Postfunctionalization of Alkyne-Linked Conjugated Carbazole Polymer by Thermal Addition Reaction of Tetracyanoethylene

Abstract
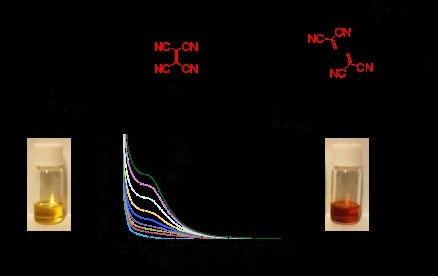
:
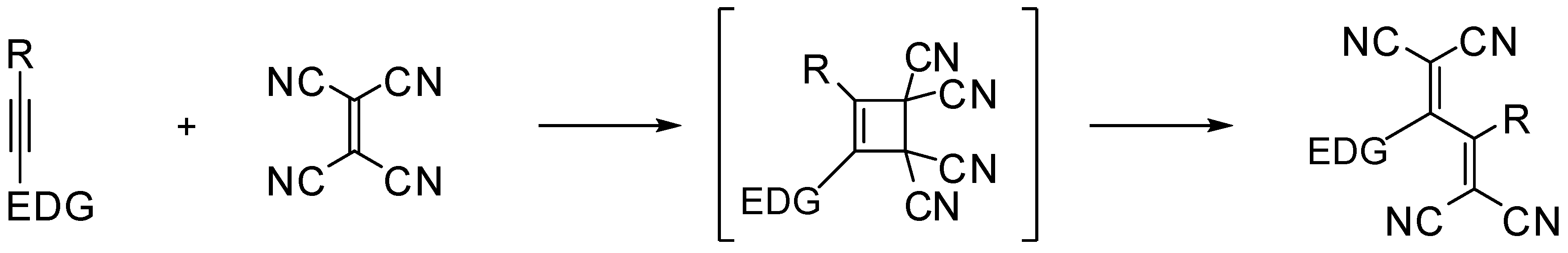
1. Introduction
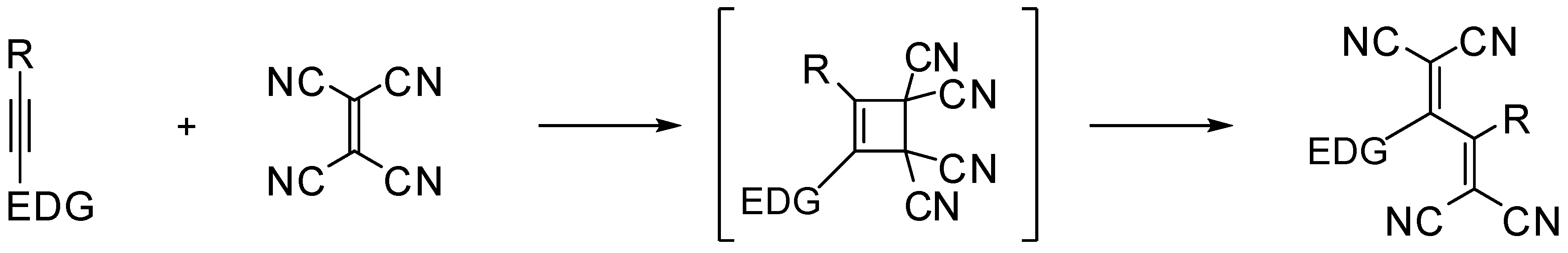
2. Results and Discussion
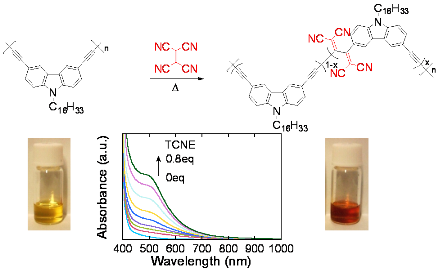
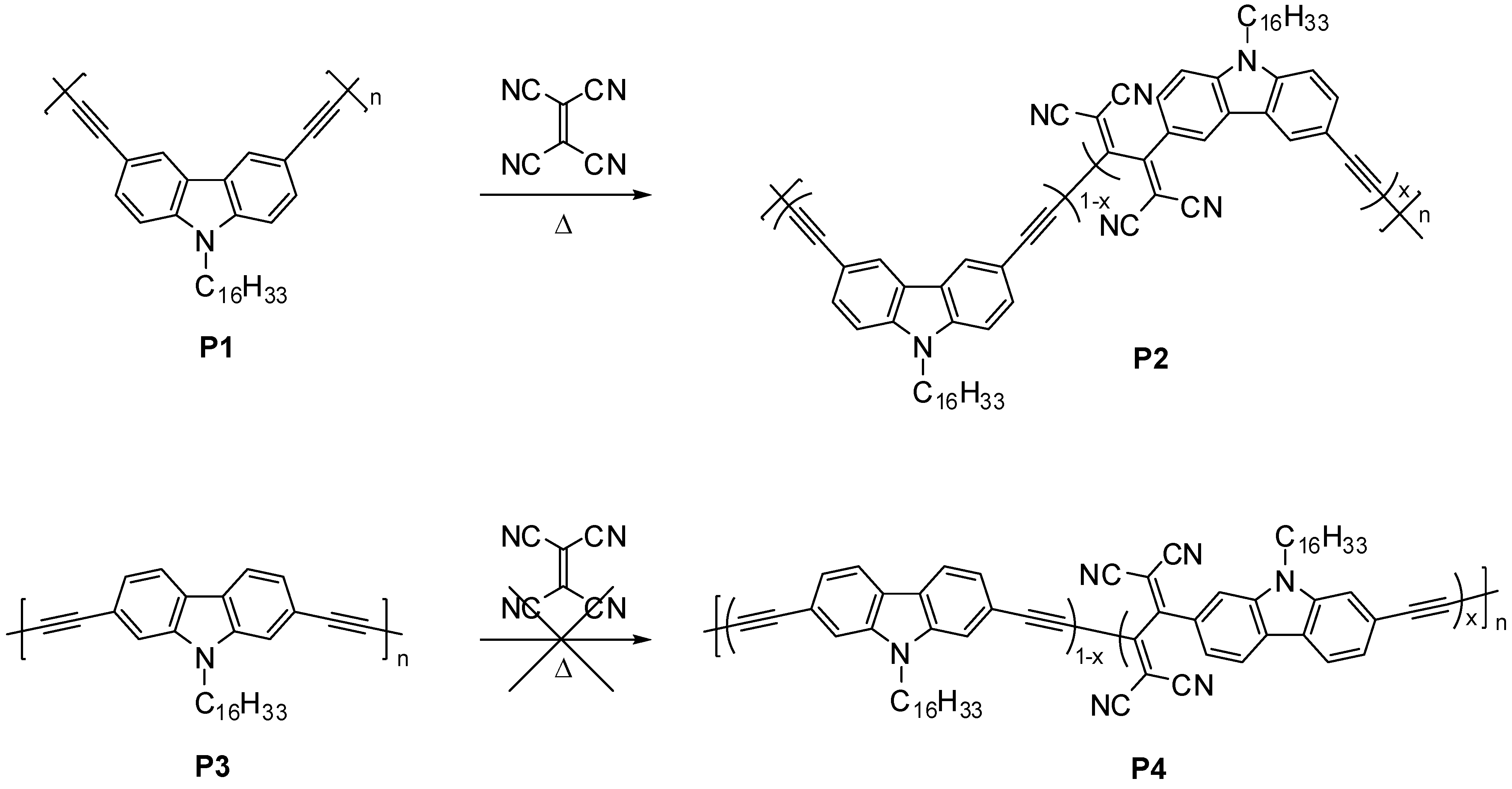
2.1. Polymer Synthesis
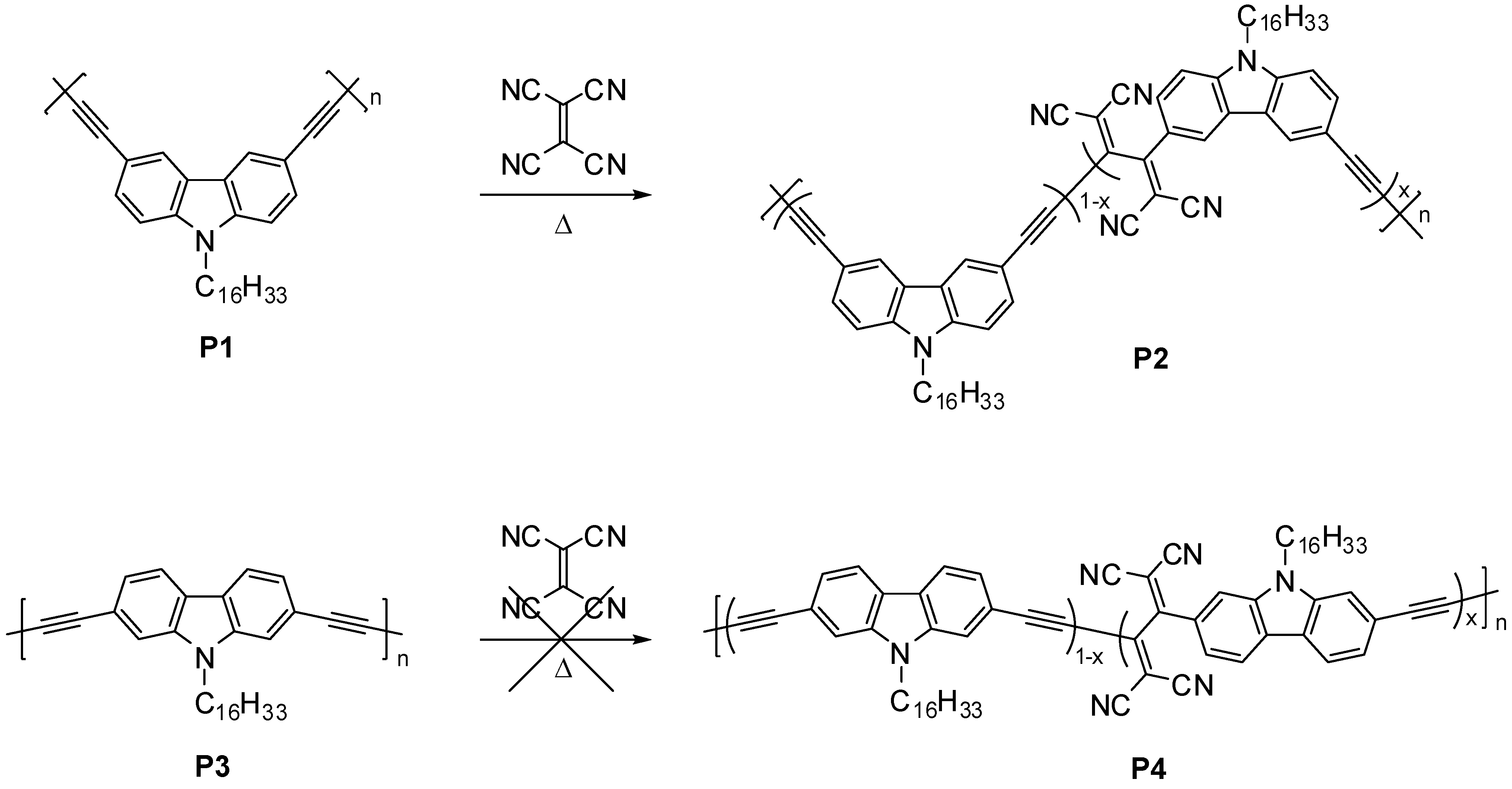

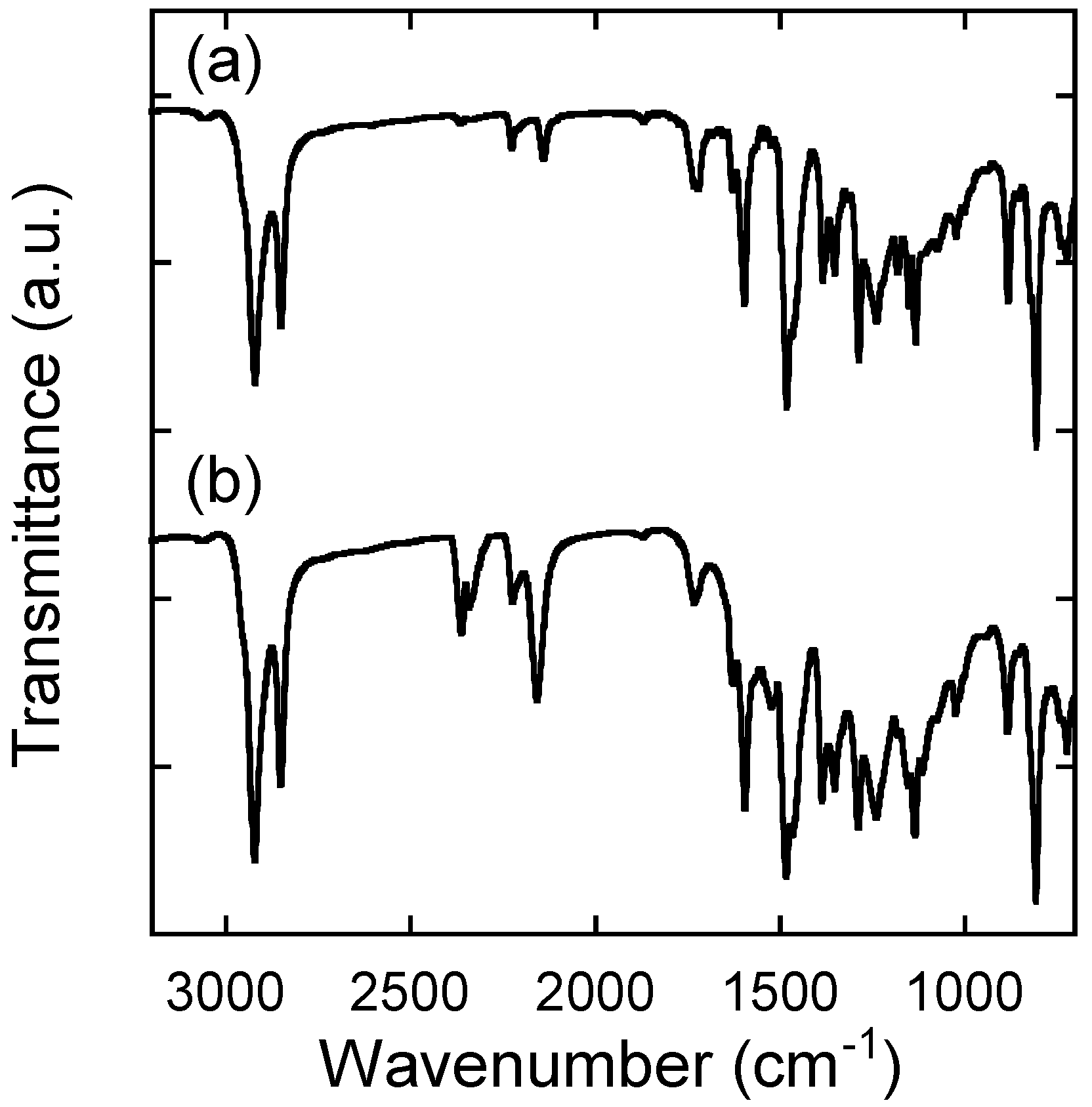
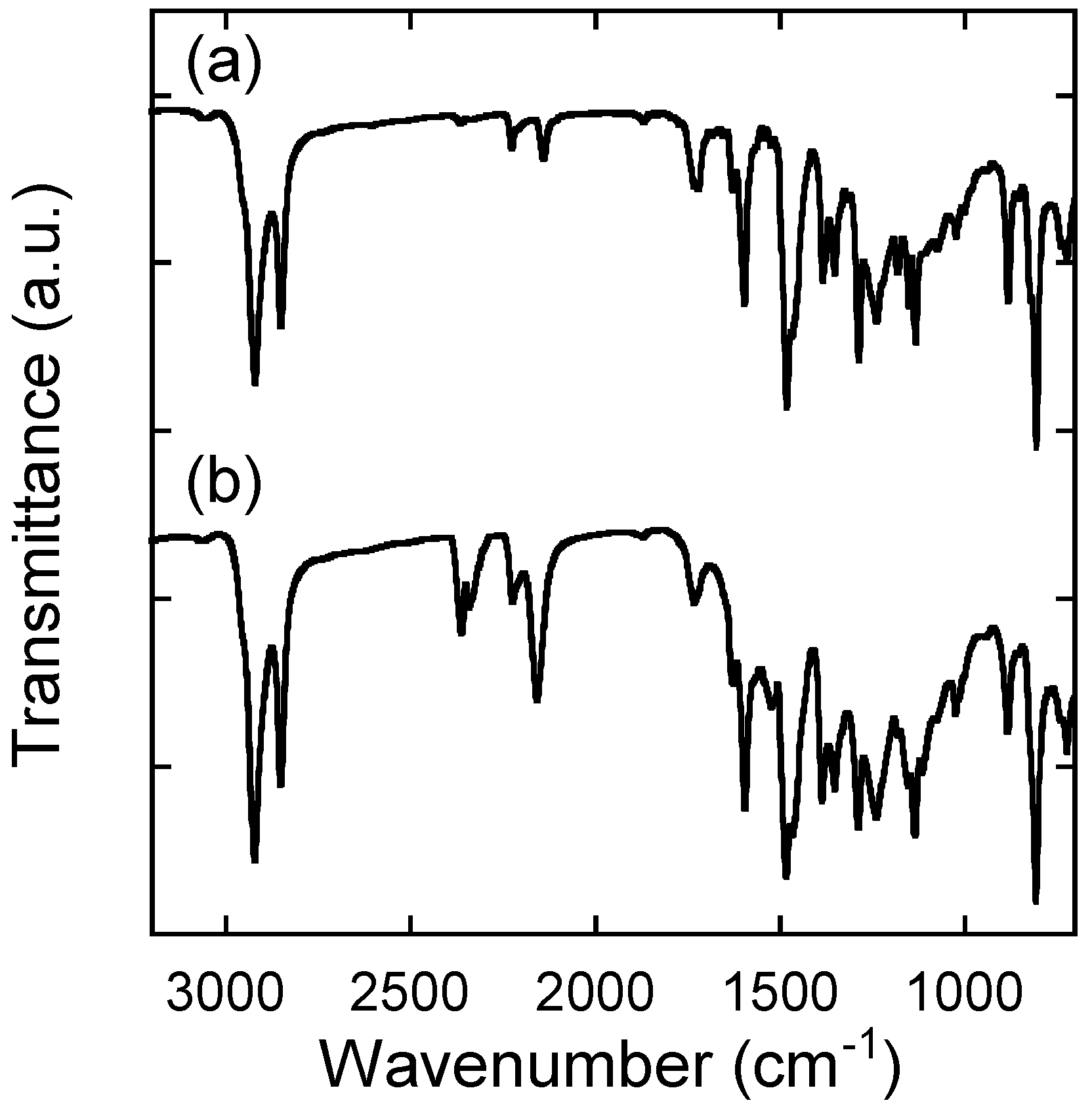
2.2. Characterization

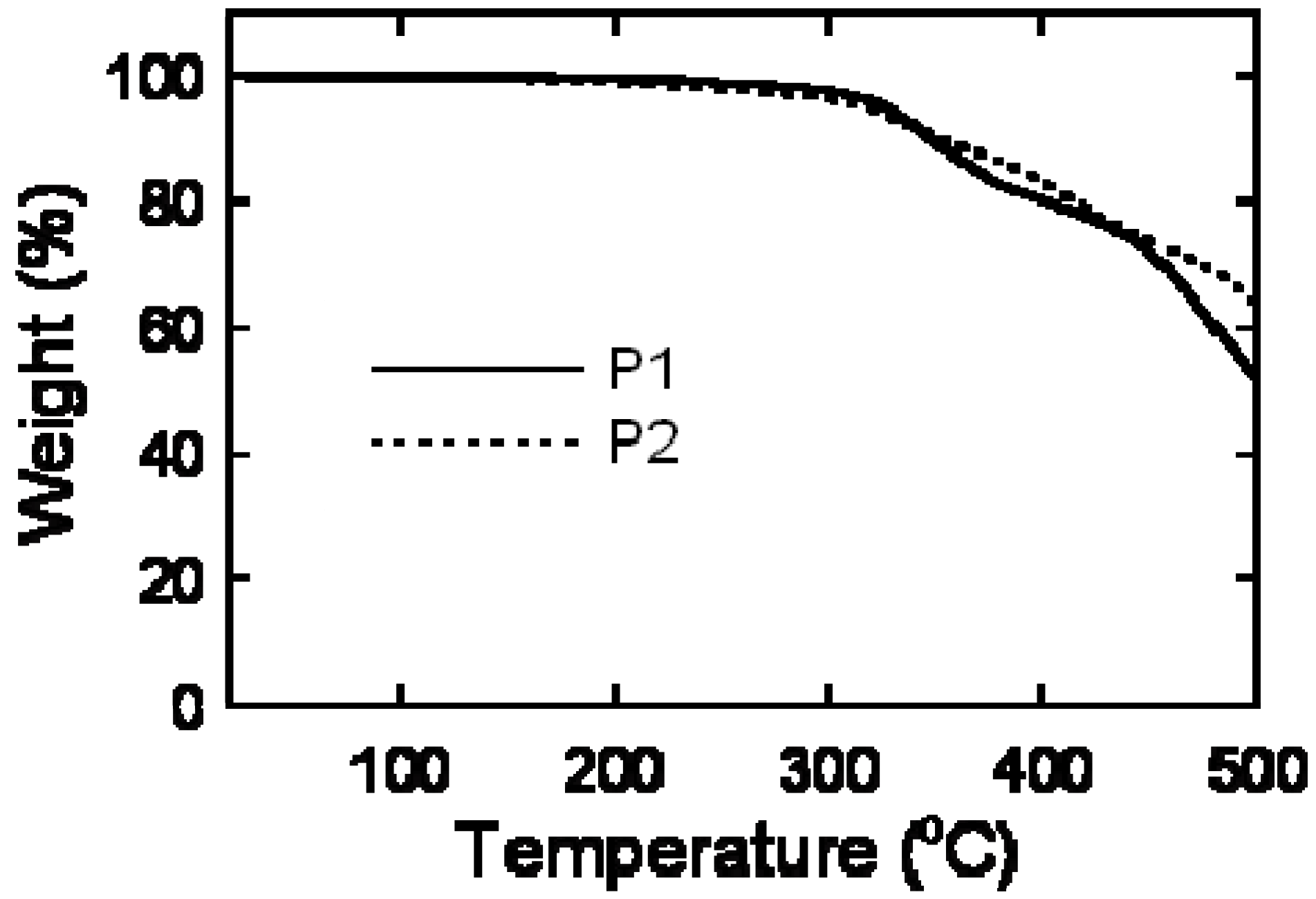
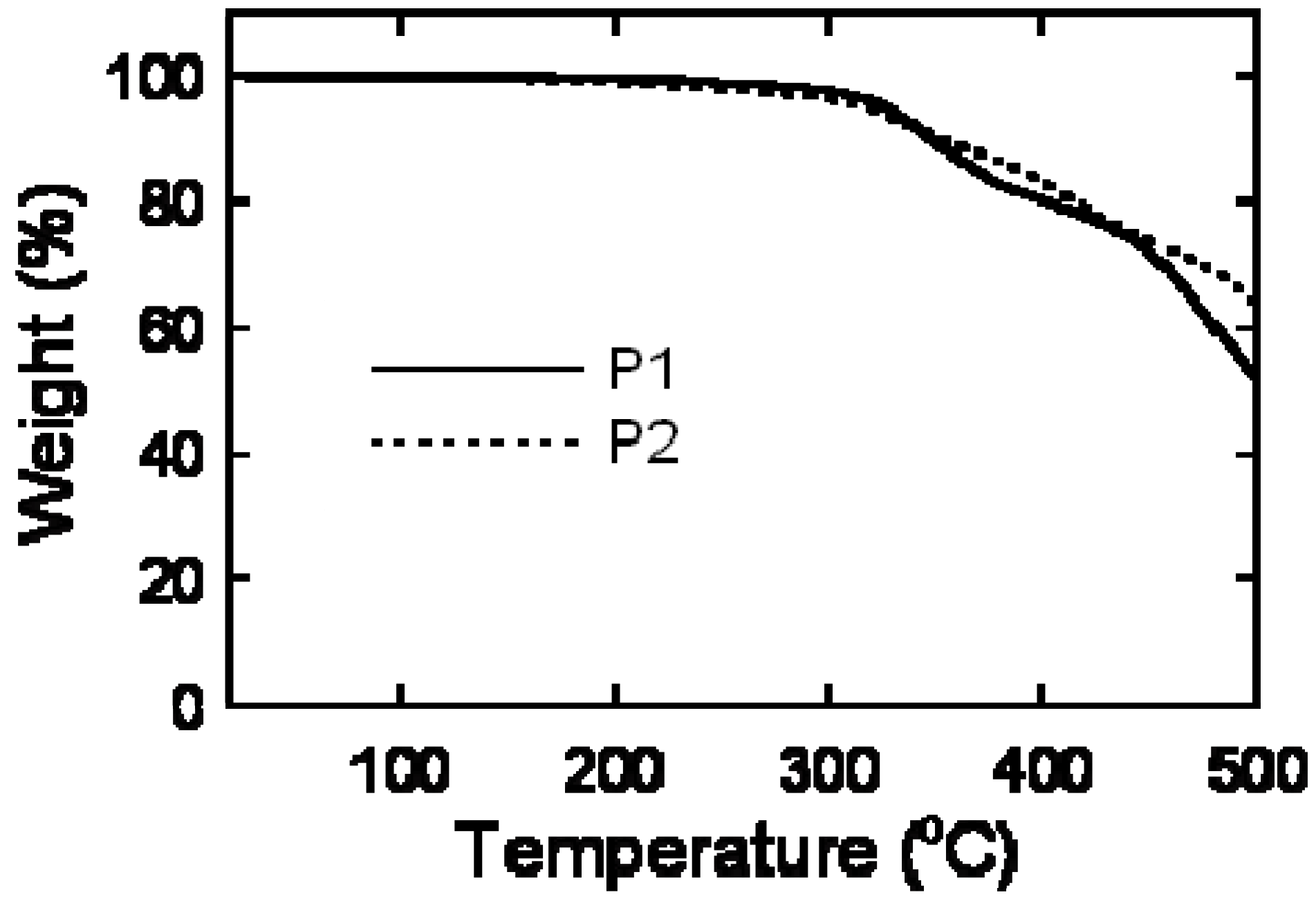
2.3. Thermal Analysis
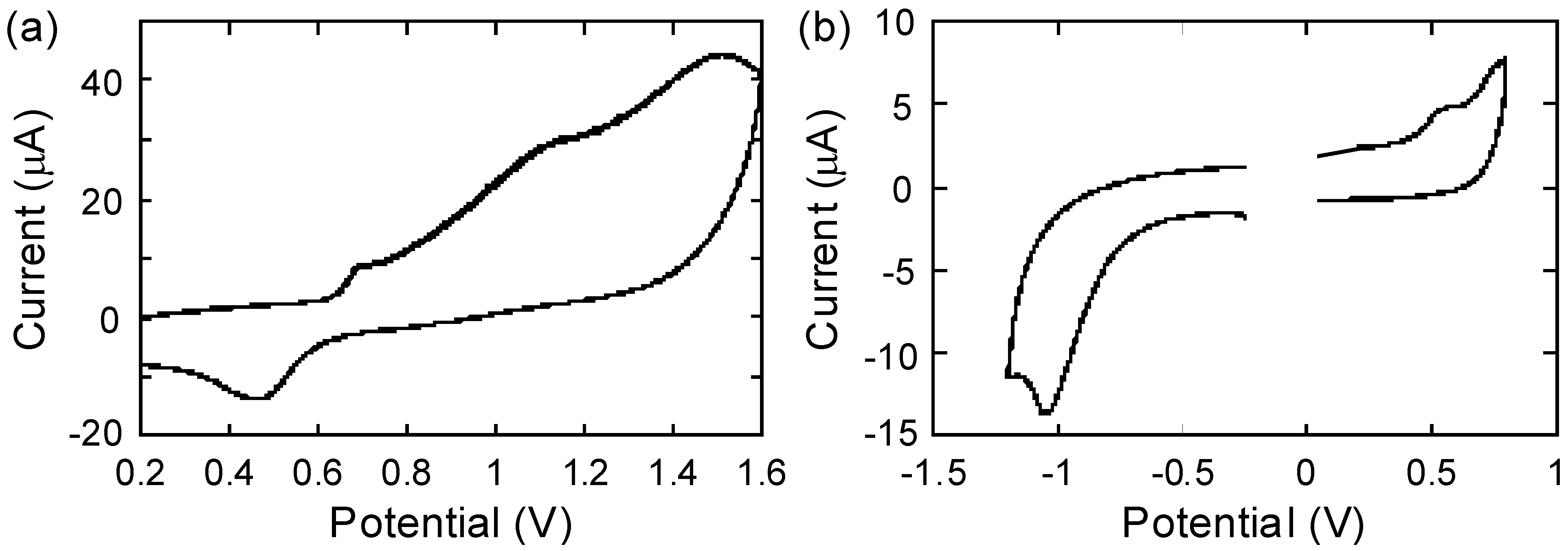
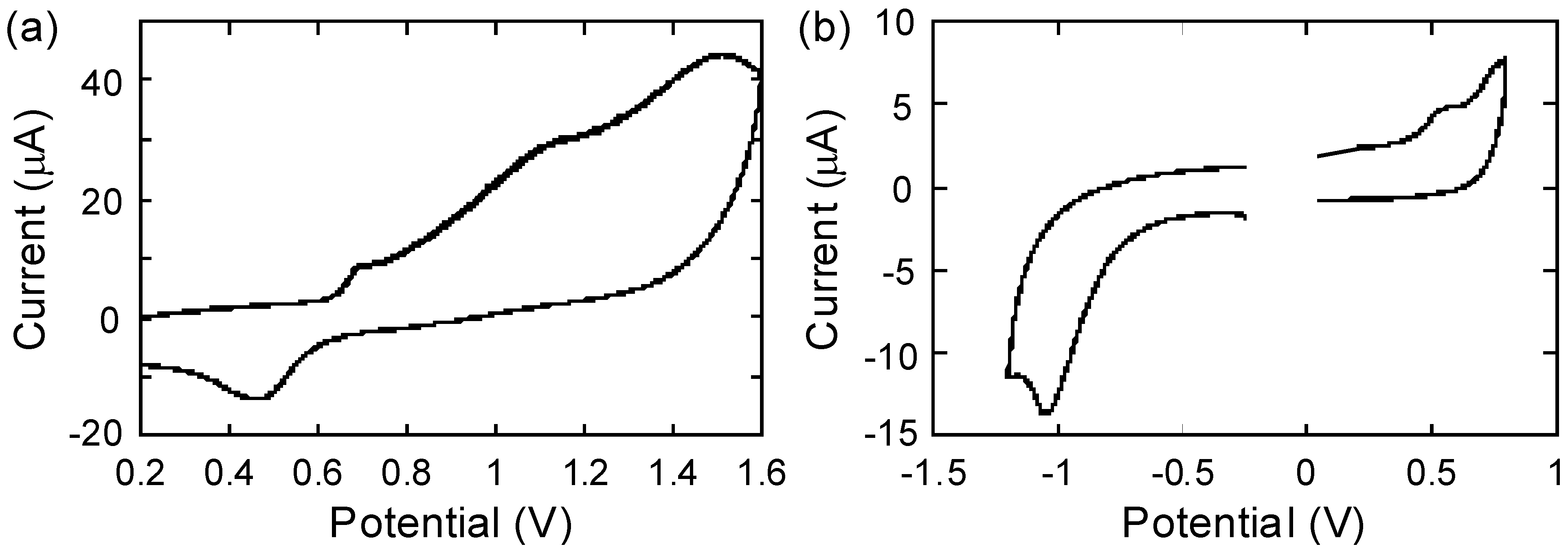
2.4. Electrochemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Eox,onset / Va | Ered,onset / Va | Δ|Eox,onset-Ered,onset| / V | λend / nmb | Opt. band gap / eVc | HOMO / eV | LUMO / eV |
|---|---|---|---|---|---|---|---|
| P1 | 0.64 | - | - | 494 | 2.51 | −5.44d | −2.93e |
| P2 | 0.42 | −0.82 | 1.24 | 950 | 1.46 | −5.22d | −3.98d |
3. Experimental Section
3.1. Materials
3.2. Measurements
3.3. Postfunctionalization with TCNE
4. Conclusions
Acknowledgements
References
- Shirakawa, H.; Louis, E.J.; MacDiarmid, A.G.; Chiang, C.K.; Heeger, A.J. Synthesis of electrically conducting organic polymers: halogen derivatives of polyacetylene, (CH)x. J. Chem. Soc. Chem. Commun. 1977, 578–580. [Google Scholar]
- Heeger, A.J. Semiconducting polymers: the third generation. Chem. Soc. Rev. 2010, 39, 2354–2371. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T. Conjugated polymers bearing electronic and optical functionalities. preparation by organometallic polycondensations, properties, and their applications. Bull. Chem. Soc. Jpn. 1999, 72, 621–638. [Google Scholar] [CrossRef]
- Michinobu, T. Click synthesis of donor-acceptor type aromatic polymers. Pure Appl. Chem. 2010, 82, 1001–1009. [Google Scholar] [CrossRef]
- Kato, S.-I.; Diederich, F. Non-planar push-pull chromophores. Chem. Commun. 2010, 46, 1994–2006. [Google Scholar] [CrossRef]
- Michinobu, T.; May, J.C.; Lim, J.H.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Gross, M.; Biaggio, I.; Diederich, F. A new class of organic donor-acceptor molecules with large third-order optical nonlinearities. Chem. Commun. 2005, 737–739. [Google Scholar]
- Michinobu, T.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Frank, B.; Moonen, N.N.P.; Gross, M.; Diederich, F. Donor-substituted 1,1,4,4-tetracyanobutadienes: new chromophores with efficient intramolecular charge-transfer interactions by atom-economic synthesis. Chem. Eur. J. 2006, 12, 1889–1905. [Google Scholar] [CrossRef] [PubMed]
- Kivala, M.; Stanoeva, T.; Michinobu, T.; Frank, B.; Gescheidt, G.; Diederich, F. One-electron reduced and oxidized stages of donor-substituted 1,1,4,4-tetracyanobuta-1,3-dienes of different molecular architectures. Chem. Eur. J. 2008, 14, 7638–7647. [Google Scholar] [CrossRef] [PubMed]
- Esembeson, B.; Scimeca, M.L.; Michinobu, T.; Diederich, F.; Biaggio, I. A high-optical quality supramolecular assembly for third-order integrated nonlinear optics. Adv. Mater. 2008, 20, 4584–4587. [Google Scholar] [CrossRef]
- Koos, C.; Vorreau, P.; Vallaitis, T.; Dumon, P.; Bogaerts, W.; Baets, R.; Esembeson, B.; Biaggio, I.; Michinobu, T.; Diederich, F.; Freude, W.; Leuthold, J. All-optical high-speed signal processing with silicon-organic hybrid slot waveguides. Nat. Photonics 2009, 3, 216–219. [Google Scholar] [CrossRef]
- Michinobu, T. Click-type reaction of aromatic polyamines for improvement of thermal and optoelectronic properties. J. Am. Chem. Soc. 2008, 130, 14074–14075. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Michinobu, T. Sequential double click reactions: a highly efficient post-functionalization method for optoelectronic polymers. Polym. Chem. 2010, 1, 72–74. [Google Scholar] [CrossRef]
- Li, Y.; Tsuboi, K.; Michinobu, T. Double click synthesis and second-order nonlinearities of polystyrenes bearing donor-acceptor chromophores. Macromolecules 2010, 43, 5277–5286. [Google Scholar] [CrossRef]
- Li, Y.; Tsuboi, K.; Michinobu, T.; Ishida, Y.; Hirai, T.; Hayakawa, T.; Kakimoto, M.-A. Efficient synthesis of block copolymers bearing donor-acceptor chromophores for second-order nonlinear optical applications. J. Photopolym. Sci. Technol. 2010, 23, 337–342. [Google Scholar] [CrossRef]
- Michinobu, T.; Kumazawa, H.; Noguchi, K.; Shigehara, K. One-step synthesis of donor-acceptor type conjugated polymers from ferrocene-containing poly(aryleneethynylene)s. Macromolecules 2009, 42, 5903–5905. [Google Scholar] [CrossRef]
- Tao, X.-T.; Zhang, Y.-D.; Wada, T.; Sasabe, H.; Suzuki, H.; Watanabe, T.; Miyata, S. Hyperbranched polymers for electroluminescence applications. Adv. Mater. 1998, 10, 226–230. [Google Scholar] [CrossRef]
- Michinobu, T.; Okoshi, K.; Osako, H.; Kumazawa, H.; Shigehara, K. Band-gap tuning of carbazole-containing donor-acceptor type conjugated polymers by acceptor moieties and -spacer groups. Polymer 2008, 49, 192–199. [Google Scholar] [CrossRef]
- Michinobu, T.; Osako, H.; Shigehara, K. Synthesis and properties of conjugated poly(1,8-carbazole)s. Macromolecules 2009, 42, 8172–8180. [Google Scholar] [CrossRef]
- Morin, J.-F.; Leclerc, M.; Adès, D.; Siove, A. Polycarbazoles: 25 years of progress. Macromol. Rapid Commun. 2005, 26, 761–778. [Google Scholar] [CrossRef]
- Blouin, N.; Leclerc, M. Poly(2,7-carbazole)s: structure-property relationships. Acc. Chem. Res. 2008, 41, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Reitzenstein, D.; Lambert, C. Localized versus Backbone Fluorescence in N-p-(Diarylboryl)phenyl-Substituted 2,7- and 3,6-Linked Polycarbazoles. Macromolecules 2009, 42, 773–782. [Google Scholar] [CrossRef]
- Michinobu, T.; Kumazawa, H.; Shigehara, K. Nitrogen-linked Aromatic Poly(2,7-carbazole)s: Partially Annulated Poly(m-aniline)s. Chem. Lett. 2007, 36, 620–621. [Google Scholar] [CrossRef]
- Michinobu, T.; Osako, H.; Shigehara, K. Alkyne-linked poly(1,8-carbazole)s: A novel class of conjugated carbazole polymers. Macromol. Rapid Commun. 2008, 29, 111–116. [Google Scholar] [CrossRef]
- Michinobu, T.; Kumazawa, H.; Otsuki, E.; Usui, H.; Shigehara, K. Synthesis and properties of nitrogen-linked poly(2,7-carbazole)s as hole-transport material for organic light emitting diodes. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 3880–3891. [Google Scholar] [CrossRef]
- Michinobu, T.; Osako, H.; Murata, K.; Shigehara, K. Blue, green, and red light emission of 1,8-carbazole-based conjugated polymers. Chem. Lett. 2010, 39, 168–169. [Google Scholar] [CrossRef]
- Michinobu, T.; Osako, H.; Shigehara, K. Synthesis and properties of 1,8-carbazole-based conjugated copolymers. Polymers 2010, 2, 159–173. [Google Scholar] [CrossRef]
- Zou, Y.; Gendron, D.; Badrou-Aïch, R.; Najari, A.; Tao, Y.; Leclerc, M. A high-mobility low-bandgap poly(2,7-carbazole) derivative for photovoltaic applications. Macromolecules 2009, 42, 2891–2894. [Google Scholar] [CrossRef]
- Tamura, K.; Shiotsuki, M.; Kobayashi, N.; Masuda, T.; Sanda, F. Synthesis and properties of conjugated polymers containing 3,9- and 2,9-linked carbazole units in the main chain. J. Polym. Sci. A 2009, 47, 3509–3517. [Google Scholar] [CrossRef]
- Thompson, B.C.; Fréchet, J.M. Polymer-fullerene composite solar cells. Angew. Chem. Int. Ed. 2008, 47, 58–77. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Michinobu, T.; Fujita, H. Postfunctionalization of Alkyne-Linked Conjugated Carbazole Polymer by Thermal Addition Reaction of Tetracyanoethylene. Materials 2010, 3, 4773-4783. https://doi.org/10.3390/ma3104773
Michinobu T, Fujita H. Postfunctionalization of Alkyne-Linked Conjugated Carbazole Polymer by Thermal Addition Reaction of Tetracyanoethylene. Materials. 2010; 3(10):4773-4783. https://doi.org/10.3390/ma3104773
Chicago/Turabian StyleMichinobu, Tsuyoshi, and Hiroyuki Fujita. 2010. "Postfunctionalization of Alkyne-Linked Conjugated Carbazole Polymer by Thermal Addition Reaction of Tetracyanoethylene" Materials 3, no. 10: 4773-4783. https://doi.org/10.3390/ma3104773