The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study

School of Materials Science and Engineering, North University of China, Taiyuan 030051, China
*
Author to whom correspondence should be addressed.
Materials 2018, 11(8), 1471; https://doi.org/10.3390/ma11081471
Submission received: 26 June 2018
/
Revised: 11 August 2018
/
Accepted: 14 August 2018
/
Published: 18 August 2018
Abstract
:The structural stability, mechanical properties, and Debye temperature of alloying elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) doped Al3Li were systematically investigated by first-principles methods. A negative enthalpy of formation ΔHf is predicted for all Al3Li doped species which has consequences for its structural stability. The Sc, Ti, Zr, Nb, and Mo are preferentially occupying the Li sites in Al3Li while the Co, Cu, and Zn prefer to occupy the Al sites. The Al–Li–X systems are mechanically stable at 0 K as elastic constants Cij has satisfied the stability criteria. The values of bulk modulus B for Al–Li–X (X = Sc, Ti, Co, Cu, Zr, Nb, and Mo) alloys (excluding Al–Li–Zn) increase with the increase of doping concentration and are larger than that for pure Al3Li. The Al6LiSc has the highest shear modulus G and Young’s modulus E which indicates that it has stronger shear deformation resistance and stiffness. The predicted universal anisotropy index AU for pure and doped Al3Li is higher than 0, implying the anisotropy of Al–Li–X alloy. The Debye temperature ΘD of Al12Li3Ti is highest among the Al–Li–X system which predicts the existence of strong covalent bonds and thermal conductivity compared to that of other systems.
1. Introduction
Lightweight structural materials such as the Al–Li based alloys have excellent comprehensive performance, such as low density, good corrosion resistance, and high elastic modulus [1,2], and it is the basic reason why Al–Li based alloys are so widely used in aviation and aerospace field. The metastable Al3Li (δ′) precipitates has an important influence on the mechanical properties of Al–Li based alloys [3,4]. The δ′ phase is highly ordered with an L12 structure and forms as spheres possessing a cube–cube orientation relationship with matrix [5]. The lattice constant of δ′ phase (4.02 Å) and dilute Al–Li solid solutions (4.04 Å) are almost equal, and the corresponding precipitate-to-matrix misfit results in an interfacial strain of approximately 0.08 ± 0.02% [6]. Due to the small lattice misfit, strong orientational habit and low interfacial strains, the δ′ phase remain crystallographically coherent with the parent solid-solution matrix and the crystallographic orientation relationship is (111)Al3Li//(111)Al [7,8]. The δ′ precipitates are considered the most important strengthening phases of Al–Li alloys [9].
As far as the importance of δ′ precipitates is concerned, its structure and electronic properties, mechanical properties, nucleation and growth mechanism, and coarsening behavior have been widely studied both experimentally and theoretically [10,11,12,13]. The solubility and stability of δ′ phase in A1–Li alloy have been reported by Mao et al. [14], which suggest that vibrational entropy is essential for the simulation of solubility. Phase-field method is applied for the investigation of coarsening kinetics of δ′ precipitates in the binary Al–Li alloys [15]. The formation enthalpy, electronic structures, and vibrational and thermodynamic properties of the δ′ phase were systematically reported by employing the first-principles methods [16,17,18,19,20,21]. Yao et al. have reported that point defects play an important role in determining the physical properties of off-stoichiometric δ′ phase [22]. The δ′ phase in binary Al–Li alloys provide limited room temperature strength due to their inability to form in high volume fractions unlike precipitates in Al–Cu or Al–Zn–Mg alloys [23]. In this regard, it is important to improve the mechanical properties of δ′ phase. For strengthening phases, doping with the additional alloying elements is an effective way to improve their specific properties [24,25,26]. The alloying elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) are often adopted to improve the specific properties of alloys. However, no one has reported the influences of alloying elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) on the mechanical properties of δ′ phase.
In this paper, first-principles methods were employed to study the effect of alloying elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) and doping concentration on the structural stability, elastic properties, hardness, elastic anisotropy, and Debye temperature of Al3Li phase.
2. Computational Studies
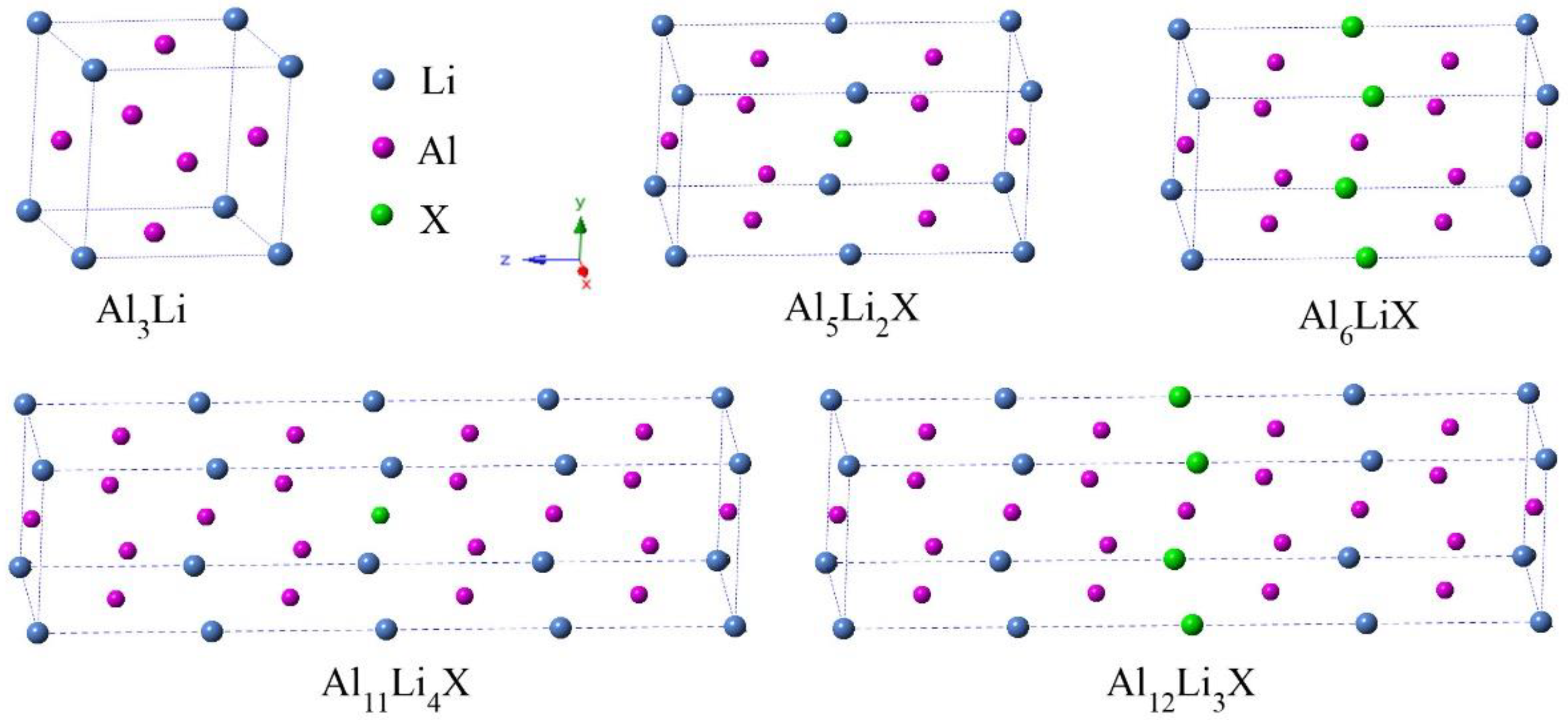
The Al3Li phase has a cubic structure with a space group of pm-3m (No. 221), which contains 3 Al atoms and 1 Li atom (see Figure 1). The Al and Li atom occupy 3c (0 0.5 0.5) and 1a (0 0 0) Wyckoff position, respectively. Based on the Al3Li phase, the supercells of 1 × 1 × 4 and 1 × 1 × 2 were constructed to study the doping effects at various alloying concentrations of 6.25 and 12.5%, respectively. In the supercells, Al or Li sites can be substituted by single alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo), while the chemical formulas of doped species can be represented as Al3Li, Al6LiX, Al5Li2X, Al12Li3X, and Al11Li4X, respectively.
All the first-principles calculations were carried out with CASTEP package [27] based on the density functional theory (DFT) [28]. The ultrasoft pseudopotential [29] in reciprocal space was performed to describe the ion-electron interactions. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) function [30] was applied to describe the exchange-correlation potential. Al 3s23p1, Li 1s22s1, Sc 3s23p63d14s2, Ti 3s23p63d24s2, Co 3d74s2, Cu 3d104s1, Zn 3d104s2, Zr 4s24p64d25s2, Nb 4s24p64d45s1, and Mo 4s24p64d55s1 were treated as valence electrons. The plane-wave energy cutoff of 500 eV was selected for all calculations. The 21 × 21 × 21 k-points mesh was set for Al3Li, and 21 × 21 × 11 and 21 × 21 × 5 k-points meshes were adopted for sampling the 1 × 1 × 4 and 1 × 1 × 2 supercells of Al3Li, respectively. The convergence threshold of 5.0 × 10−6 eV/atom was chosen for maximum energy change.
3. Results and Discussion
3.1. Site Preference and Phase Stability
The predicted lattice constants, volume, mass density, and formation enthalpy of pure Al3Li phase are listed in Table 1. The DFT is one of the calculation methods to simplify the solution of the Schrodinger equation [31]. Thus, the discrepancies between the calculated and experimental values are inevitable. In Table 1, the obtained lattice constant and mass density is consistent with the experimental values, illustrating the reliability of the present computational model.
The structural stability of doped Al3Li phase can be predicted by the simulated value of enthalpy of formation (ΔHf). The ΔHf can be calculated with the help of Formula (1) [33].
where Etot represents the total energy of doped Al3Li phase, , , and denote the energies per atom of Al, Li, and X in solid states, n stands for the total number of atoms in the Al–Li–X system while a and b are the number of Al and Li atoms. The predicted ΔHf and site occupancy behaviors of doping elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) in Al3Li are listed in Table 2. A negative ΔHf is predicted for all doped Al3Li, indicating its structural stability. Generally, a higher negative value of the ΔHf means the material is more stable. Thus, the Sc, Ti, Zr, Nb, and Mo are preferentially occupying the Li sites in Al3Li while the Co, Cu, and Zn prefer to occupy the Al sites. Moreover, the ΔHf of Al6LiZr phase is smaller than other systems, which led us to predict that the occupancy of alloying elements Zr in Li site can substantially improve the stability of alloy.
3.2. Mechanical Properties
Elastic constants (Cij) is an important parameter which can be used to predict the physical properties and mechanical stability of materials [34,35]. In this paper, the Cij is obtained by employing the strain–stress method, based on the general Hooke’s law [36,37]. The pure cubic crystal of Al3Li has three independent elastic constants, i.e., C11, C12, and C44. As shown in Figure 1, the doped Al3Li has tetragonal structure, which is slightly distorted from the pure Al3Li crystal [24,38]. Hence, there are six independent elastic constants (C11, C12, C13, C33, C44, and C66) for doped Al3Li. Moreover, Cij can be used as an important criterion to judge the mechanical stability of pure and doped Al3Li. For the cubic crystals this criterion can be used as:
C11 − C12 > 0, C11 > 0, C44 > 0, C11 + 2C12 > 0
For tetragonal crystals:
The predicted Cij for pure and doped Al3Li at 0 K are summarized in Table 3. The simulated Cij of pure Al3Li has a strong correlation with the already reported experimental and calculated results [17,39], implying the accuracy of our simulations. As shown in Table 3, all the predicted Cij satisfied the stability criteria, indicating that the pure and doped Al3Li are mechanically stable at 0 K. All the Cij of Al–Li–X (X =Sc, Ti, Zr, and Nb) systems are higher than those of pure Al3Li, which indicate that the doping elements X (X =Sc, Ti, Zr, and Nb) can effectively improve the Cij of pure Al3Li. At high concentration (12.5 at %), the compression for Al6LiX (X = Sc, Zr, and Nb) and Al5Li2X (X = Co, Cu, and Zn) systems in the direction of the x-axis was more difficult than other axis owing the largest C11, and the deformation resistance for Al–Li–X (X =Ti and Mo) systems along z-axis are higher than others due to higher C33. When the doping concentration drops to 6.25 at %, the C33 for Al12Li3X (X = Ti, Zr, Nb, and Mo) systems are higher than other Cij demonstrating that z-axis exhibits incompressibility.
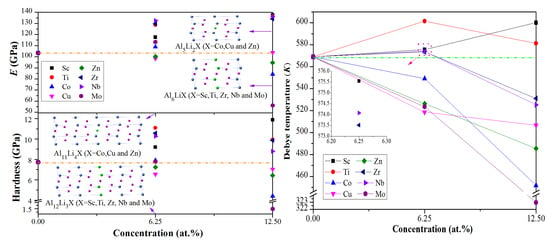
The elastic moduli (bulk modulus B, shear modulus G, and Young’s modulus E) of pure and doped Al3Li were estimated by Voigt–Reuss–Hill method [40,41]. Generally, B describes the resistance to volume change. As shown in Table 3 and Figure 2, the values of B for Al–Li–X (X = Sc, Ti, Co, Cu, Zr, Nb, and Mo) alloys increase with the increase of doping concentration which is higher than that of pure Al3Li. The greater B is, the better the ability to resist to volume change is. When the doping concentration is constant, the values of B for Al–Li–Nb are greater than other counterpart species, indicating that Al–Li–Nb has the stronger resistance to volume change. Thus, we inferred that the addition of Nb can effectively improve the resistance to volume change in Al–Li–X. The G and E measure the resistance to shape change and stiffness of Al–Li–X system, respectively. At low concentration (6.25 at %), the addition of Cu and Zn elements can reduce the G and E of Al3Li. On the other hand, the Al12Li3Nb has higher values of G and E. when the doping concentration up to 12.5 at %, the Sc, Ti, Zr, and Nb elements can play an important role in enhancing the G and E of Al3Li. The Al6LiSc has higher G and E which consequently gives stronger shear deformation resistance and stiffness. Comparative analysis of these parameters led us to conclude that the values of E (G) decrease with the higher concentration (from 6.25 to 12.5%) of Co, Zn, and Mo (Co, Zn, Nb, and Mo). In short, this high doping concentration may decrease the overall performance of the material.
Hardness (H) is an important parameter of materials which can be estimated by the ability to resist localized deformation [42]. The defects (i.e., dislocations) and grain sizes of materials have great influence on the hardness [43] and it is very difficult to get the exact value of H through empirical method. In this paper, the H was roughly predicted by the following semi-empirical formulas [44]:
As described in Figure 2, the H of Al–Li–Zn follow a descending trend with the increase of doping concentration, but it increases in case of Al-Li-Sc. For Al–Li–X (X = Ti, Co, Zr, Nb, and Mo), the H reached to the maximum when 6.25% doping concentration is considered. Besides, the Al6LiSc and Al6LiMo have maximum and minimum values of H, respectively. However, the difference between the hardness of Al6LiSc and Al12Li3Ti is small (about 0.78 GPa). Hence, considering high cost of Sc, the addition of Ti may be the best choice to improve the hardness of pure Al3Li.
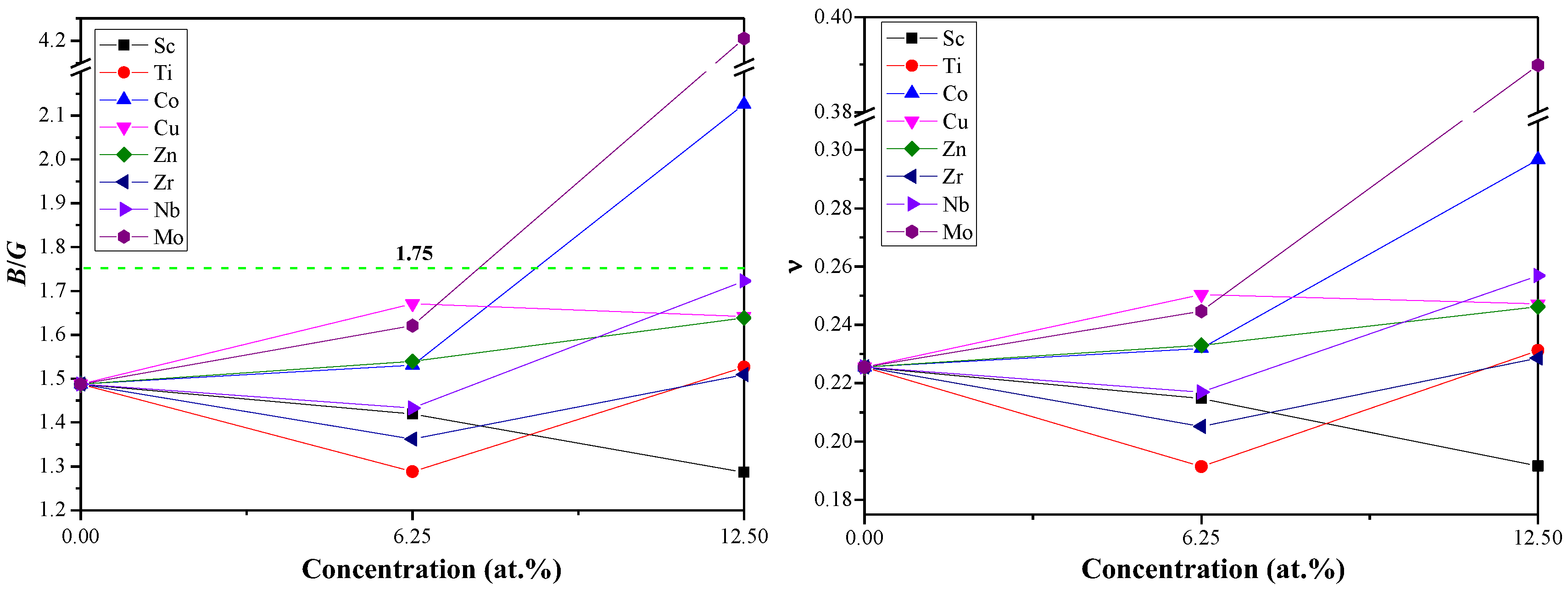
The brittle or ductile behavior of Al–Li–X system can be roughly evaluated from the ratio of B/G. Hence, the materials tend to brittle (ductile) if the ratios of B/G is smaller (larger) than 1.75 [45]. As shown in Figure 3, all the Al–Li–X systems present brittle behavior with a doping concentration of 0 to 6.25 at.%. The Al–Li–X (X = Co and Mo) systems tend to be ductile due to the higher B/G ratios while other Al–Li–X systems still possess brittle behavior at high concentration (12.5 at.%). Comparative analysis of this behavior led us to suggest that the existence of Co and Mo can transform the intrinsic brittleness of Al3Li into ductility. Moreover, the B/G ratios for the Al–Li–Sc system decrease with the increase of Sc concentration (from 0 to 12.5%), while an increasing trend of B/G ratios is found in the Al–Li–X (X = Co, Zn, and Mo) systems. In short, it is necessary to choose an appropriate doping element and its concentration for the desired ductility or brittleness of materials.
The Poisson’s ratio ν is defined as ν = (3B − 2G)/(6B + 2G) and adopted to reveal the stability of the crystal against shear stress. As shown in Figure 3, the values of for Al6LiMo and Al5Li2Co are bigger than those of other Al–Li–X alloys, indicating that Al6LiMo and Al5Li2Co have a higher structural plasticity [46,47]. Besides, the typical values of ν for ionic and metallic materials are 0.25 and 0.33, respectively [48,49]. The predicted ν for Al–Li–X systems (excluding Al5Li2Co and Al6LiMo) are close to 0.25, which shows that main chemical bonding is ionic bonding. The calculated ν for Al6LiMo (0.39) and Al5Li2Co (0.30) are closer to 0.33 than 0.25, implying that the metallic bonding plays the dominant position. Thus, the main chemical bonds of Al6LiMo and Al5Li2Co are different from that of other Al–Li–X alloys, and this may be the reason why Al6LiMo and Al5Li2Co tend to be ductile.
Furthermore, the elastic anisotropy plays a vital role in the mechanical/physical processes such as crack behavior and phase transformations [50]. The elastic anisotropy of pure and doped Al3Li can be predicted from the universal anisotropy index (AU) and its formula is defined as follows [51]:
where GV and GR represent the Voigt and Reuss shear modulus, BV and BR are the Voigt and Reuss bulk modulus, respectively.
As listed in Table 3, the predicted AU for pure and doped Al3Li is higher than 0, implying the anisotropy of the Al–Li–X alloy. In case of Al–Li–X alloy, the AU shows an upward trend with the increase of doping concentration (6.25 to 12.5%). The AU for most of the Al–Li–X (except Al5Li2Co, Al6LiMo, and Al12Li3Mo) systems are near zero and smaller than that for pure Al3Li, which indicates that most doping elements can reduce the anisotropy of materials. The AU of Al6LiMo is much larger than other species. The reason behind this is that a large difference between C44 (C11) and C66 (C33) in Al6LiMo alloy [52].
3.3. Debye Temperature
The Debye temperature ΘD is an important parameter of a solid and it is associated with thermodynamic properties of materials, such as entropy, thermal expansion, and vibrational internal energy. One of the standard methods of calculating the Debye temperature is from elastic constant data. Thus, the ΘD was predicted from averaged sound velocity by employing the following formula [53]:
where h, kB, n, NA, ρ, M, and vm stand for Planck’s constant, Boltzmann’s constant, total number of atoms, Avogadro’s number, density, molecular weight, and average wave velocity, respectively. vs and vl represent the shear and longitudinal sound velocities of materials, respectively.
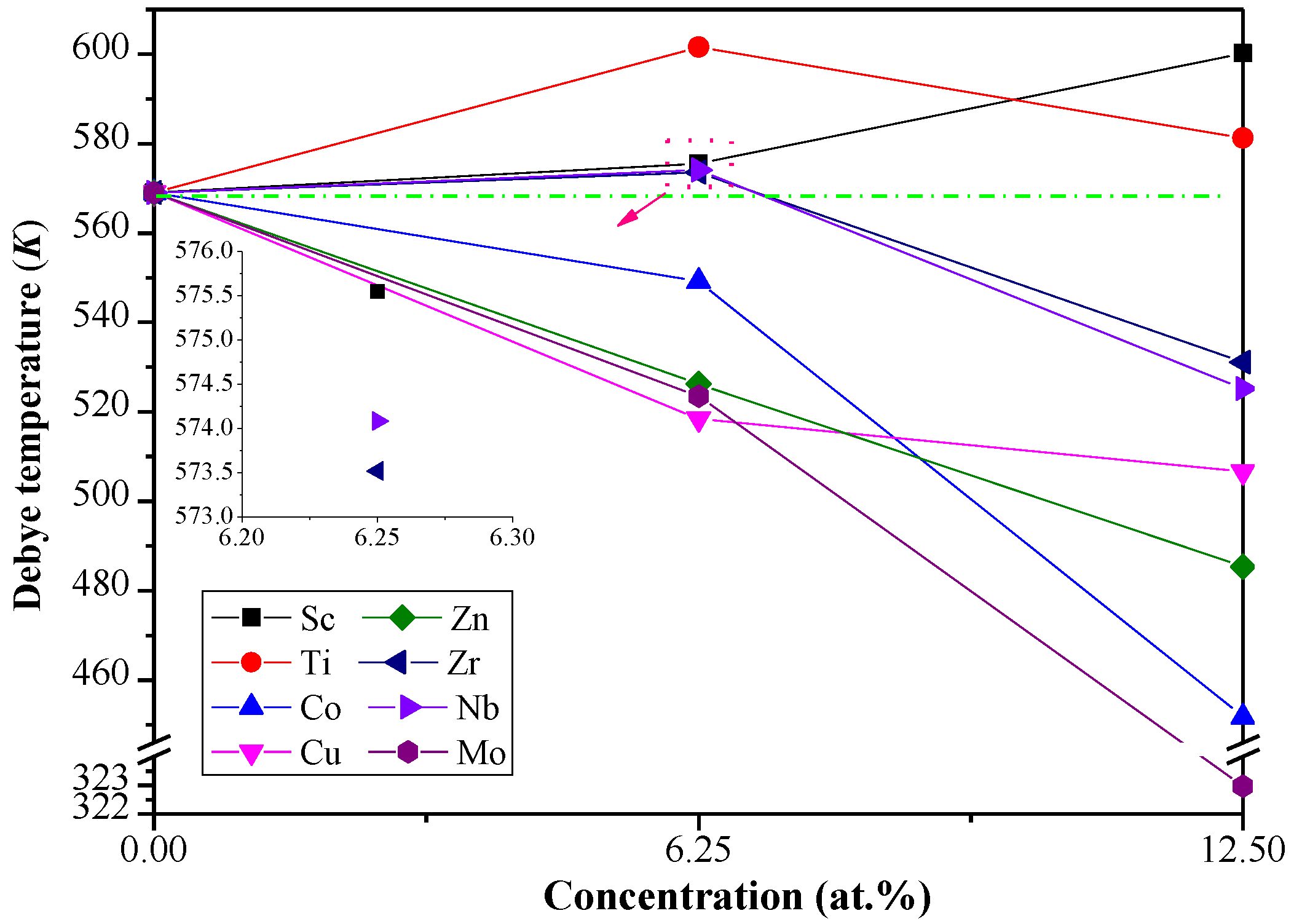
As depicted in Figure 4, the predicted value of ΘD for pure Al3Li is 568.9 K at 0 K, which is in consistent with the already reported data (573 K) [19]. The ΘD decreases (increases) with the increase of Co, Cu, Zn, and Mo (Sc) concentration in the Al–Li–X systems. In general, a higher ΘD implies that the materials have higher thermal conductivity and stronger covalent bonds. The ΘD of Al12Li3Ti is higher among the Al–Li–X systems, which illustrates that the strength of covalent bonds and thermal conductivity of Al12Li3Ti are better than others. Furthermore, the predicted density of Al12Li3Ti (2.5 g/cm3) is close to the density of Al3Li (2.2 g/cm3), which demonstrates thatAl12Li3Ti may be a better reinforcement phase in aerospace materials.
4. Conclusions
The effect of alloying elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) and doping concentration on the structural stability, mechanical properties, and Debye temperature of Al3Li were systematically investigated through density functional theory. The main contents of this work can be summarized as follows:
(1) All doped Al3Li systems are structural stability. The Sc, Ti, Zr, Nb, and Mo preferentially occupied the Li sites in Al3Li while the Co, Cu, and Zn prefer to occupy the Al sites rather than Li sites.
(2) All the Cij of Al–Li–X (X = Sc, Ti, Zr, and Nb) systems are higher than those of pure Al3Li, which indicate that the doping elements X (X = Sc, Ti, Zr, and Nb) can effectively improve the Cij of pure Al3Li. The values of B for Al–Li–X (X = Sc, Ti, Co, Cu, Zr, Nb, and Mo) alloys (excluding Al-Li-Zn) increase with the increase of doping concentration and are higher than that for pure Al3Li. The Al6LiSc has higher G and E, which consequences stronger shear deformation resistance and stiffness.
(3) All the Al–Li–X systems present brittle behavior with the increase in doping concentration (from 0 to 6.25 at %). The Al–Li–X (X = Co and Mo) systems tend to ductile while other Al–Li–X systems still possess brittle behavior at high concentration (12.5 at %), which suggests that the existence of Co and Mo can transform the intrinsic brittleness of Al3Li into ductility. Moreover, the Al6LiSc and Al6LiMo have maximum values of H and AU, respectively.
(4) A higher ΘD is observed for Al12Li3Ti, responsible for strong covalent bonds and higher thermal conductivity, compared to other Al–Li–X systems.
Author Contributions
Investigation, Y.H.Z and J.Z.T.; Data Curation, Y.H.Z. and H.H.; Writing-Original Draft Preparation, J.Z.T.; Writing-Review & Editing, Y.H.Z., H.H. and B.W.
Funding
This research was funded by the National Natural Science Foundation of China [Nos. 51774254, 51774253, 51701187, U1610123, 51674226, 51574207, and 51574206], and the Science and Technology Major Project of Shanxi Province [No. MC2016-06].
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ning, J.; Zhang, L.J.; Bai, Q.L.; Yin, X.Q.; Niu, J.; Zhang, J.X. Comparison of the microstructure and mechanical performance of 2A97 Al-Li alloy joints between autogenous and non-autogenous laser welding. Mater. Des. 2017, 120, 144–156. [Google Scholar] [CrossRef]
- Bairwa, M.L.; Date, P.P. Effect of heat treatment on the tensile properties of Al-Li alloys. J. Mater. Process. Technol. 2004, 153–154, 603–607. [Google Scholar] [CrossRef]
- Ovri, H.; Lilleodden, E.T. New insights into plastic instability in precipitation strengthened Al-Li alloys. Acta Mater. 2015, 89, 88–97. [Google Scholar] [CrossRef]
- Rösner, H.; Kalogeridis, A.; Liu, W.; Pesicka, J.; Nembach, E. Dislocation mechanisms in Al-rich Al-Li alloys. Mater. Sci. Eng. A 1997, 234–236, 298–301. [Google Scholar] [CrossRef]
- Flower, H.; Gregson, P. Solid state phase transformations in aluminium alloys containing lithium. Mater. Sci. Technol. 1987, 3, 81–90. [Google Scholar] [CrossRef]
- Williams, D.; Edington, J. The precipitation of δ′(Al3Li) in dilute aluminium–lithium alloys. Met. Sci. 1975, 9, 529–532. [Google Scholar] [CrossRef]
- Laverock, J.; Dugdale, S.B.; Alam, M.A.; Roussenova, M.V.; Wensley, J.R.; Kwiatkowska, J.; Shiotani, N. Fermi surface of an important nanosized metastable phase: Al3Li. Phys. Rev. Lett. 2010, 105, 236401. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, B.A.; Wang, K.G.; Glicksman, M.E. Experimental, computational and theoretical studies of δ′ phase coarsening in Al-Li alloys. Acta Mater. 2012, 60, 5803–5817. [Google Scholar] [CrossRef]
- Mogucheva, A.; Kaibyshev, R. Microstructure and Mechanical Properties of an Al-Li-Mg-Sc-Zr Alloy Subjected to ECAP. Metals 2016, 6, 254. [Google Scholar] [CrossRef]
- Chabala, J.M.; Levi-Setti, R.; Soni, K.K.; Williams, D.B.; Newbury, D.E. Secondary ion imaging of the distribution of δ′ (Al3Li) in Al-Li alloys. Appl. Surf. Sci. 1991, 51, 185–192. [Google Scholar] [CrossRef]
- Gu, B.P.; Liedl, G.L.; Kulwicki, J.H.; Sanders, T.H., Jr. Coarsening of δ′ (Al3Li) precipitates in an Al-2.8Li0.3Mn alloy. Mater. Sci. Eng. 1985, 70, 217–228. [Google Scholar] [CrossRef]
- Hoyt, J.J.; Spooner, S. The surface energy of metastable Al3Li precipitates from coarsening kinetics. Acta Metall. Et Mater. 1991, 39, 689–693. [Google Scholar] [CrossRef]
- Lee, B.C.; Park, J.K. Effect of the addition of Ag on the strengthening of Al3Li phase in Al-Li single crystals. Acta Mater. 1998, 46, 4181–4187. [Google Scholar] [CrossRef]
- Mao, Z.; Seidman, D.N.; Wolverton, C. The effect of vibrational entropy on the solubility and stability of ordered Al3Li phases in Al-Li alloys. APL Mater. 2016, 4, 144202. [Google Scholar] [CrossRef]
- Poduri, R.; Chen, L.Q. Computer simulation of morphological evolution and coarsening kinetics of δ′ (Al3Li) precipitates in Al-Li alloys. Acta Mater. 1998, 46, 3915–3928. [Google Scholar] [CrossRef]
- Li, Z.; Tse, J.S. Ab initio studies on the vibrational and thermal properties of Al3Li. Phys. Rev. B 2000, 61, 14531–14536. [Google Scholar] [CrossRef]
- Yu, H.; Duan, X.; Ma, Y.; Zeng, M. First Principles Study of Al-Li Intermetallic Compounds. Chin. J. Chem. Phys. 2012, 25, 659–665. [Google Scholar] [CrossRef]
- Sluiter, M.; De, F.D.; Guo, X.Q.; Podloucky, R.; Freeman, A.J. First-principles calculation of phase equilibria in the aluminum lithium system. Phys. Rev. B 1990, 42, 10460–10476. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. Mechanical and thermodynamic properties of Al3Sc and Al3Li precipitates in Al-Li-Sc alloys from first-principles calculations. Phys. B Condens. Matter 2013, 427, 85–90. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliņš, V. First-principles aluminum database: Energetics of binary Al alloys and compounds. Phys. Rev. B 2006, 73, 144104. [Google Scholar] [CrossRef]
- Guo, X.; Podloucky, R.; Xu, J.; Freeman, A.J. Cohesive, electronic, and structural properties of Al3Li: An important metastable phase. Phys. Rev. B 1990, 41, 12432. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, C.; Jiang, Y.; Tao, H.; Yin, D. Prediction on elastic properties of off-stoichiometric L12-Al3Li intermetallic due to point defects. Comput. Mater. Sci. 2015, 107, 184–189. [Google Scholar] [CrossRef]
- Makineni, S.K.; Sugathan, S.; Meher, S.; Banerjee, R.; Bhattacharya, S.; Kumar, S.; Chattopadhyay, K. Enhancing elevated temperature strength of copper containing aluminium alloys by forming L12 Al3Zr precipitates and nucleating θ″ precipitates on them. Sci. Rep. 2017, 7, 11154. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wu, X.; Wang, R.; Jia, Z.; Li, W.; Liu, Q. Structural stability, mechanical properties and stacking fault energies of TiAl3 alloyed with Zn, Cu, Ag: First-principles study. J. Alloys Compd. 2016, 666, 185–196. [Google Scholar] [CrossRef]
- Gu, J.; Bai, J.; Zhu, Y.; Qin, Y.; Gu, H.; Zhai, Y.; Ma, P. First-principles study of the influence of doping elements on phase stability, crystal and electronic structure of Al2Cu (θ) phase. Comput. Mater. Sci. 2016, 111, 328–333. [Google Scholar] [CrossRef]
- Kubouchi, M.; Hayashi, K.; Miyazaki, Y. Electronic structure and thermoelectric properties of boron doped Mg2Si. Scr. Mater. 2016, 123, 59–63. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.; Hasnip, P.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, T.; Hasebe, K.; Mannami, M. Al3Li Superlattice in Al-4.5 wt % Li Alloy. J. Phys. Soc. Jpn. 2007, 25, 908. [Google Scholar] [CrossRef]
- Sahu, B.R. Electronic structure and bonding of ultralight LiMg. Mater. Sci. Eng. B 1995, 49, 74–78. [Google Scholar] [CrossRef]
- Wang, J.-H.; Lu, Y.; Zhang, X.-L.; Shao, X.-H. The elastic behaviors and theoretical tensile strength of γ-TiAl alloy from the first principles calculations. Intermetallics 2018, 101, 1–7. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Wang, B.; Hou, H.; Zhang, Y. The structural, mechanical and thermodynamic properties of Ti-B compounds under the influence of temperature and pressure: First-principles study. Mater. Chem. Phys. 2018, 209, 200–207. [Google Scholar] [CrossRef]
- Xiao, B.; Feng, J.; Zhou, C.T.; Jiang, Y.H.; Zhou, R. Mechanical properties and chemical bonding characteristics of Cr7C3 type multicomponent carbides. J. Appl. Phys. 2011, 109, 083521. [Google Scholar] [CrossRef]
- Qi, Y.Y.; Mu, Y.; Cheng, Y.; Ji, G.F. Pressure effect on electronic, elastic and optical properties of Eu:CaF2 crystal: A first-principles study. Philos. Mag. 2015, 95, 2974–2989. [Google Scholar] [CrossRef]
- Mao, Z.; Chen, W.; Seidman, D.N.; Wolverton, C. First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys. Acta Mater. 2011, 59, 3012–3023. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Watt, J.P.; Peselnick, L. Clarification of the Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. J. Appl. Phys. 1980, 51, 1525–1531. [Google Scholar] [CrossRef]
- Gao, F.M.; Gao, L.H. Microscopic models of hardness. J. Superhard Mater. 2010, 32, 148–166. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Yousef, E.S.; El-Adawy, A.; El-Kheshkhany, N. Effect of rare earth (Pr2O3, Nd2O3, Sm2O3, Eu2O3, Gd2O3 and Er2O3) on the acoustic properties of glass belonging to bismuth–borate system. Solid State Commun. 2006, 139, 108–113. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Song, Y.; Fan, Q.; Yang, Y. Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure. Materials 2018, 11, 740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shang, S.; Wang, Y.; Saengdeejing, A.; Chen, L.; Liu, Z. First-principles calculations of the elastic, phonon and thermodynamic properties of Al12Mg17. Acta Mater. 2010, 58, 4012–4018. [Google Scholar] [CrossRef]
- Nong, Z.-S.; Zhu, J.-C.; Yang, X.-W.; Cao, Y.; Lai, Z.-H.; Liu, Y.; Sun, W. First-principles calculations of the stability and hydrogen storage behavior of C14 Laves phase compound TiCrMn. Solid State Sci. 2014, 32, 1–7. [Google Scholar] [CrossRef]
- Haines, J.; Leger, J.; Bocquillon, G. Synthesis and design of superhard materials. Ann. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Huang, J.; Wang, W.; Ye, Z.; Chen, S.; Zhao, Y. First-principles calculations on physical properties of Ni3Snx binary system intermetallic compounds and Ni/Ni3Sn interfaces in Nickel-Tin TLPS bonding layer. Intermetallics 2018, 101, 27–38. [Google Scholar] [CrossRef]
- Ledbetter, H.; Migliori, A. A general elastic-anisotropy measure. J. Appl. Phys. 2006, 100, 063516. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wu, X.; Wang, R.; Li, W.; Liu, Q. Phase stability, mechanical properties and electronic structure of TiAl alloying with W, Mo, Sc and Yb: First-principles study. J. Alloys Compd. 2016, 658, 689–696. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
Figure 1.
Crystalline structures of Al3Li doped with alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) at different alloying concentrations of 6.25 and 12.5%.
Figure 1.
Crystalline structures of Al3Li doped with alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) at different alloying concentrations of 6.25 and 12.5%.

Figure 2.
The predicted elastic moduli B, G, and E (GPa) and hardness H of Al3Li doped with alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo).
Figure 2.
The predicted elastic moduli B, G, and E (GPa) and hardness H of Al3Li doped with alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo).

Figure 3.
Simulated B/G and ν of Al3Li doped with alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) as a function of doping concentration.
Figure 3.
Simulated B/G and ν of Al3Li doped with alloying element X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) as a function of doping concentration.

Figure 4.
The calculated ΘD for Al3Li doped with alloying element X (X= Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) with different doping concentration.
Figure 4.
The calculated ΘD for Al3Li doped with alloying element X (X= Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) with different doping concentration.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Simulated lattice constant a (Å), volume V (Å3), mass density (Kg/m3), and formation enthalpy Hform (eV) of pure Al3Li phase at 0 K.
Table 1.
Simulated lattice constant a (Å), volume V (Å3), mass density (Kg/m3), and formation enthalpy Hform (eV) of pure Al3Li phase at 0 K.
| Species | a | V | Mass Density | Hform |
|---|---|---|---|---|
| Present | 4.034 | 65.65 | 2.223 | −0.097 |
| Cal. [17] | 4.030 | 65.45 | 2.221 | - |
| Cal. [14] | 4.029 | 65.40 | - | −0.100 |
| Exp. [32] | 4.01 | 64.48 | 2.260 | - |
Table 2.
Simulated enthalpy of formation ΔHf (eV) and site occupancy behaviors of doping elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) in Al3Li at 0 K.
Table 2.
Simulated enthalpy of formation ΔHf (eV) and site occupancy behaviors of doping elements X (X = Sc, Ti, Co, Cu, Zn, Zr, Nb, and Mo) in Al3Li at 0 K.
| Element X | ΔHf | Site Preference | Element X | ΔHf | Site Preference | ||
|---|---|---|---|---|---|---|---|
| Al6LiX | Al5Li2X | Al12Li3X | Al11Li4X | ||||
| Sc | −0.267 | −0.154 | Li | Sc | −0.180 | −0.127 | Li |
| Ti | −0.232 | −0.127 | Li | Ti | −0.169 | −0.108 | Li |
| Co | −0.185 | −0.202 | Al | Co | −0.140 | −0.152 | Al |
| Cu | −0.057 | −0.128 | Al | Cu | −0.075 | −0.111 | Al |
| Zn | −0.017 | −0.099 | Al | Zn | −0.057 | −0.098 | Al |
| Zr | −0.276 | −0.148 | Li | Zr | −0.191 | −0.118 | Li |
| Nb | −0.188 | −0.098 | Li | Nb | −0.150 | −0.094 | Li |
| Mo | −0.100 | −0.059 | Li | Mo | −0.107 | −0.075 | Li |
Table 3.
Simulated elastic constants Cij (GPa), elastic moduli B, G, and E (GPa), hardness H and universal anisotropy index AU for pure and doped Al3Li at 0 K and 0 GPa.
Table 3.
Simulated elastic constants Cij (GPa), elastic moduli B, G, and E (GPa), hardness H and universal anisotropy index AU for pure and doped Al3Li at 0 K and 0 GPa.
| Phase | Species | C11 | C33 | C44 | C66 | C12 | C13 | B | G | E | H | AU |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al3Li | Present | 129.7 | - | 37.7 | - | 29.4 | - | 62.8 | 42.2 | 103.5 | 7.73 | 0.099 |
| Cal. [17] | 128 | - | 39 | - | 30 | - | 63.3 | 42.8 | 116.8 | - | - | |
| Exp. [39] | 123.6 | - | 42.8 | - | 37.2 | - | 66 | 43 | 105.9 | - | - | |
| Al12Li3Sc | Present | 136.2 | 130.4 | 46.1 | 51.7 | 31.0 | 38.3 | 68.7 | 48.4 | 117.6 | 9.20 | 0.019 |
| Al12Li3Ti | Present | 141.3 | 147.7 | 52.4 | 54.4 | 35.2 | 31.2 | 69.5 | 54.0 | 128.6 | 11.10 | 0.007 |
| Al11Li4Co | Present | 138.0 | 118.9 | 39.4 | 48.1 | 29.0 | 39.2 | 67.7 | 44.2 | 108.9 | 7.90 | 0.085 |
| Al11Li4Cu | Present | 126.7 | 116.2 | 37.0 | 37.5 | 37.7 | 37.0 | 65.8 | 39.4 | 98.5 | 6.55 | 0.032 |
| Al11Li4Zn | Present | 124.3 | 121.8 | 36.9 | 38.9 | 33.8 | 31.2 | 62.5 | 40.6 | 100.1 | 7.23 | 0.049 |
| Al12Li3Zr | Present | 143.4 | 147.5 | 53.1 | 55.9 | 42.2 | 35.2 | 73.3 | 53.8 | 129.7 | 10.58 | 0.008 |
| Al12Li3Nb | Present | 147.8 | 163.5 | 53.1 | 54.9 | 50.8 | 35.5 | 78.1 | 54.5 | 132.6 | 10.28 | 0.041 |
| Al12Li3Mo | Present | 118.7 | 147.0 | 52.0 | 51.5 | 66.9 | 36.6 | 73.8 | 45.4 | 113.3 | 7.77 | 0.426 |
| Al6LiSc | Present | 146.6 | 136.8 | 61.3 | 62.0 | 35.2 | 41.3 | 74.3 | 57.7 | 137.6 | 11.88 | 0.040 |
| Al6LiTi | Present | 153.7 | 157.5 | 55.0 | 62.0 | 51.8 | 48.2 | 84.6 | 55.4 | 136.4 | 9.93 | 0.020 |
| Al5Li2Co | Present | 144.9 | 76.5 | 35.9 | 34.0 | 15.6 | 58.2 | 69.1 | 32.5 | 84.3 | 4.41 | 1.534 |
| Al5Li2Cu | Present | 134.6 | 122.0 | 37.3 | 41.8 | 39.3 | 36.6 | 68.4 | 41.6 | 103.8 | 7.03 | 0.055 |
| Al5Li2Zn | Present | 123.2 | 116.4 | 33.5 | 36.4 | 37.0 | 30.9 | 62.2 | 38.0 | 94.7 | 6.42 | 0.082 |
| Al6LiZr | Present | 153.9 | 143.0 | 55.7 | 60.5 | 51.6 | 47.2 | 82.4 | 54.6 | 134.2 | 9.88 | 0.026 |
| Al6LiNb | Present | 162.0 | 156.2 | 54.2 | 64.0 | 56.4 | 61.9 | 93.4 | 54.2 | 136.3 | 8.88 | 0.047 |
| Al6LiMo | Present | 90.3 | 146.3 | 39.6 | 50.9 | 86.8 | 67.9 | 85.0 | 20.2 | 56.1 | 1.48 | 17.33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tian, J.; Zhao, Y.; Hou, H.; Wang, B. The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study. Materials 2018, 11, 1471. https://doi.org/10.3390/ma11081471
AMA Style
Tian J, Zhao Y, Hou H, Wang B. The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study. Materials. 2018; 11(8):1471. https://doi.org/10.3390/ma11081471
Chicago/Turabian StyleTian, Jinzhong, Yuhong Zhao, Hua Hou, and Bing Wang. 2018. "The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study" Materials 11, no. 8: 1471. https://doi.org/10.3390/ma11081471
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.