Cooperativity of Spin Crossover Complexes: Combining Periodic Density Functional Calculations and Monte Carlo Simulation


Abstract
:1. Introduction
1.1. Slichter–Drickamer Model
1.2. Periodic Density Functional Calculations
1.3. Ising-Like Models
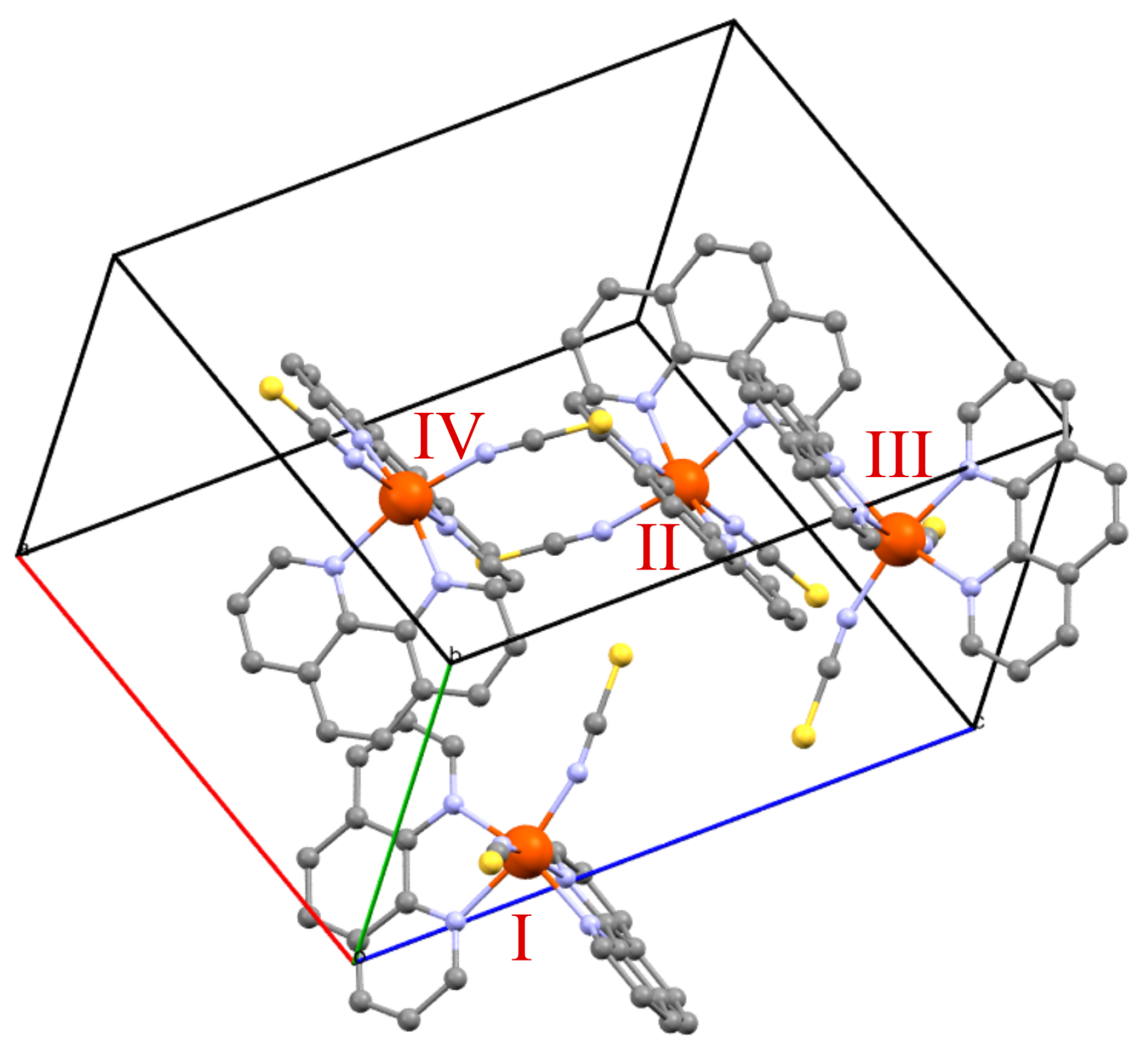
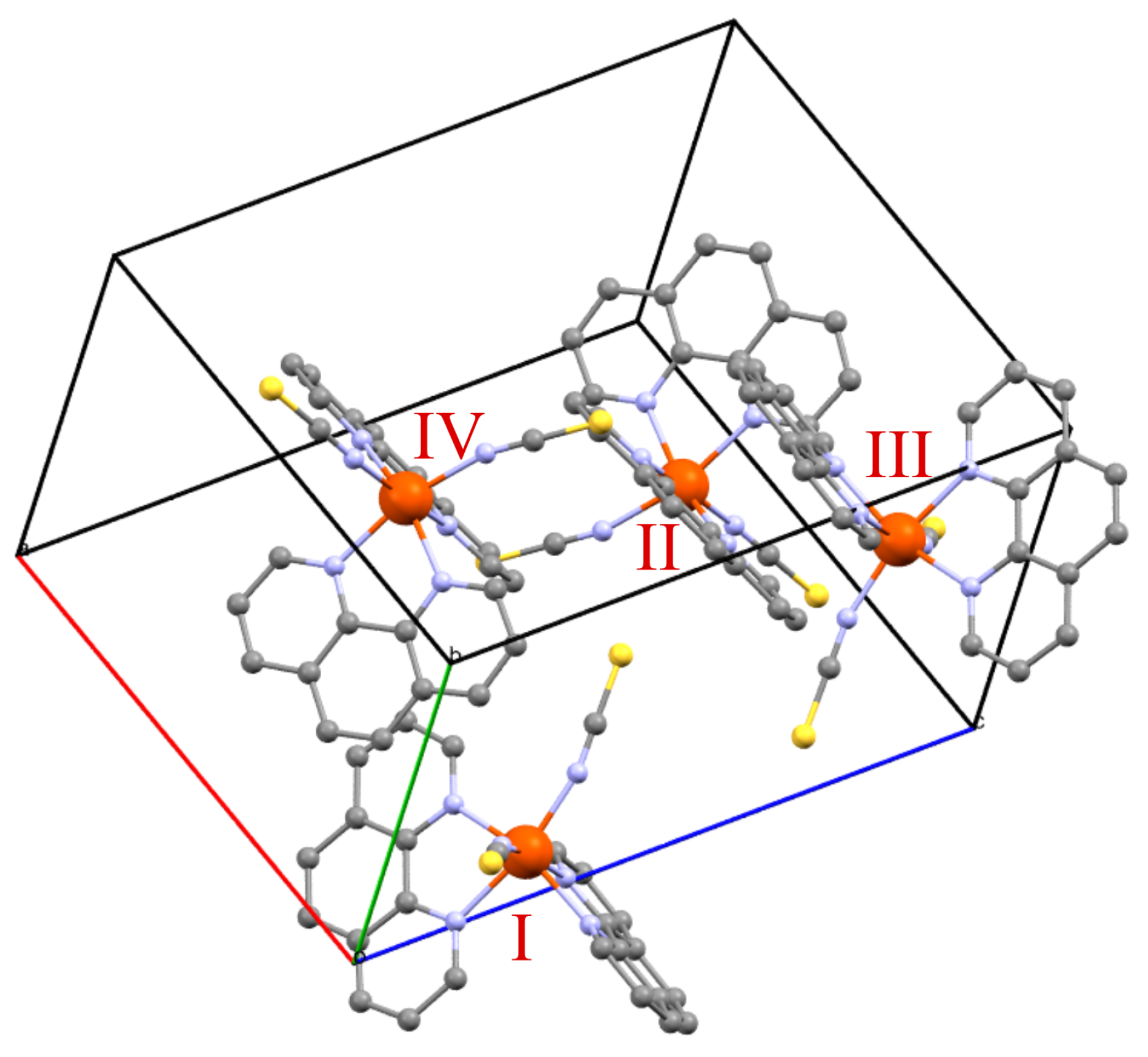
1.4. [Fe(phen)(NCS)]
2. Results and Discussion
2.1. DFT Calculations
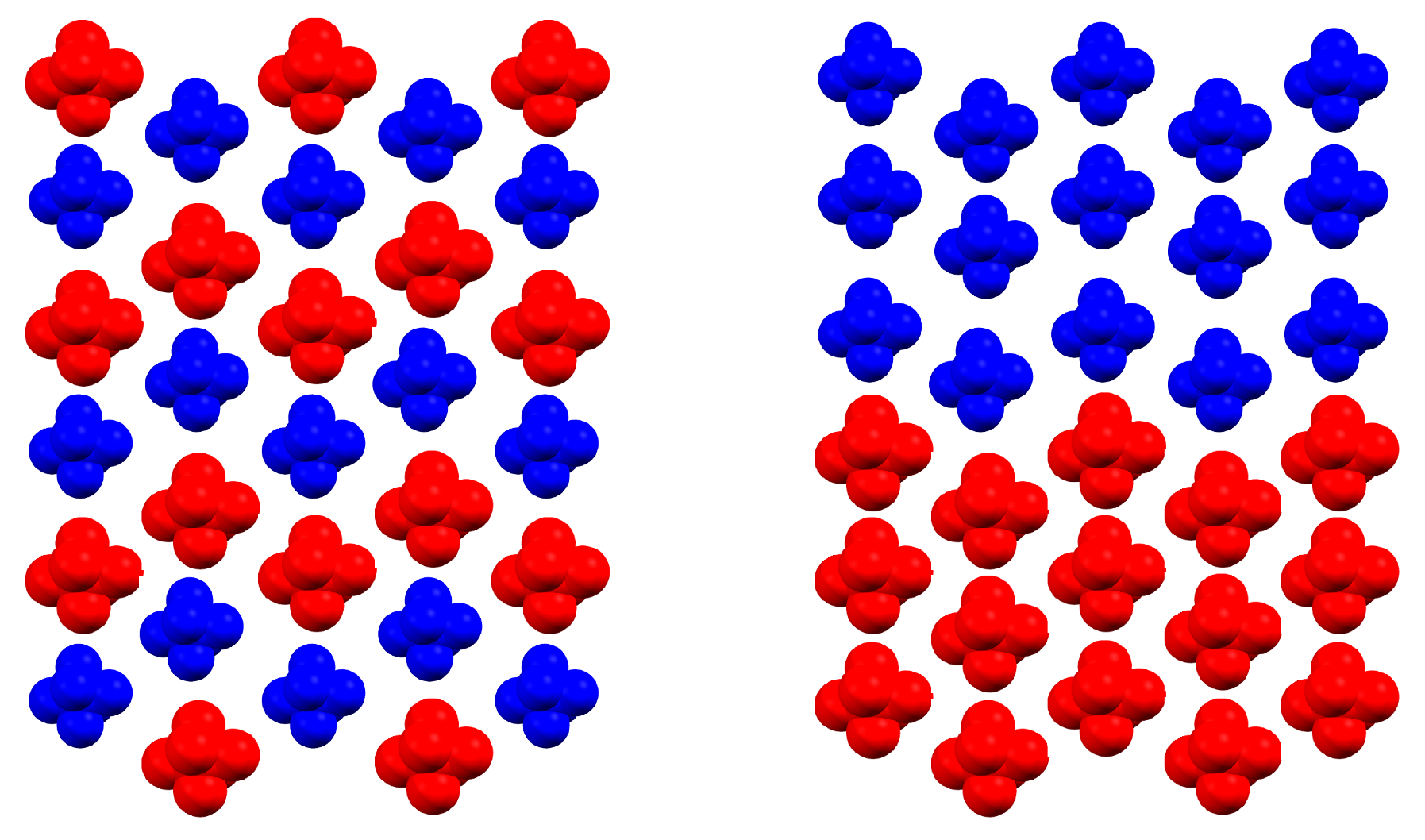
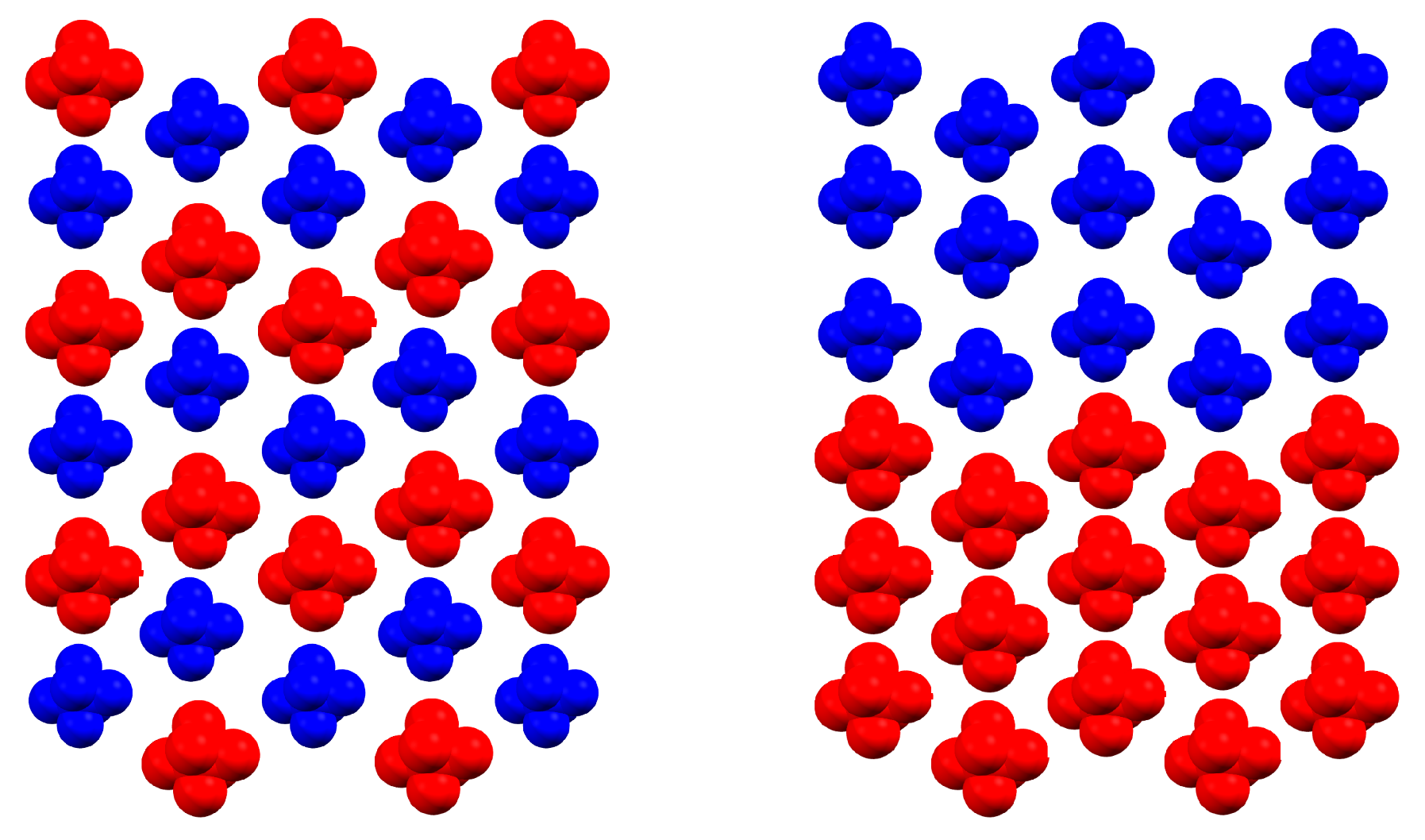
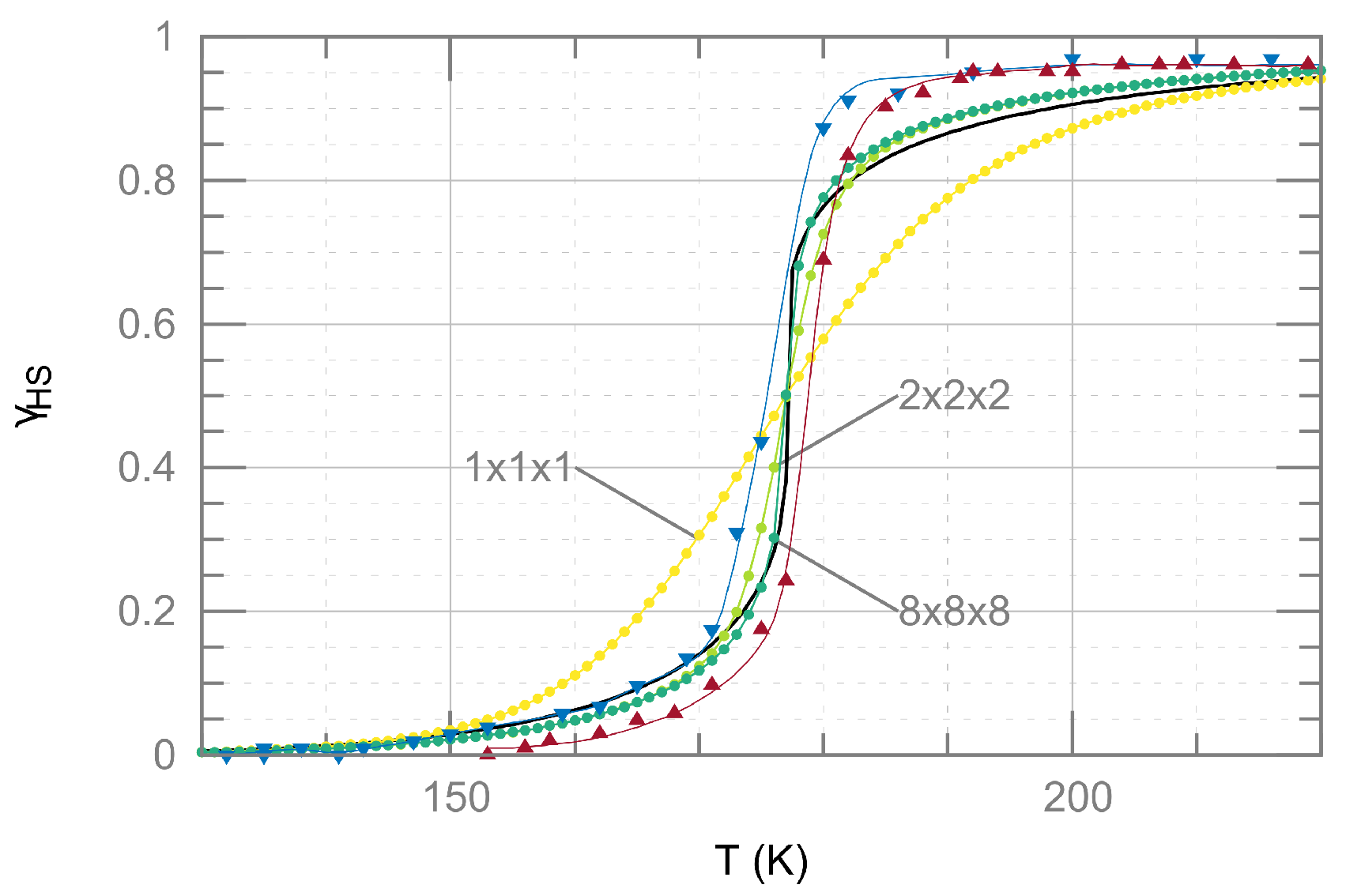
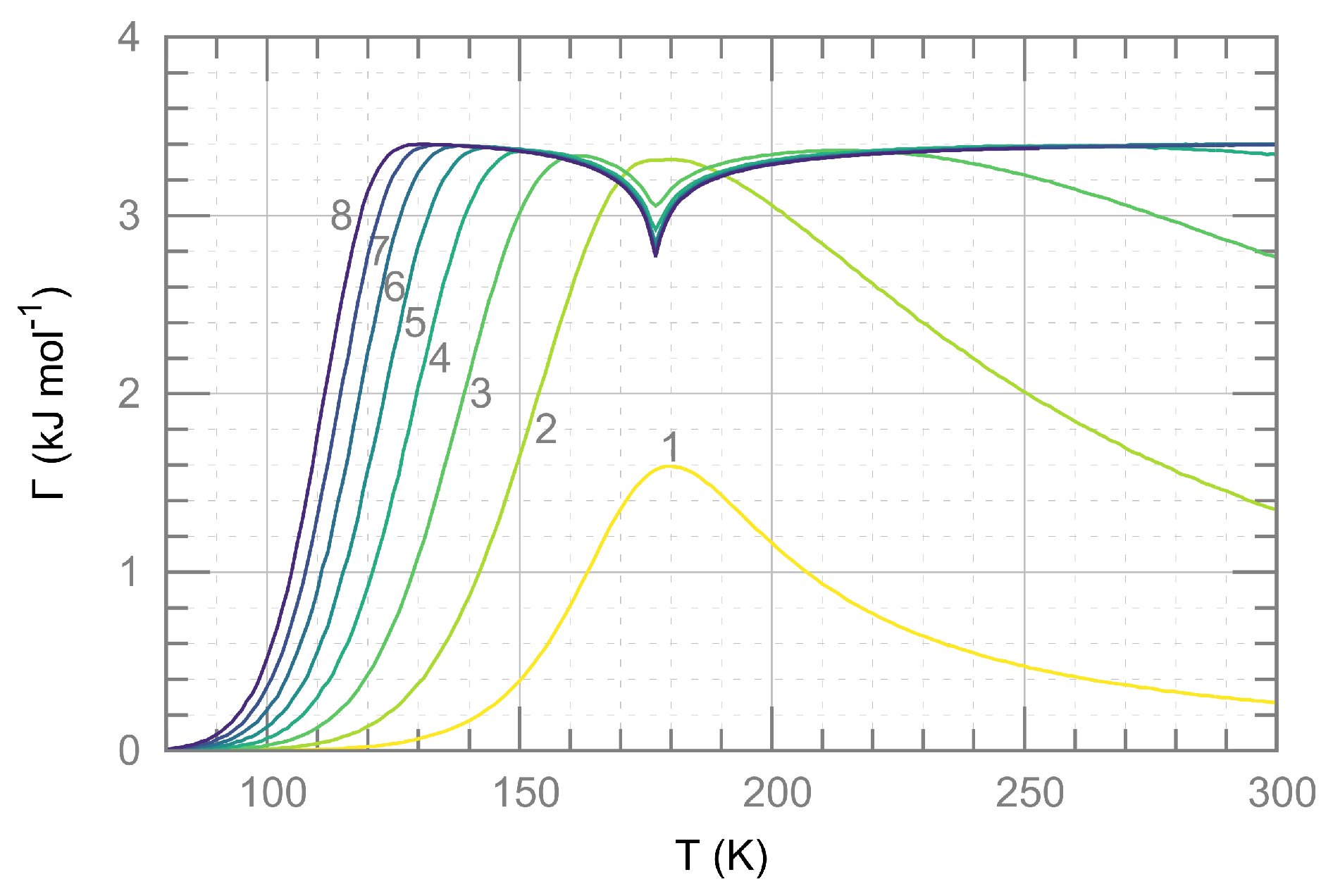
2.2. Ising-Like Model
3. Computational Details
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds I; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 1.
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds II; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 2.
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds III; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 3.
- Gütlich, P. Spin Crossover—Quo Vadis? Eur. J. Inorg. Chem. 2013, 2013, 581–591. [Google Scholar] [CrossRef]
- Decurtins, S.; Gütlich, P.; Köhler, C.; Spiering, H.; Hauser, A. Light-induced excited spin state trapping in a transition-metal complex: The hexa-1-propyltetrazole-iron (II) tetrafluoroborate spin-crossover system. Chem. Phys. Lett. 1984, 105, 1–4. [Google Scholar] [CrossRef]
- Paulsen, H.; Trautwein, A. Density Functional Theory Calculations for Spin Crossover Complexes. Top. Curr. Chem. 2004, 235, 197–219. [Google Scholar]
- Paulsen, H.; Schünemann, V.; Wolny, J.A. Progress in Electronic Structure Calculations on Spin-Crossover Complexes. Eur. J. Inorg. Chem. 2013, 5-6, 628–641. [Google Scholar] [CrossRef]
- Kepp, K.P. Consistent description of metal-ligand bonds and spin-crossover in inorganic chemistry. Coord. Chem. Rev. 2013, 257, 196–209. [Google Scholar] [CrossRef]
- Deeth, R.J. Molecular Discovery in Spin Crossover. In Spin States in Biochemistry and Inorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 85–102. [Google Scholar]
- Kepenekian, M.; Guennic, B.L.; Robert, V. Primary Role of the Electrostatic Contributions in a Rational Growth of Hysteresis Loop in Spin-Crossover Fe(II) Complexes. J. Am. Chem. Soc. 2009, 131, 11498–11502. [Google Scholar] [CrossRef] [PubMed]
- Sinitskiy, A.V.; Tchougréeff, A.L.; Dronskowski, R. Phenomenological model of spin crossover in molecular crystals as derived from atom-atom potentials. Phys. Chem. Chem. Phys. 2011, 13, 13238–13246. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, H. Periodic Density Functional Calculations in Order to Assess the Cooperativity of the Spin Transition in Fe(phen)2(NCS)2. Magnetochemistry 2016, 2, 14. [Google Scholar] [CrossRef]
- Slichter, C.P.; Drickamer, H.G. Pressure-Induced Electronic Changes in Compounds of Iron. J. Chem. Phys. 1972, 56, 2142–2160. [Google Scholar] [CrossRef]
- Cirera, J.; Babin, V.; Paesani, F. Theoretical Modeling of Spin Crossover in Metal-Organic Frameworks: [Fe(pz)2Pt(CN)4] as a Case Study. Inorg. Chem. 2014, 53, 11020–11028. [Google Scholar] [CrossRef]
- Pham, C.H.; Cirera, J.; Paesani, F. Molecular Mechanisms of Spin Crossover in the Fe(pz)[Pt(CN)4] Metal-Organic Framework upon Water Adsorption. J. Am. Chem. Soc. 2016, 138, 6123–6126. [Google Scholar] [CrossRef]
- Pham, C.H.; Paesani, F. Spin Crossover in the Fe(pz)[Pt(CN)4] Metal-Organic Framework upon Pyrazine Adsorption. J. Phys. Chem. Lett. 2016, 7, 4022–4026. [Google Scholar] [CrossRef] [PubMed]
- Bousseksou, A.; Nasser, J.; Linares, J.; Boukheddaden, K.; Varret, F. Ising-like model for the two-step spin-crossover. J. Phys. I 1992, 2, 1381–1403. [Google Scholar] [CrossRef]
- Bousseksou, A.; Varret, F.; Nasser, J. Ising-like model for the two step spin-crossover of binuclear molecules. J. Phys. I 1993, 3, 1463–1473. [Google Scholar] [CrossRef]
- Bolvin, H.; Kahn, O. Ising model for low-spin high-spin transitions in molecular compounds; within and beyond the mean-field approximation. Chem. Phys. 1995, 192, 295–305. [Google Scholar] [CrossRef]
- Pavlik, J.; Nicolazzi, W.; Molnár, G.; Boǒa, R.; Bousseksou, A. Coupled magnetic interactions and the Ising-like model for spin crossover in binuclear compounds. Eur. Phys. J. B 2013, 86, 292. [Google Scholar] [CrossRef]
- Linares, J.; Jureschi, C.; Boukheddaden, K. Surface Effects Leading to Unusual Size Dependence of the Thermal Hysteresis Behavior in Spin-Crossover Nanoparticles. Magnetochemistry 2016, 2, 24. [Google Scholar] [CrossRef]
- Lebègue, S.; Pillet, S.; Ángyán, J.G. Modeling spin-crossover compounds by periodic DFT+U approach. Phys. Rev. B 2008, 78, 024433. [Google Scholar] [CrossRef]
- Matar, S.F.; Létard, J.F. Ab initio Molecular and Solid-state Studies of the Spin Crossover System [Fe(phen)2(NCS)2]. Z. Naturforsch. B 2010, 65b, 565–570. [Google Scholar] [CrossRef]
- Bučko, T.; Hafner, J.; Lebègue, S.; Ángyán, J.G. Spin crossover transition of Fe(phen)2(NCS)2: Periodic dispersion-corrected density-functional study. Phys. Chem. Chem. Phys. 2012, 14, 5389–5396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Predicting critical temperatures of iron(II) spin crossover materials: Density functional theory plus U approach. J. Chem. Phys. 2014, 141, 214703. [Google Scholar] [CrossRef] [PubMed]
- Vela, S.; Fumanal, M.; Ribas-Arino, J.; Robert, V. Towards an accurate and computationally-efficient modelling of Fe(ii)-based spin crossover materials. Phys. Chem. Chem. Phys. 2015, 17, 16306–16314. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Millis, A.J.; Marianetti, C.A. Density functional plus dynamical mean-field theory of the spin-crossover molecule Fe(phen)2(NCS)2. Phys. Rev. B 2015, 91, 241111(R). [Google Scholar] [CrossRef]
- Gallois, B.; Real, J.A.; Hauw, C.; Zarembowitch, J. Structural Changes Associated with the Spin Transition in Fe(phen)2(NCS)2: A Single-Crystal X-ray Investigation. Inorg. Chem. 1990, 29, 1152–1158. [Google Scholar] [CrossRef]
- Legrand, V.; Pillet, S.; Weber, H.P.; Souhassou, M.; Létard, J.F.; Guionneau, P.; Lecomte, C. On the precision and accuracy of structural analysis of light-induced metastable states. J. Appl. Cryst. 2007, 40, 1076–1088. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. On the accuracy of density functional theory in transition metal chemistry. Annu. Rep. Prog. Chem. Sect. C Phys. Chem. 2006, 102, 203–226. [Google Scholar] [CrossRef]
- Sorai, M.; Seki, S. Phonon Coupled Cooperative Low-Spin 1A1 High-Spin 5T2 Transition in [Fe(phen)2(NCS)2] and [Fe(phen)2(NCSe)2] Crystals. J. Phys. Chem. Solids 1974, 35, 555–570. [Google Scholar] [CrossRef]
- Reiher, M. Theoretical Study of the Fe(phen)2(NCS)2 Spin-Crossover Complex with Reparametrized Density Functionals. Inorg. Chem. 2002, 41, 6928–6935. [Google Scholar] [CrossRef] [PubMed]
- Reiher, M.; Brehm, G.; Schneider, S. Assignment of Vibrational Spectra of 1,10-Phenanthroline by Comparison with Frequencies and Raman Intensities from Density Functional Calculations. J. Phys. Chem. A 2004, 108, 734–742. [Google Scholar] [CrossRef]
- Ronayne, K.L.; Paulsen, H.; Höfer, A.; Dennis, A.C.; Wolny, J.A.; Chumakov, A.I.; Schünemann, V.; Winkler, H.; Spiering, H.; Bousseksou, A.; et al. Vibrational spectrum of the spin-crossover complex [Fe(phen)2(NCS)2] studied by IR and Raman spectroscopy, nuclear inelastic scattering and DFT calculations. Phys. Chem. Chem. Phys. 2006, 8, 4685–4693. [Google Scholar] [CrossRef] [PubMed]
- Palfi, V.K.; Guillona, T.; Paulsen, H.; Molnar, G.; Bousseksou, A. Isotope effects on the vibrational spectra of the Fe(Phen)2(NCS)2 spin-crossover complex studied by density functional calculation. C. R. Chim. 2005, 8, 1317–1325. [Google Scholar] [CrossRef]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y.; Ksenofontov, V. Pressure effect studies in molecular magnetism. C. R. Chim. 2007, 10, 21–36. [Google Scholar] [CrossRef]
- Bolvin, H. The Neel point for spin-transition systems: toward a two-step transition. Chem. Phys. 1996, 211, 101–114. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Pseudopotentials: Fe.pbe-sp-van_ak.upf, S.pbe-van_bm.upf, N.pbe-van_ak.upf, C.pbe-van_ak.upf, H.pbe-van_ak.upf. Available online: www.quantum-espresso.org/pseudopotentials (accessed on 8 February 2017).
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA + U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | c | LDA | PBE | Ising | |||
|---|---|---|---|---|---|---|---|
| 1 | LLLL | 0.00 | 0.0 | 0.0 | 0.0 | ||
| 2 | LHLL | 0.25 | 4.4 | 10.4 | 4.3 | 9.2 | 4.3 |
| 3 | LLHL | 0.25 | 4.6 | 11.5 | 4.3 | 9.2 | 4.3 |
| 4 | LLLH | 0.25 | 4.6 | 11.5 | 4.3 | 9.2 | 4.3 |
| 5 | HLLL | 0.25 | 4.7 | 12.0 | 4.3 | 9.2 | 4.3 |
| 6 | HLHL | 0.50 | 6.2 | 5.2 | 6.8 | 6.6 | 6.8 |
| 7 | LLHH | 0.50 | 6.3 | 5.6 | 6.8 | 6.6 | 6.8 |
| 8 | LHLH | 0.50 | 7.0 | 8.4 | 7.5 | 9.4 | 7.5 |
| 9 | HLLH | 0.50 | 7.3 | 9.6 | 7.5 | 9.4 | 7.5 |
| 10 | HHLL | 0.50 | 7.6 | 10.8 | 7.8 | 10.6 | 7.8 |
| 11 | LHHL | 0.50 | 7.7 | 11.2 | 7.8 | 10.6 | 7.8 |
| 12 | HLHH | 0.75 | 8.2 | 4.5 | 9.3 | 8.4 | 9.4 |
| 13 | HHHL | 0.75 | 8.3 | 5.1 | 9.3 | 8.4 | 9.4 |
| 14 | HHLH | 0.75 | 8.3 | 5.1 | 9.3 | 8.4 | 9.4 |
| 15 | LHHH | 0.75 | 8.4 | 5.6 | 9.3 | 8.4 | 9.4 |
| 16 | HHHH | 1.00 | 9.8 | 10.3 | 10.2 | ||
| I | II | III | IV | |
|---|---|---|---|---|
| I | – | |||
| II | – | |||
| III | – | |||
| IV | – |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreutzburg, L.; Hübner, C.G.; Paulsen, H. Cooperativity of Spin Crossover Complexes: Combining Periodic Density Functional Calculations and Monte Carlo Simulation. Materials 2017, 10, 172. https://doi.org/10.3390/ma10020172
Kreutzburg L, Hübner CG, Paulsen H. Cooperativity of Spin Crossover Complexes: Combining Periodic Density Functional Calculations and Monte Carlo Simulation. Materials. 2017; 10(2):172. https://doi.org/10.3390/ma10020172
Chicago/Turabian StyleKreutzburg, Lars, Christian G. Hübner, and Hauke Paulsen. 2017. "Cooperativity of Spin Crossover Complexes: Combining Periodic Density Functional Calculations and Monte Carlo Simulation" Materials 10, no. 2: 172. https://doi.org/10.3390/ma10020172