borophene can adsorb up to 14 H2 molecules and the minimum average adsorption energy is −0.220 eV. The hydrogen storage capacity can reach up to 10.85 wt %, which is larger than the hydrogen storage capacity with a gravimetric hydrogen density of 9.5 wt % of the Ca-β12-borophene/H2 system [17]. The optimized structure is shown in Figure 5h–n. The average adsorption energy () is in the range of −0.381 to −0.220 eV/H2, which is necessary for practical application [3,4]. In addition, the calculated adsorption energy and average energy of H2 adsorption by DFT-D method are larger than those calculated by the GGA PBE functional, but the overall adsorption method has not changed.
3.2.3. Electronic Properties of Li-β12-Borophene/H2
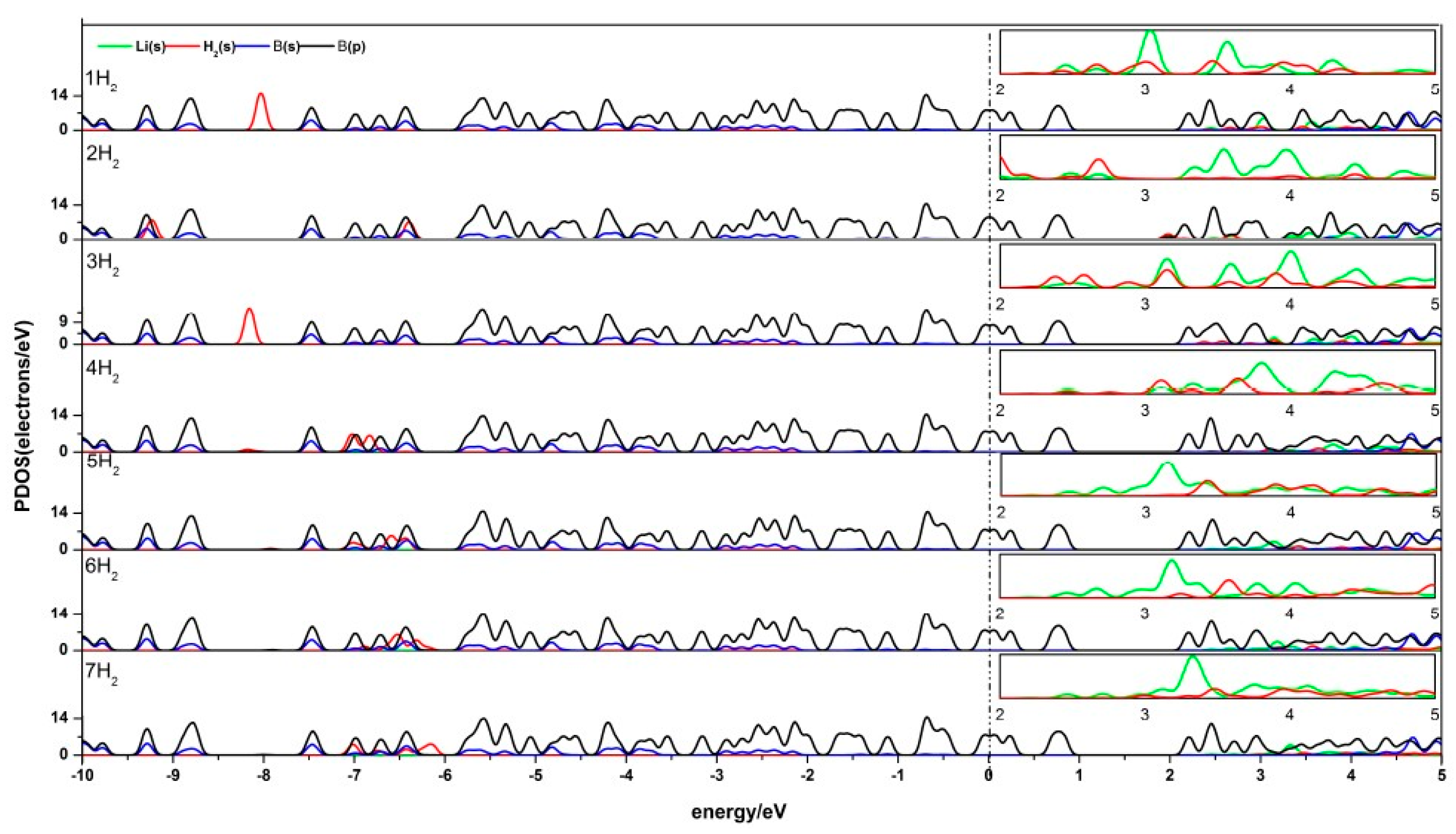
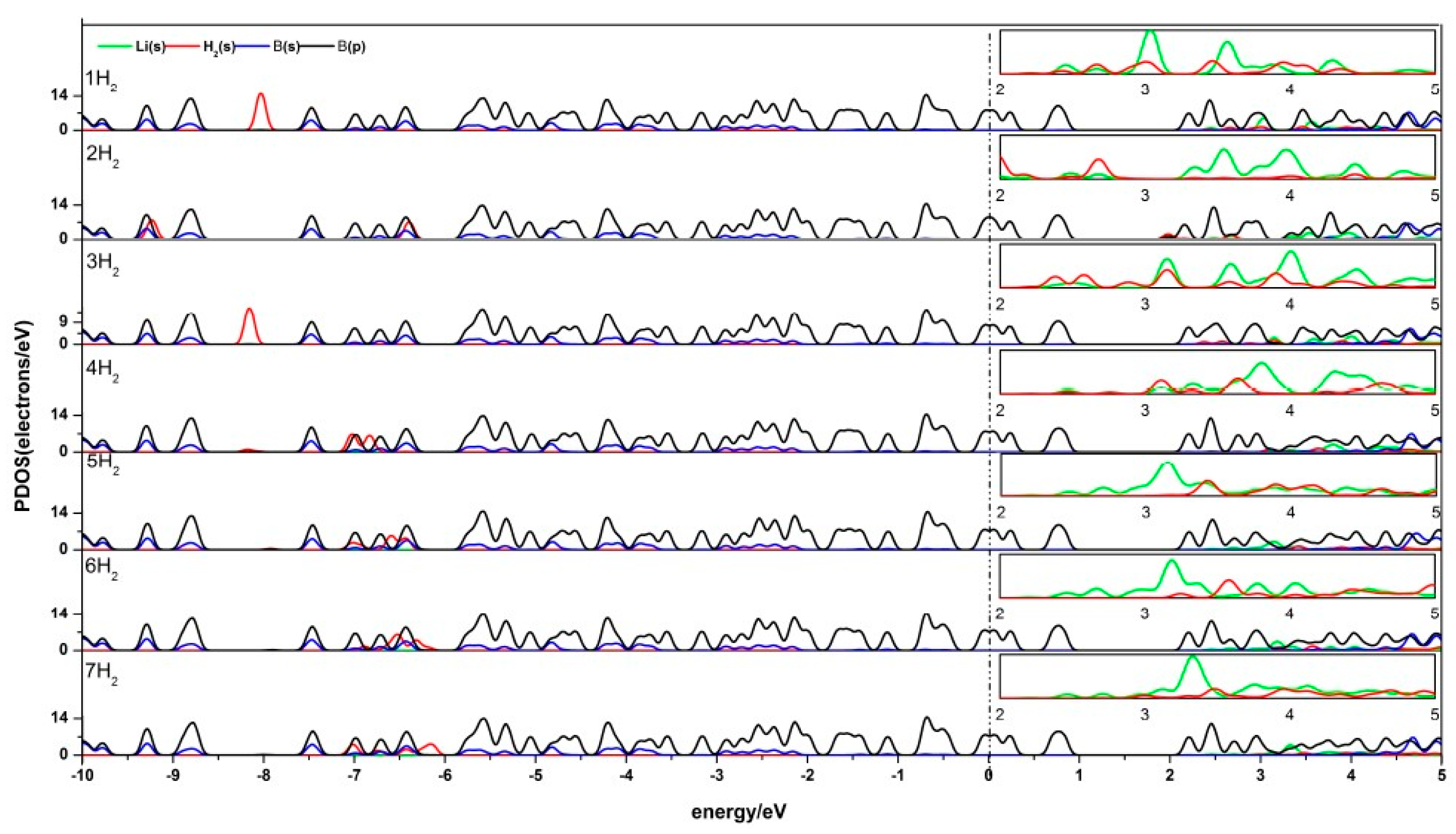
The density of states (DOS) reflects the number of states of the unit energy, which is important in further understanding the interaction between H2 and Li-β12-borophene. The partial density of states (PDOS) of Li-β12-borophene/H2 is shown in Figure 6. Obvious hybridizations between the Li-s orbit and H-s orbit can be found in 2.0 eV~5.0 eV, which demonstrates a strong interaction between H2 and Li atoms. With an increase in the number of H2 molecules, the peak values of H2 molecules become smaller and further away from the fermi level. This indicates that the interaction between H2 molecules and Li-β12-borophene weakens, which is consistent with the average adsorption energy becoming smaller. Another overlap between the B-p and H-s orbits was found at –10.0 eV~5.0 eV. Upon the adsorption of the second H2 molecule, the H 1s orbit peaks move to the lift, implying an increased stability in the system. This is consistent with the increase in the Eads value after the second H2 addition. With an increase in the number of H2 molecules (an expected in the second H2), the H-s orbits move to the right and the peak values become smaller, which indicates that the interaction between H2 and β12-borophene becomes increasingly weaker. This conclusion is consistent with the decrease in the average adsorption energy ). The B-p and Li-s orbits also have hybridization, which implies an interaction between the Li and B atoms. The peaks near and below the fermi surface are mostly contributed by the B-s orbits, which means that the H2 molecules and Li atom have less influence on the β12-borophene. The comparison of the PDOS of a single Li-β12-borophene show that the interaction between Li atom and β12-borophene is weakened due to the adsorption of H2 molecules. The PDOS of two Li-β12-borophene/H2 consistent with this analysis.
The bonding strength between atoms can be quantitatively analyzed based on the Mulliken charge population and bond population. Table 2 shows the Mulliken charge population before and after one H2 moleculebecomes absorbed on the Li-β12-borophene. H (1) and H (2) represent the two H atoms of the adsorbed H2 molecule; while B1, B5 and B6 are three B atoms that transfer the greatest amount of charge in the β12-borophene (as shown in Figure 1). The two H atoms have charges of 0.06 e and 0.05 e, respectively. In contrast, the Li atom loses 1.40 e, which occurs mainly in the H and Li atomic orbits. The Li atom transfers charge to the H2 molecules, resulting in the H2 molecules carrying more negative charge and Li atom showing positive charge. The interaction between the H2 molecules and the Li atom is consistent with the conclusion of the PDOS analysis. In addition, the B atoms obtains charge, with this charge transfer mainly occurring in the B-2p orbits and H-s orbits. This is in contrast with the Mulliken charge population of the β12-borophene/H2, in which the charge transfer mainly occurs in H and B atoms forming a covalent bond of H–B that is not favorable for the desorption of H2. Due to the β12-borophene being modified by Li atoms, H2 molecules and B atoms only have small interactions, resulting in the H2 molecules physically adsorbing on the Li-β12-borophene. This is conducive for H2 desorption and increases the hydrogen storage capacity.
4. Conclusions
In summary, we performed a study on hydrogen storage properties of pure β12-borophene and Li-decorated β12-borophene through DFT calculations. It is found that H2 molecules are mainly adsorbed on pure β12-borophene as chemical adsorption with an adsorption energy of −0.536 eV. The H2 molecules are dissociated into two H atoms, which tend to the bridge of two B site and the H–B bond to form covalent bond. In order to improve the hydrogen storage performance of pure β12-borophene and increase the hydrogen storage capacity, we use the Li atom to modify the β12-borophene. It is found that a single Li atom adsorbed on the center of Boron ring with the adsorption energy −3.088 eV, the Li-β12-borophene can adsorb up to 7 H2 molecules with the average adsorption energy of −0.210 eV/H2. The charge transfer of the Li-β12-borophene/H2 is that H and B atoms lose electron, Li atom get electron. We use two Li atoms to modify β12-borophene to increase its hydrogen storage capacity. It is find that the two Li atoms are located at the same position on both sides of the same boron hole. 2Li-β12-borophene system can adsorb up to 14 H2 molecules and the hydrogen storage capacity up to 10.85 wt %. The average adsorption energy is range of −0.381 to −0.220 eV/H2, which is necessary for practical application [3,4].
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 51562022), the fund for the State Key Laboratory of Advanced Processing and Recycling of Non-Ferrous Metals, Lanzhou University of Technology (grant number SKLAB02014004), the Basic Scientific Research Foundation for Gansu Universities of China (grant number 05-0342), the Science and Technology Project of Lanzhou City (grant number 2011-1-10), the Natural Science Foundation of Gansu Province (Grant No. 17JR5RA123), and the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (second phase).
Author Contributions
Yuhong Chen designed the project, Tingting Liu performed the calculations, Yuhong Chen and Tingting Liu prepared the manuscript, Haifeng Wang and Cairong Zhang revised the paper, Meiling Zhang and Lihua Yuan analyzed the data, and all authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Song, Y.; Guo, Z.X.; Yang, R. Influence of selected alloying elements on the stability of magnesium dihydride storage applications: A first-principles investigation. Phys. Rev. B 2004, 69, 094205. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.T.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Hydrogen storage in microporous metal-organic frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Goddard, W.A. Lighium-doped metal-organic frameworks for reversible H2 storage at ambient temperature. J. Am. Chem. Soc. 2007, 129, 8422–8423. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Energy. Hydrogen, Fuel Cells Program: FY Annual Progress Report; U.S. Department of Energy: Washington, DC, USA, 2014.
- Seenithurai, S.; Pandyan, R.K.; Kumar, S.V.; Saranya, C.; Mahendran, M. Al-decorated carbon nanotube as the molecular hydrogen storage medium. Int. J. Hydrog. Energy 2014, 39, 11990–11998. [Google Scholar] [CrossRef]
- Wu, M.H.; Gao, Y.; Zhang, Z.Y.; Zeng, X.C. Edge-decorated graphene nanoribbons by scandium as hydrogen storage media. Nanoscale 2012, 4, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, Two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Yang, Y.; Gao, G.Y.; Yakobson, B.I. Two-Dimensional Boron Monolayers Mediated by metal Substrates. Angew. Chem. Int. Ed. Engl. 2015, 127, 13022–13026. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.J.; Zhang, J.; Zhong, Q.; Li, W.B.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K.H. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.J.; Zhang, J.; Liu, R.Y.; Iimori, T.; Lian, C.; Li, H.; Chen, L.; Wu, K.H.; Meng, S.; Komori, F.; et al. Direct evidence of metallic bands in a monolayer boron sheet. Phys. Rev. B 2016, 94, 041408. [Google Scholar] [CrossRef]
- Padilha, J.E.; Miwa, R.H.; Fazzio, A. Directional dependence of the electronic and transport properties of 2D borophene and borophane. Phys. Chem. Chem. Phys. 2016, 18, 25491–25496. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Rahaman, O.; Dianat, A.; Rabczuk, T. Mechanical responses of borophene sheets: A first-principles study. Phys. Chem. Chem. Phys. 2016, 18, 27405. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Li, Q.F.; Gao, Y.; Miao, F.; Zhou, X.F.; Wan, X.G. Strain effects on borophene: Ideal strength, negative Possion’s ration and phonon instability. New J. Phys. 2016, 18, 073016. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.J.; Tang, Z.; Wang, X.F.; Wang, L.; Hou, T.G.; Lin, H.P.; Li, Y.Y. Stable and metallic borophene nanoribbons from first-principles calculations. J. Mater. Chem. C 2016, 4, 6380–6385. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Wang, L.; Zhang, W.T.; Zhang, J.L.; Yuan, Y.Q. Ca-decorated borophene as potential candidates for hydrogen storage: A first-principle study. Int. J. Hydrog. Energy 2017, 42, 20036–20045. [Google Scholar] [CrossRef]
- Wang, Y.H.; Meng, Z.S.; Liu, Y.Z.; You, D.; Wu, K.; Lv, J.; Wang, X.Z.; Deng, K.M.; Rao, D.; Lu, R.F. Lithium decoration of three dimensional boron-doped graphene frameworks for high-capacity storage. Appl. Phys. Lett. 2015, 106, 2721. [Google Scholar] [CrossRef]
- Reunchan, P.; Jhi, S.H. Metal-dispersed porous graphene for hydrogen storage. Appl. Phys. Lett. 2011, 98, 93103. [Google Scholar] [CrossRef]
- Ao, Z.M.; Jiang, Q.; Zhang, R.Q.; Tan, T.T.; Li, S. Al doped graphene: A promising material for hydrogen storage at room temperature. J. Appl. Phys. 2009, 105, 074307. [Google Scholar] [CrossRef]
- Faye, O.; Eduok, U.; Szpunar, J.; Szpunar, B.; Samoura, A.; Beye, A. Hydrogen Storage on bare Cu atom and Cu-functionalized boron-doped graphene: A first principles study. Int. J. Hydrog. Energy 2017, 42, 4233–4243. [Google Scholar] [CrossRef]
- Faye, O.; Szpunar, J.A.; Szpunar, B.; Beye, A.C. Hydrogen adsorption and storage on Palladium-functionalized graphene with NH-dopant: A first principles calculation. Appl. Surf. Sci. 2017, 392, 362–374. [Google Scholar] [CrossRef]
- Yuan, L.H.; Chen, Y.H.; Kang, L.; Zhang, C.R.; Wang, D.B.; Wang, C.N.; Zhang, M.L.; Wu, X.J. First-principles investigation of hydrogen storage capacity of Y-decorated porous graphene. Appl. Surf. Sci. 2017, 399, 463–468. [Google Scholar] [CrossRef]
- Zhao, Y.; Kim, Y.H.; Dillion, A.C.; Heben, M.J.; Zhang, S.B. Hydrogen storage in novel organometallic buckyballs. Phys. Rev. Lett. 2005, 94, 155504. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Yang, S.Y.; Hicke, C.; Wang, E.; Geohegan, D.; Zhang, Z.Y. Calcium as the superior coating metal in functionalization of carbon fullerenes for high-capacity hydrogen storage. Phys. Rev. Lett. 2008, 100, 206806. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, Z.G.; Liu, S.Q.; Zhao, X.H.; Huang, K.L. High-capacity hydrogen storage medium: Ti doped fullerene. Appl. Phys. Lett. 2011, 98, 023107. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.; Refson, K.R.; Payne, M.C. First Principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, T.; Gulans, A.; Krasheninnikov, A.V.; Nieminen, R.M. Van der Waals Bonding in Layered Compounds from Advanced Density-Functional First-Principles Calculations. Phys. Rev. Lett. 2012, 108, 235502. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Hu, W.; Xia, N.; Wu, X.; Li, Z.; Yang, J. Silicene as a highly sensitive molecule sensor for NH3, NO and NO2. Phys. Chem. Chem. Phys. 2014, 16, 6957–6962. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Wang, J.; Yuan, L.H.; Zhang, M.L.; Zhang, C.R. Sc-Decorated Porous Graphene for High-Capacity Hydrogen Storage: First-Principles Calculations. Materials 2017, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dai, J.; Zhuo, Z.; Yang, J.; Zeng, X. Two-Dimensional Boron Monolayer Sheets. ACS Nano 2012, 6, 7443–7453. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Zhang, H.; Shao, H.; Ning, Z.; Xu, Y.; Ni, G.; Lu, H.; Zhang, D.; Zhu, H. Stability and strength of atomically thin borophene from first principles calculations. Mater. Res. Lett. 2017, 5, 399–407. [Google Scholar] [CrossRef]
- Peng, B.; Zhang, H.; Shao, H.Z.; Xu, Y.F.; Zhang, R.J.; Zhua, H.Y. Electronic, Optical, and thermodynamic properties of borophene from first-principle calculations. J. Mater. Chem. C 2016, 4, 3592–3598. [Google Scholar] [CrossRef]
- Pan, C.C.; Chen, Y.H.; Wu, N.; Zhang, M.L.; Yuan, L.H.; Zhang, C.R. A First Principles Study of H2 Adsorption on LaNiO3(001) Surfaces. Materials 2017, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Ataca, C.; Akturk, E.; Ciraci, S.; Ustunel, H. High-capacity hydrogen storage by metallized graphene. Appl. Phys. Lett. 2008, 93. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Wang, Q.; Jena, P.; Kawazoe, Y. Clustering of Ti on a C60 surface and its effect on hydrogen storage. J. Am. Chem. Soc. 2005, 127, 14582–14583. [Google Scholar] [CrossRef] [PubMed]
- Doll, K.; Harrison, N.M.; Saunders, V.R. A density functional study of lithium bulk and surfaces. J. Phys.-Condens. Matter 1999, 11, 5007–5019. [Google Scholar] [CrossRef]
- Mulliken, R.S. Molecular Compounds and Their Spectra. V. Orientation in Molecular Complexes. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- An, H.; Liu, C.S.; Zeng, Z.; Fan, C.; Ju, X. Li-doped B2C graphene as potential hydrogen storage medium. Appl. Phys. Lett. 2011, 98, 173101–173103. [Google Scholar] [CrossRef]
Figure 1.
The optimized atomic structure of pure β12-borophene. The alphanumeric characters on the graph represent the corresponding atoms.
Figure 1.
The optimized atomic structure of pure β12-borophene. The alphanumeric characters on the graph represent the corresponding atoms.
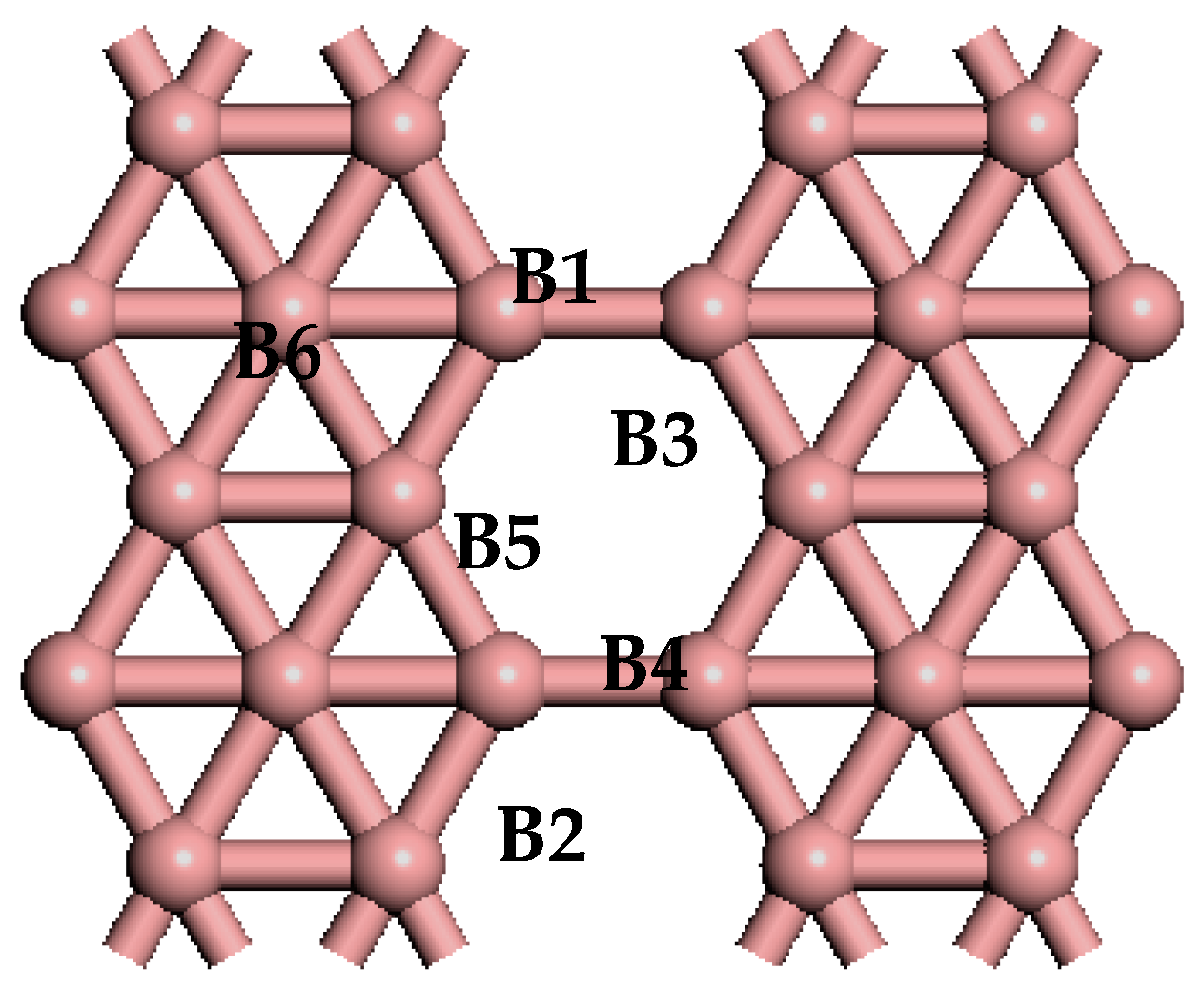
Figure 2.
(a–e) show the five stable optimized geometrical structures of β12-borophene/H2.
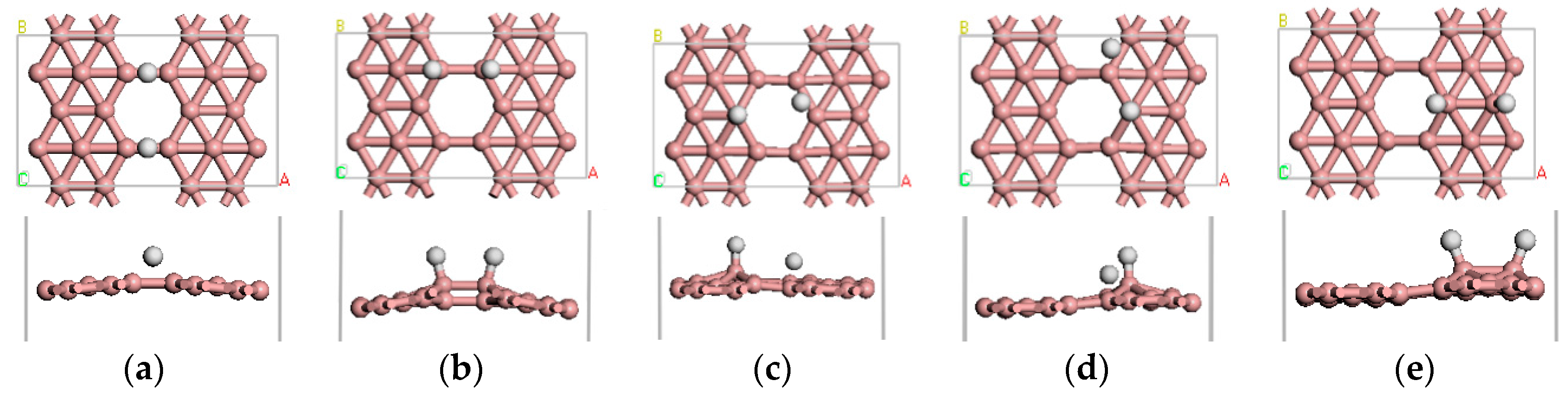
Figure 3.
The optimized atomic structure of Li atom decorated β12-borophene. (a–c) show the one Li atom decorated single-sided β12-borophene, respectively. (d–f) show the two Li atoms decorated double-sided β12-borophene, respectively.
Figure 3.
The optimized atomic structure of Li atom decorated β12-borophene. (a–c) show the one Li atom decorated single-sided β12-borophene, respectively. (d–f) show the two Li atoms decorated double-sided β12-borophene, respectively.

Figure 4.
Partial density of states (PDOS) of Li-decorated β12-borophene system.
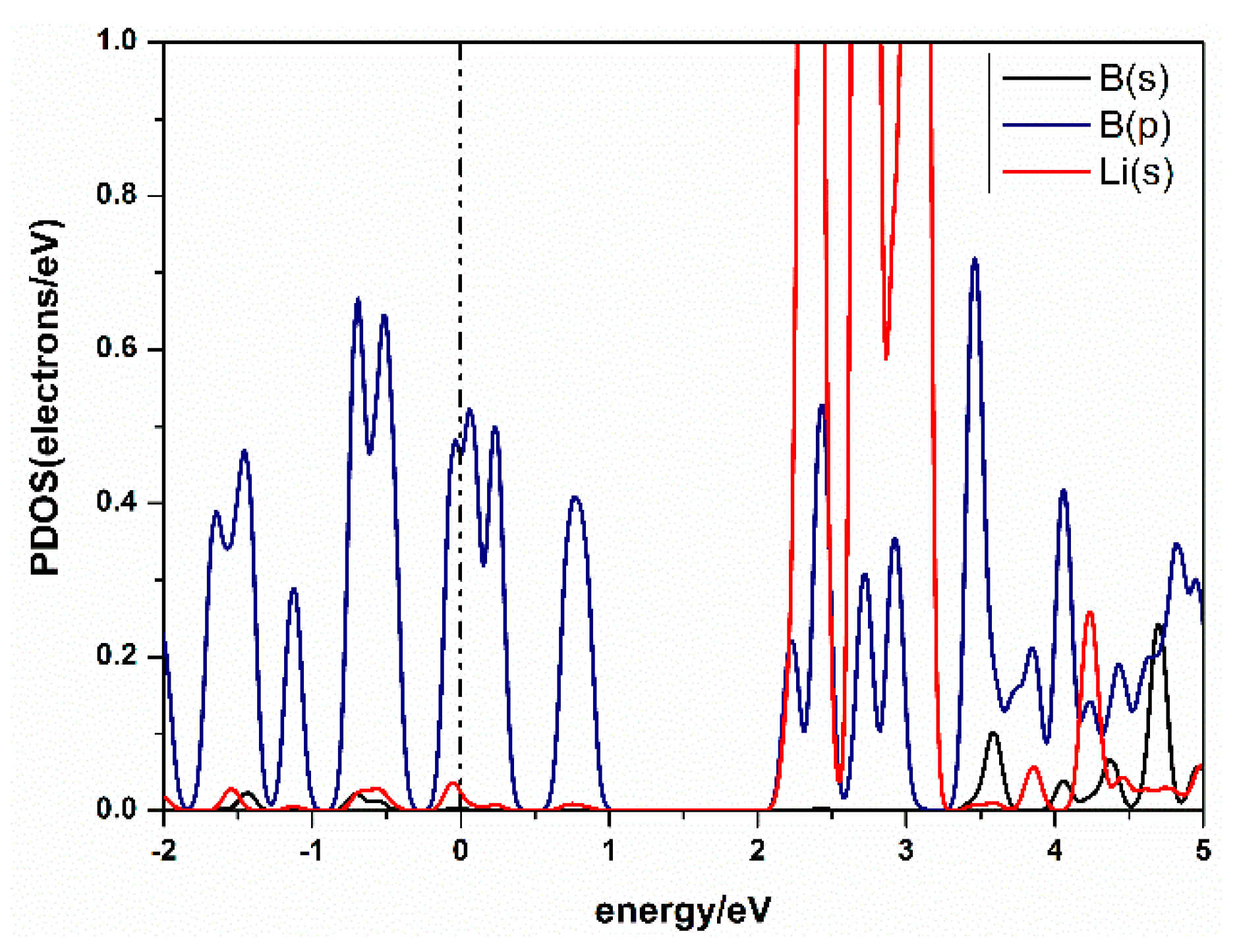
Figure 5.
The optimized atomic structures of the Li-β12-borophene/H2. (a–g) are 1~7 H2 molecules adsorption on Li-β12-borophene system. (h–n) are 2~14 H2 molecules adsorption on 2Li-β12-borophene system. The pink, purple and white balls in this and aforementioned figures express B, Li and H atoms, respectively.
Figure 5.
The optimized atomic structures of the Li-β12-borophene/H2. (a–g) are 1~7 H2 molecules adsorption on Li-β12-borophene system. (h–n) are 2~14 H2 molecules adsorption on 2Li-β12-borophene system. The pink, purple and white balls in this and aforementioned figures express B, Li and H atoms, respectively.
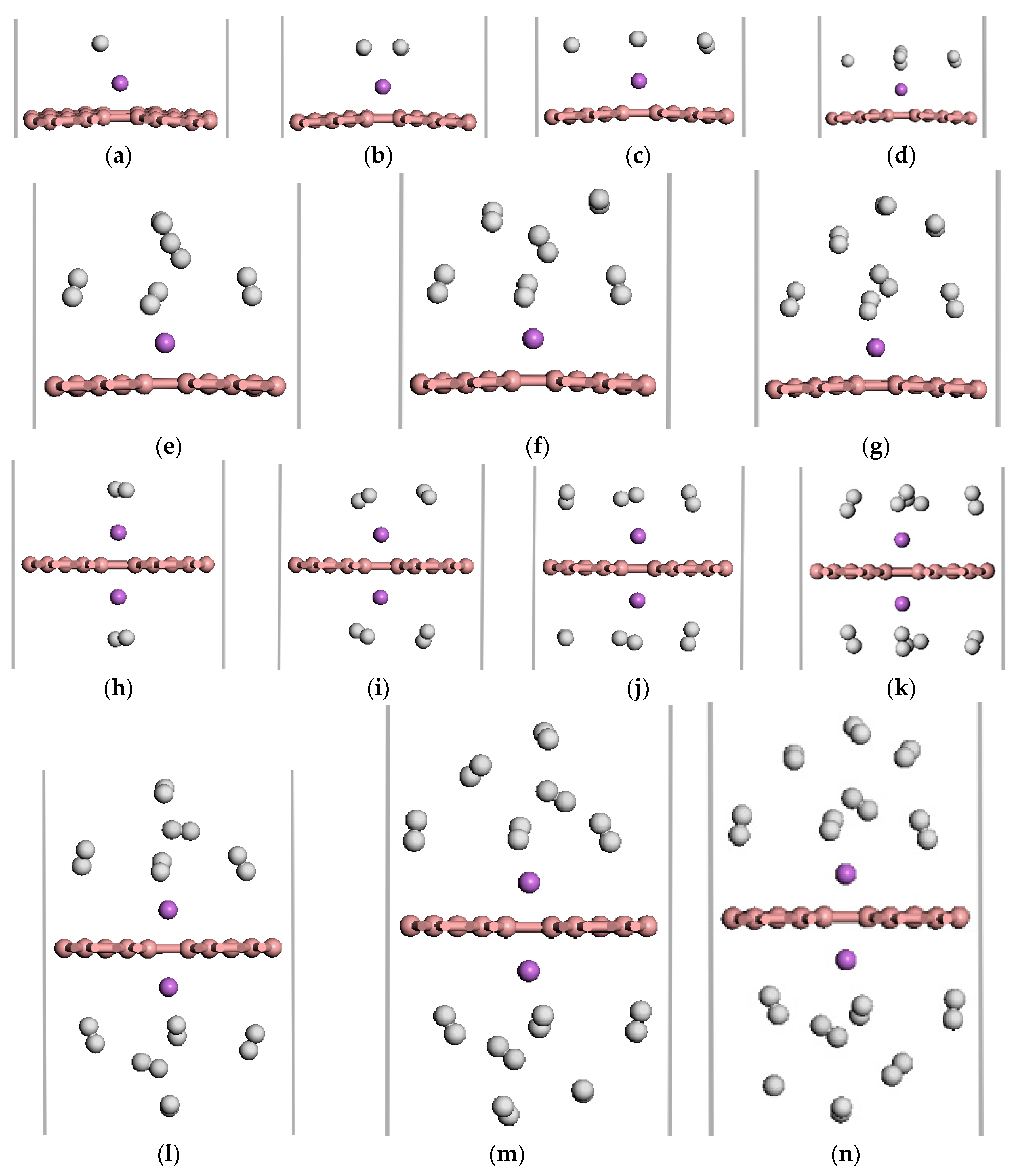
Figure 6.
PDOS of Li-β12-borophene with 1–7 H2 molecules adsorbing. (The PDOS of Li-s orbit and H-s orbit in the range of 2.0 eV~5.0 eV is enlarged as shown in the small box above each corresponding figure.)
Figure 6.
PDOS of Li-β12-borophene with 1–7 H2 molecules adsorbing. (The PDOS of Li-s orbit and H-s orbit in the range of 2.0 eV~5.0 eV is enlarged as shown in the small box above each corresponding figure.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The adsorption energy, average adsorption energy, the distance between H and H (rH-H), the distance between H and Li of Li-β12-borophene system (rH-Li).
Table 1.
The adsorption energy, average adsorption energy, the distance between H and H (rH-H), the distance between H and Li of Li-β12-borophene system (rH-Li).
| Li-β12-borophene | Number of H2 | 1 H2 | 2 H2 | 3 H2 | 4 H2 | 5 H2 | 6 H2 | 7 H2 |
| /eV | −0.247 | −0.281 | −0.154 | −0.179 | −0.139 | −0.169 | −0.134 | |
| (DFT-D)/eV | −0.385 | −0.388 | −0.251 | −0.147 | −0.160 | −0.167 | −0.142 | |
| /eV/H2 | −0.247 | −0.213 | −0.194 | −0.190 | −0.181 | −0.178 | −0.173 | |
| (DFT-D)/eV/H2 | −0.385 | −0.387 | −0.286 | −0.251 | −0.233 | −0.222 | −0.210 | |
| rH-H/Å | 0.756 | 0.757 | 0.753 | 0.755 | 0.753 | 0.753 | 0.753 | |
| rH-Li/Å | 2.164 | 2.169 | 3.813 | 3.810 | 4.661 | 5.667 | 6.368 | |
| 2Li-β12-borophene | Number of H2 | 2 H2 | 4 H2 | 6 H2 | 8 H2 | 10 H2 | 12 H2 | 14 H2 |
| (DFT-D)/eV/H2 | −0.381 | −0.298 | −0.274 | −0.262 | −0.230 | −0.226 | −0.220 |
Table 2.
Mulliken population analysis of the Li-β12-borophene before and after one H2 molecule adsorption.
Table 2.
Mulliken population analysis of the Li-β12-borophene before and after one H2 molecule adsorption.
| Atom | Mulliken | |||||
|---|---|---|---|---|---|---|
| Before Adsorption/e | After Adsorption/e | |||||
| s | p | Charge | s | p | Charge | |
| H (1) | 1.0 | 1.06 | −0.06 | |||
| H (2) | 1.0 | 1.05 | −0.05 | |||
| B1 | 0.82 | 2.18 | 0 | 0.83 | 2.36 | −0.19 |
| B5 | 0.74 | 2.23 | 0.03 | 0.75 | 2.40 | −0.15 |
| B6 | 0.65 | 2.40 | −0.05 | 0.65 | 2.40 | −0.05 |
| Li | 3 | 0 | 0 | 1.60 | 1.40 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, T.; Chen, Y.; Wang, H.; Zhang, M.; Yuan, L.; Zhang, C. Li-Decorated β12-Borophene as Potential Candidates for Hydrogen Storage: A First-Principle Study. Materials 2017, 10, 1399. https://doi.org/10.3390/ma10121399
AMA Style
Liu T, Chen Y, Wang H, Zhang M, Yuan L, Zhang C. Li-Decorated β12-Borophene as Potential Candidates for Hydrogen Storage: A First-Principle Study. Materials. 2017; 10(12):1399. https://doi.org/10.3390/ma10121399
Chicago/Turabian StyleLiu, Tingting, Yuhong Chen, Haifeng Wang, Meiling Zhang, Lihua Yuan, and Cairong Zhang. 2017. "Li-Decorated β12-Borophene as Potential Candidates for Hydrogen Storage: A First-Principle Study" Materials 10, no. 12: 1399. https://doi.org/10.3390/ma10121399
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.