Tissue Response to, and Degradation Rate of, Photocrosslinked Trimethylene Carbonate-Based Elastomers Following Intramuscular Implantation

Abstract
:1. Introduction
2. Results and Discussion
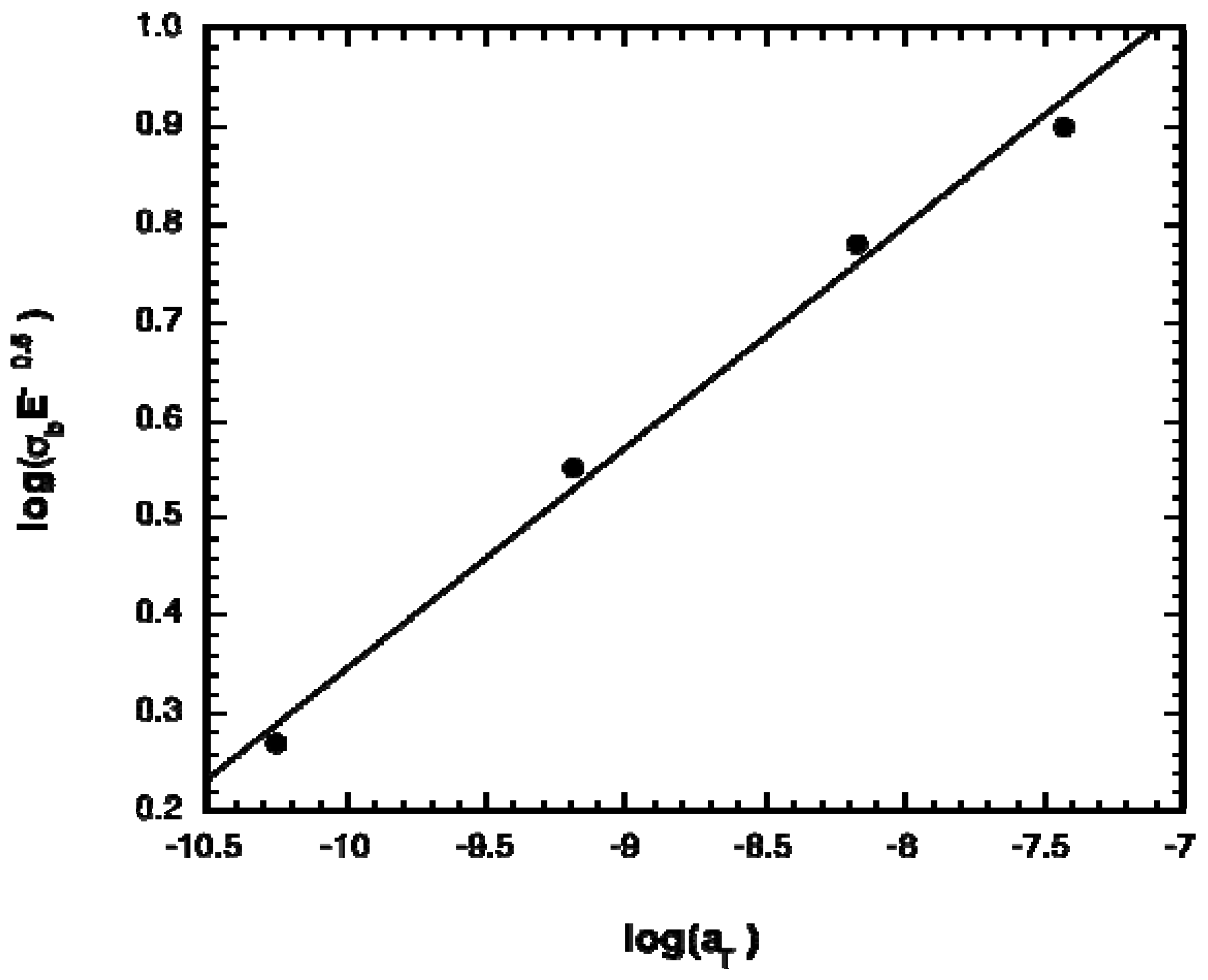
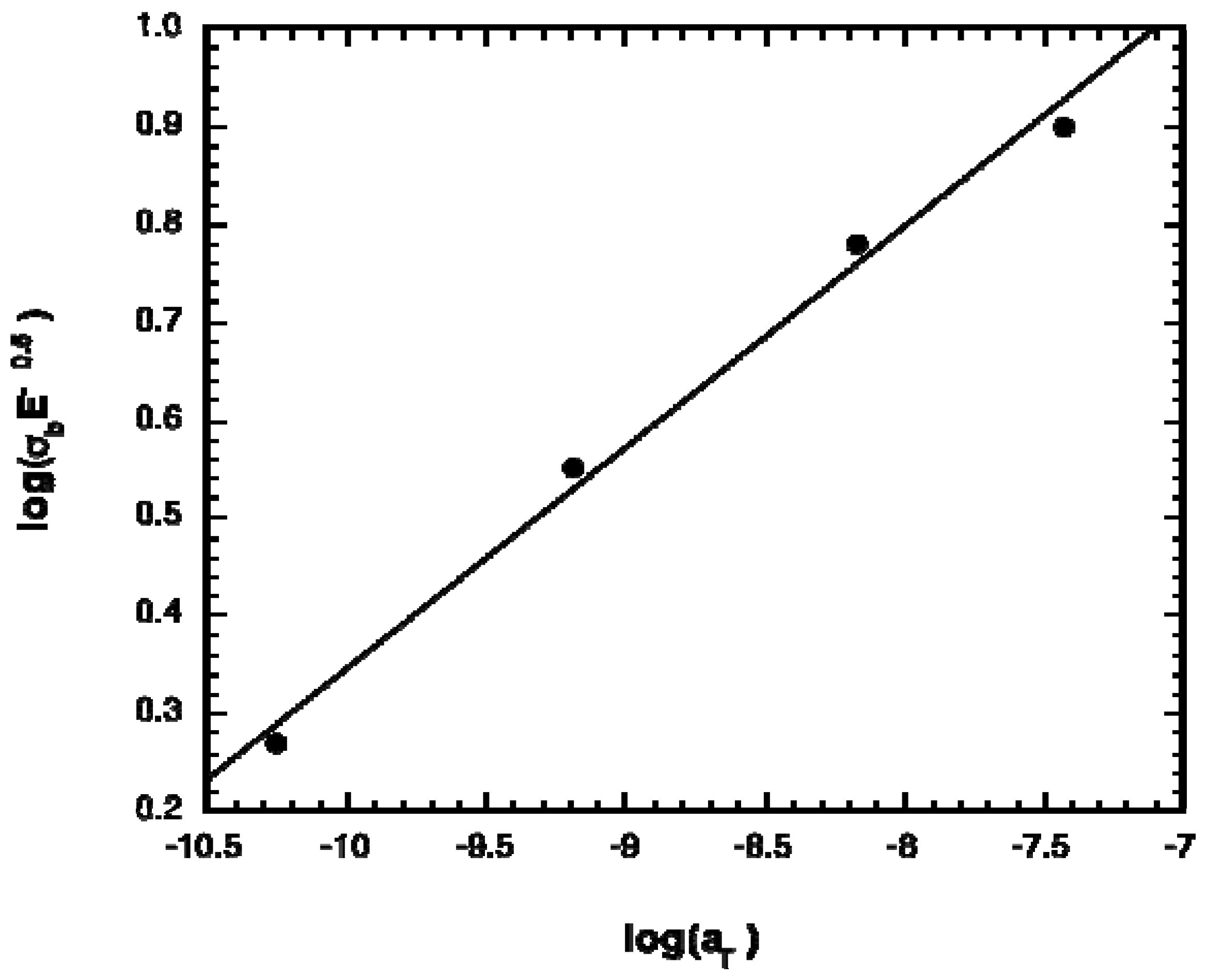
2.1. Polymer Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nature | % TMC | Mn (g/mol) | DA (%) | Tg (°C) |
| TMC:CL | 67.1 | 9,300 | 80.6 | −32.5 |
| TMC:DLLA | 66.7 | 8,770 | 83.8 | −13.3 |
| Comonomer | % TMC | E (MPa) | σb (MPa) | εb (mm/mm) | Tg (°C) |
| TMC* | 100 | 1.44 ± 0.15 | 7.23 ± 0.16 | 11.8 ± 0.5 | − 12 |
| CL | 67 | 0.16 ± 0.04 | 1.43 ± 0.60 | 10.9 ± 0.78 | − 23 |
| CL* | 50 | 0.83 ± 0.14 | 1.73 ± 0.58 | 5.2 ± 1.5 | − 42 |
| DLLA | 66 | 0.42 ± 0.09 | 5.2 ± 0.93 | 14.8 ± 1.2 | 4 |
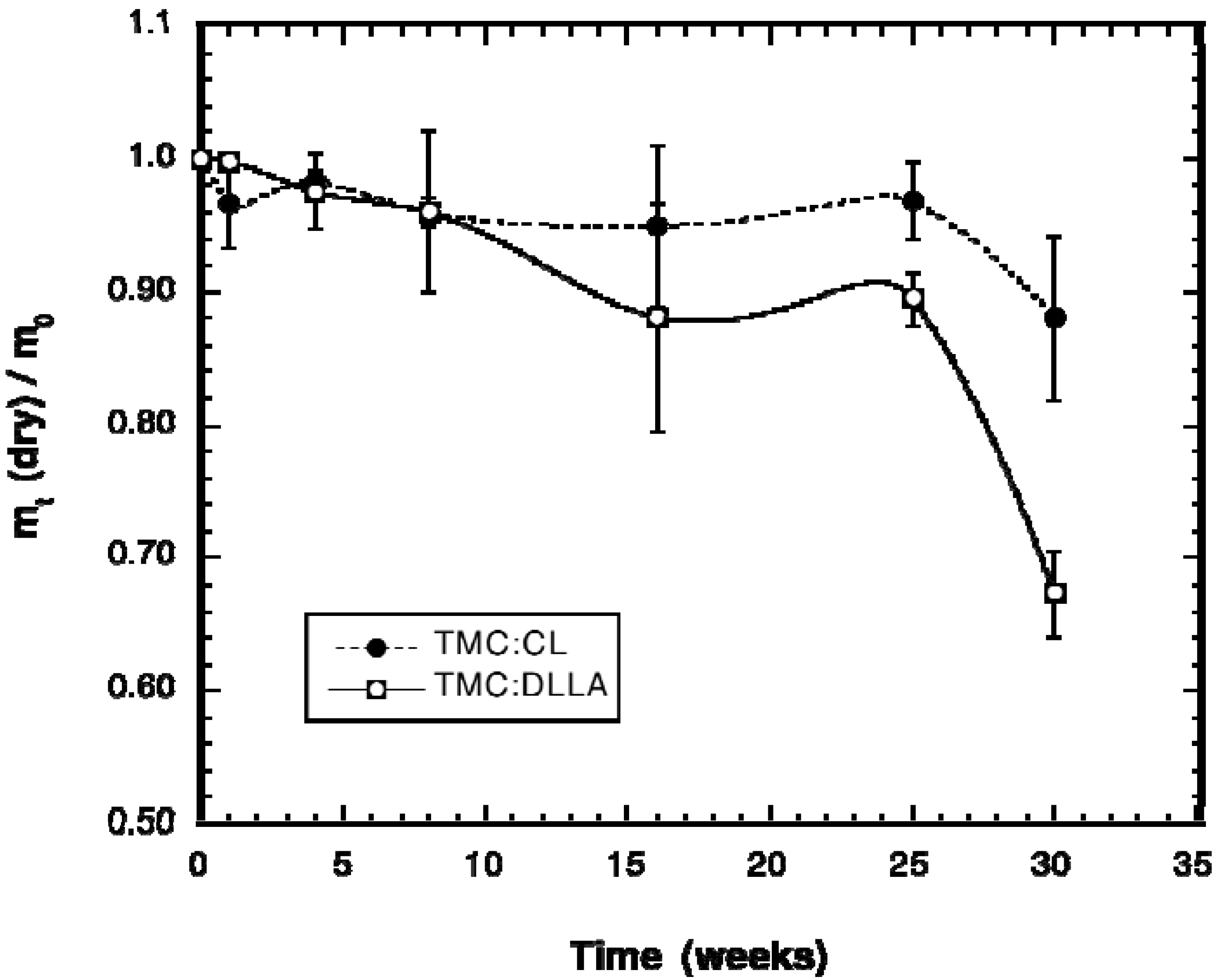
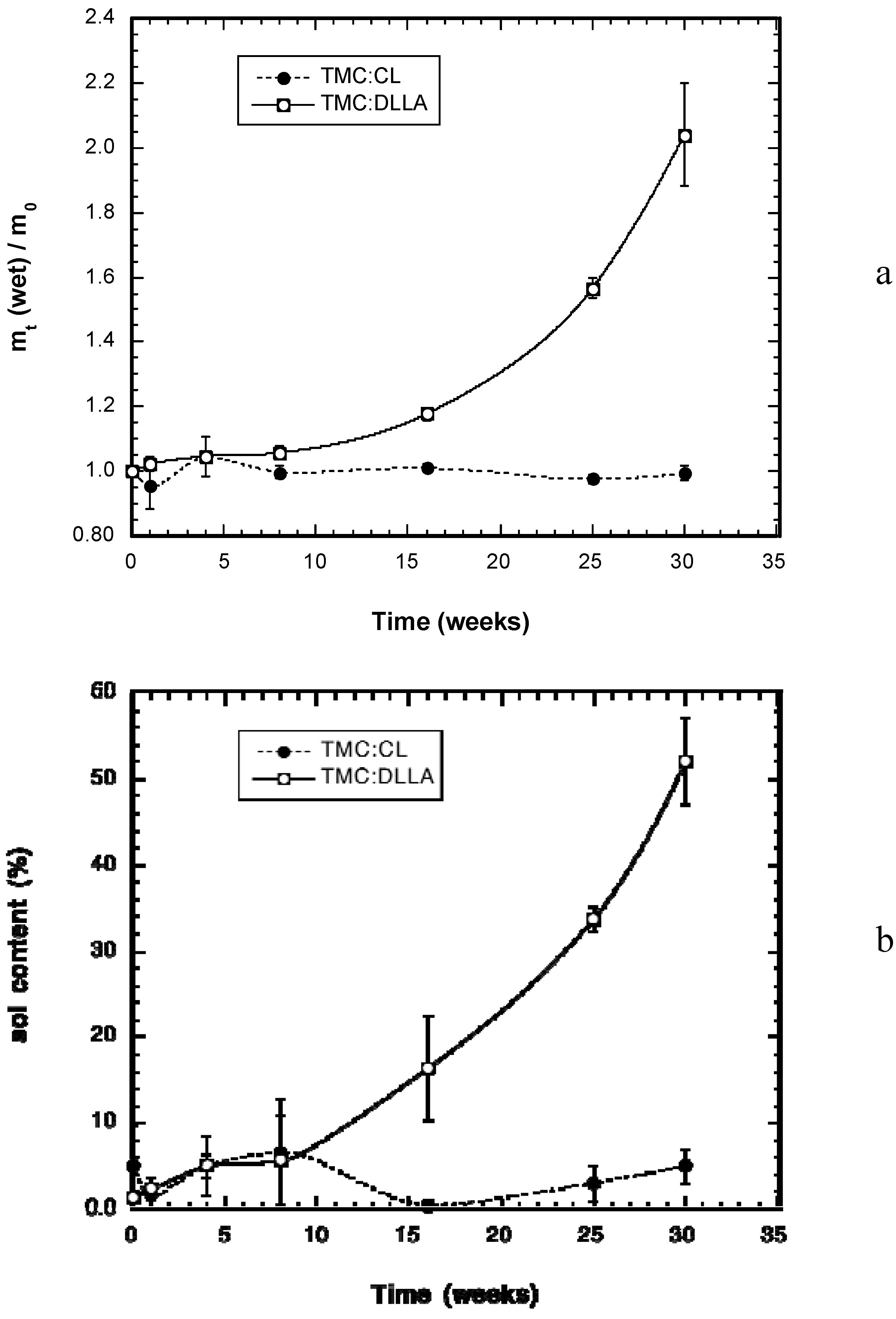
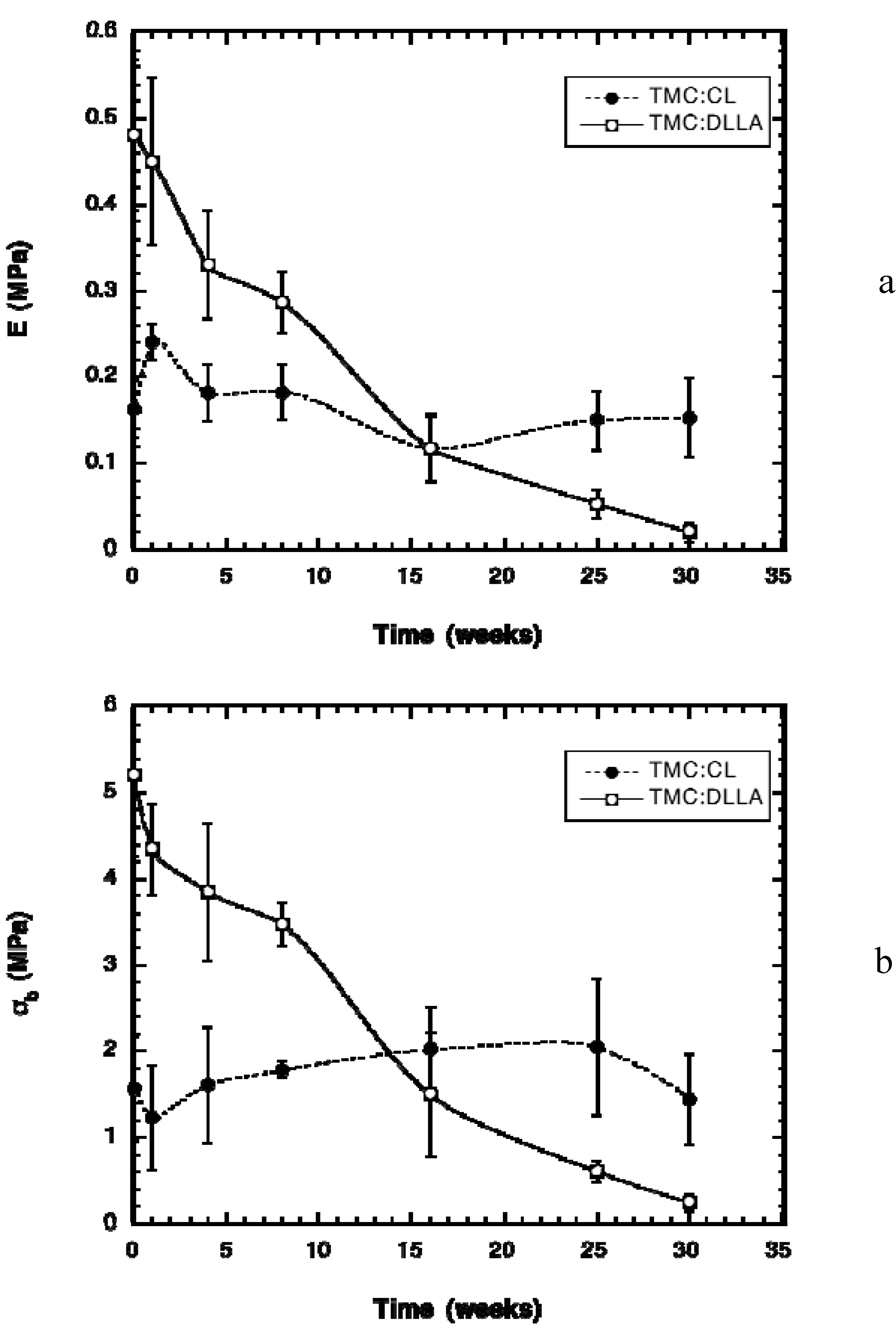
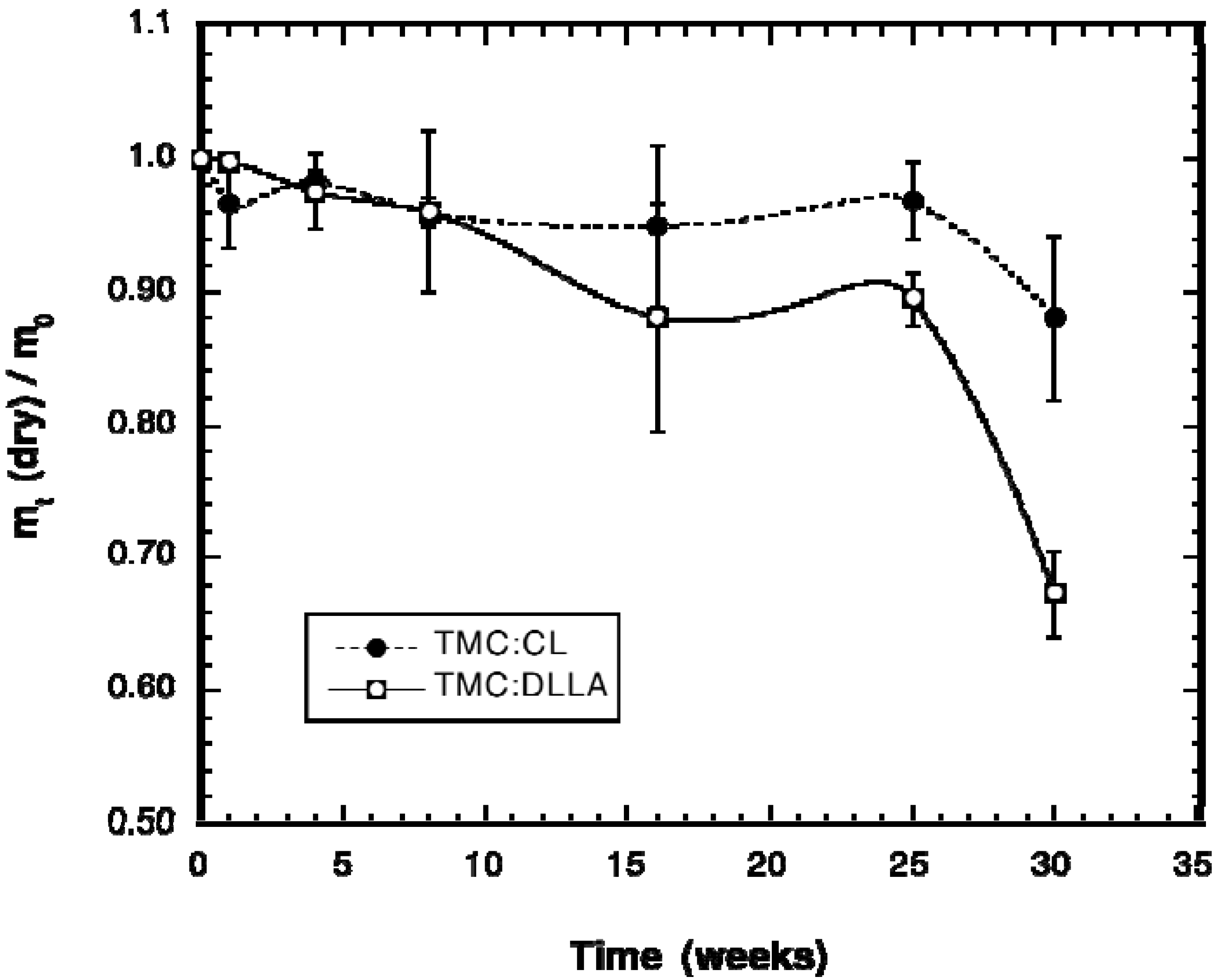
2.2. Change in Polymer Properties during in Vivo Degradation
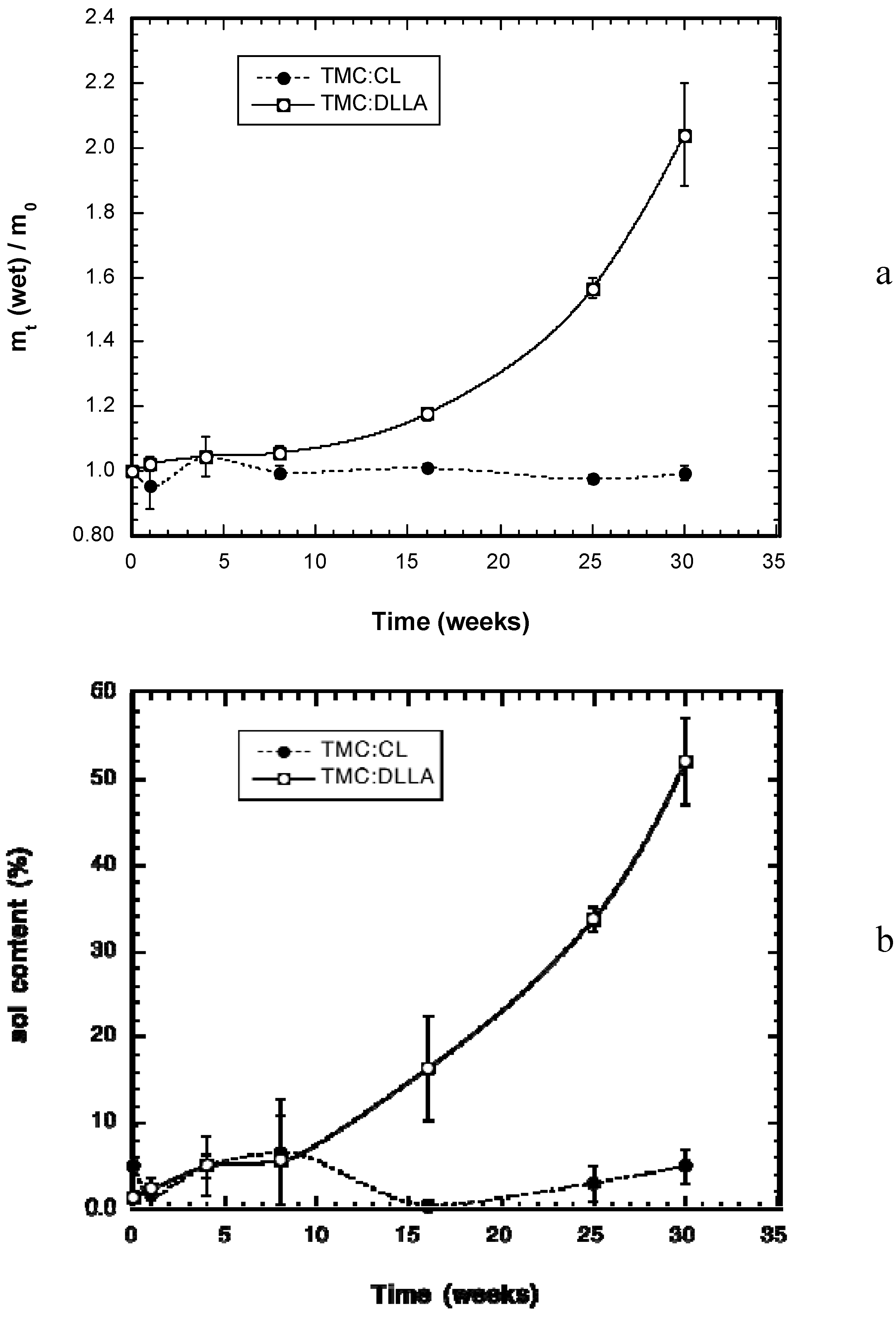

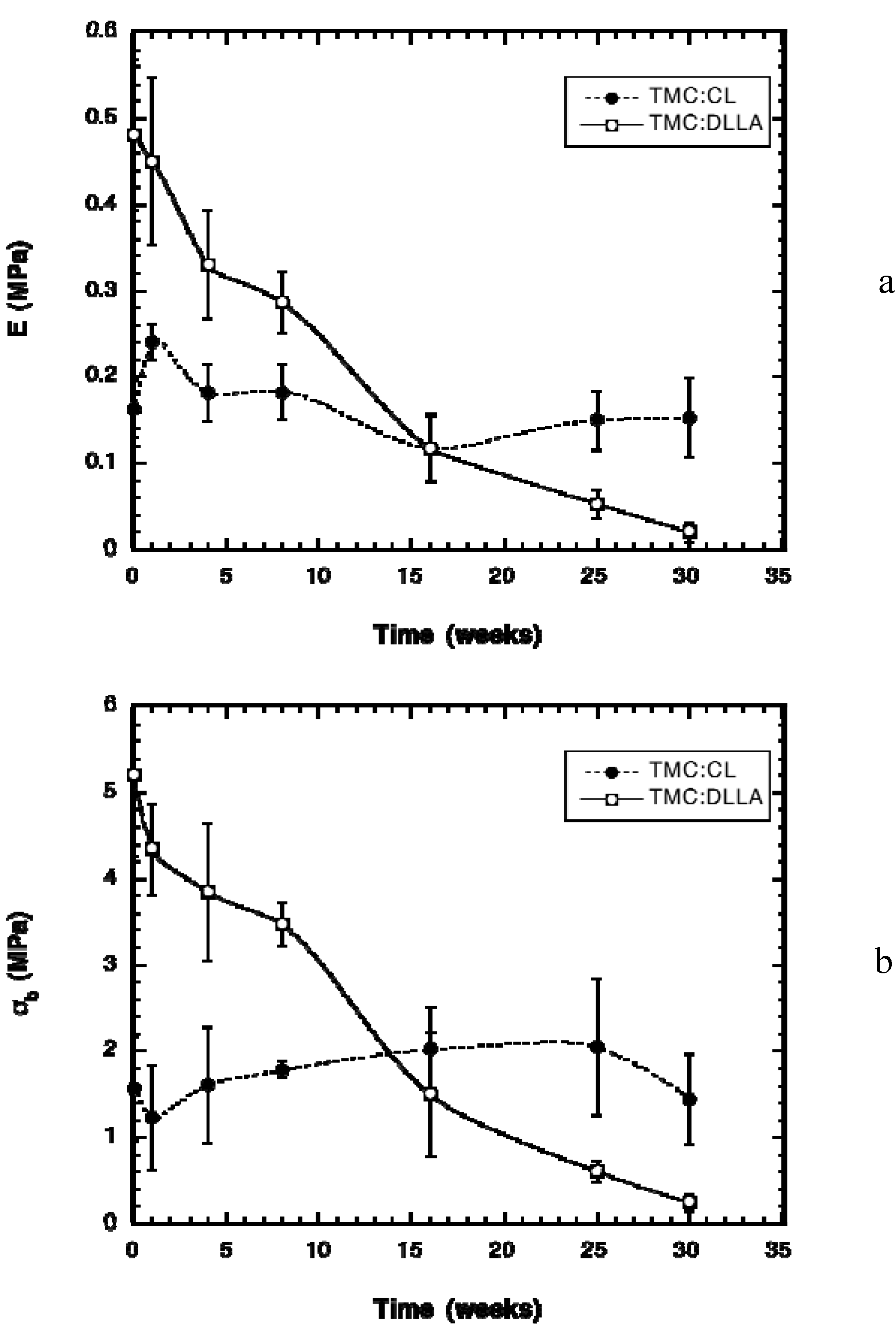
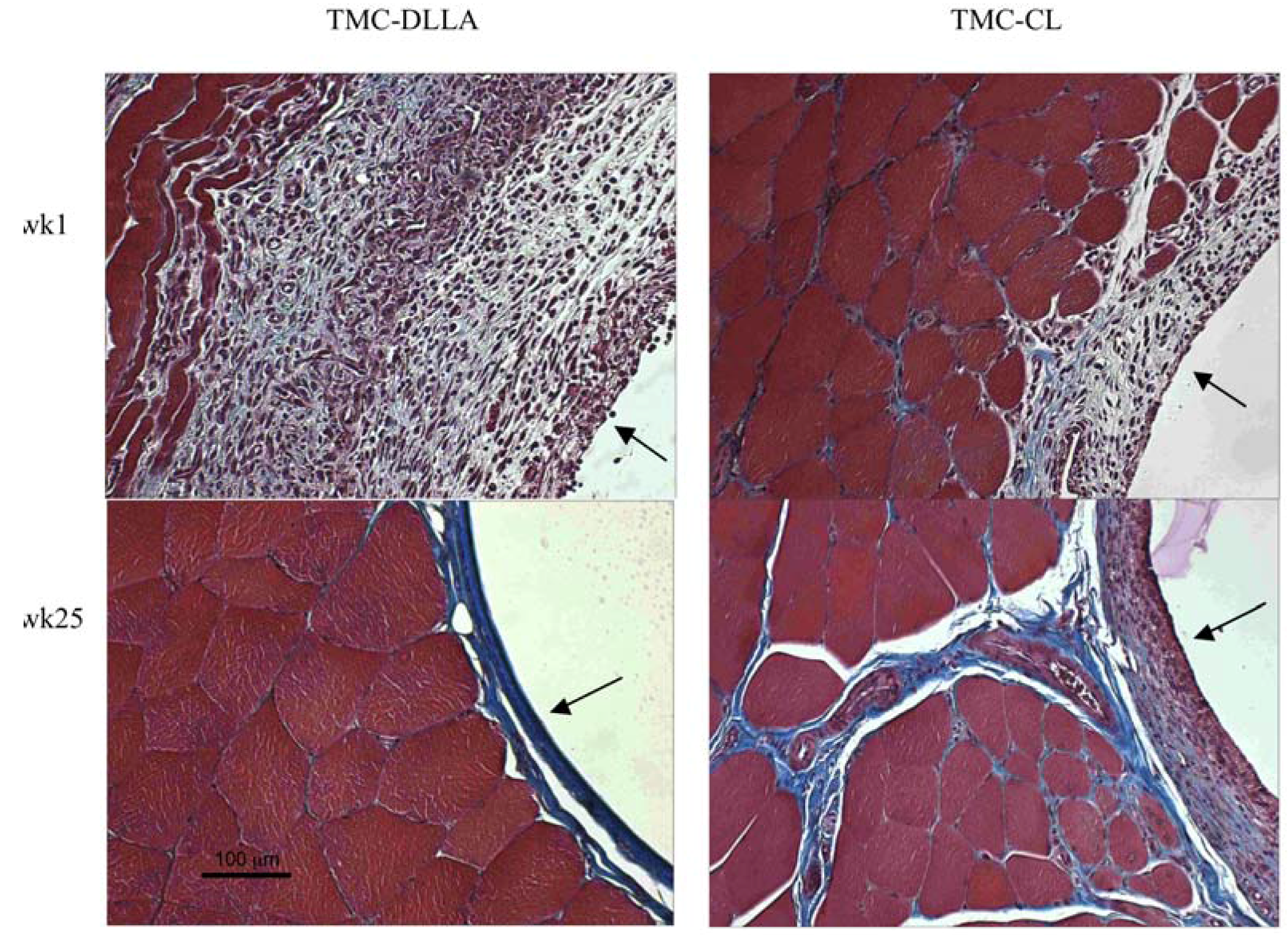
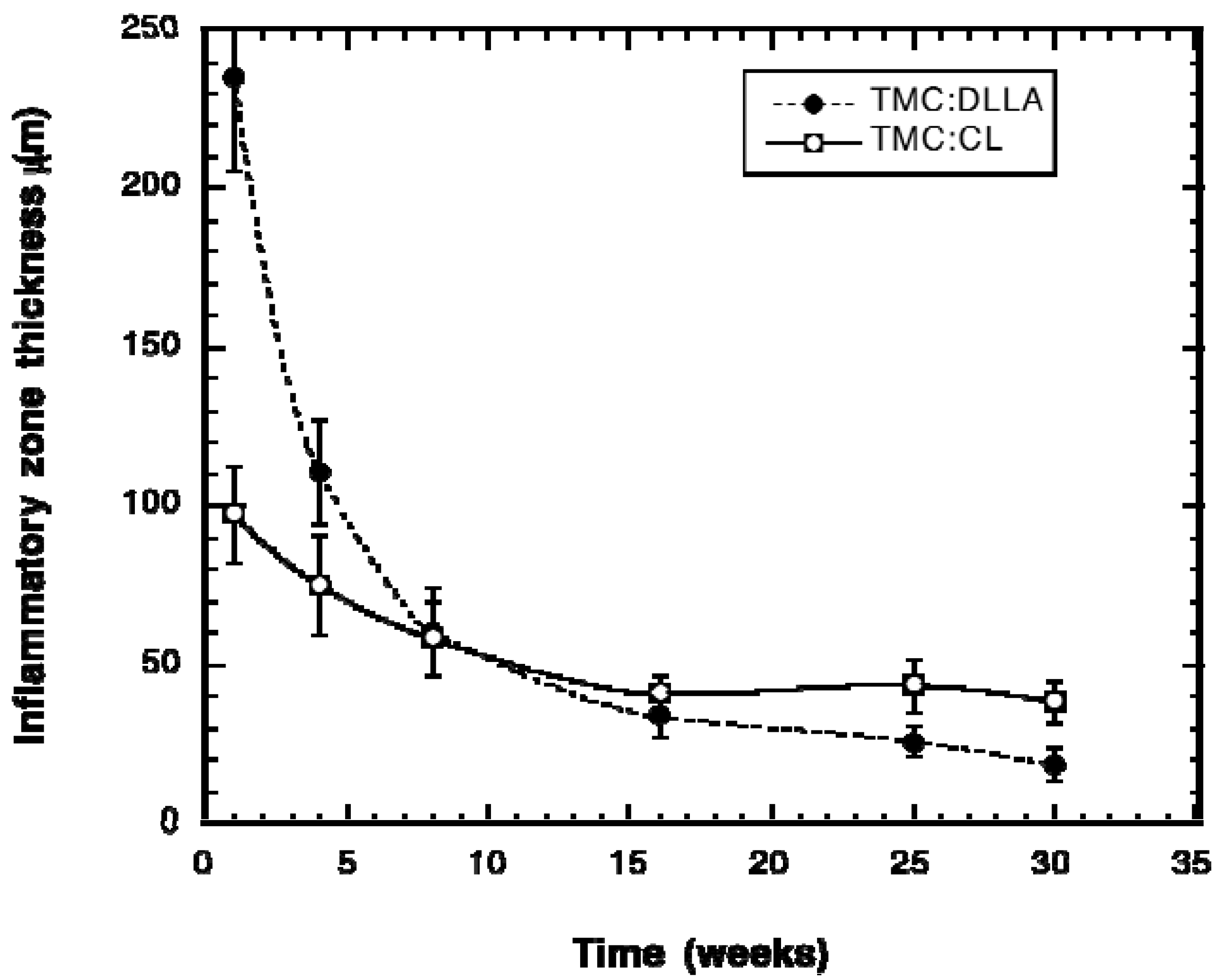
2.3. Tissue Response
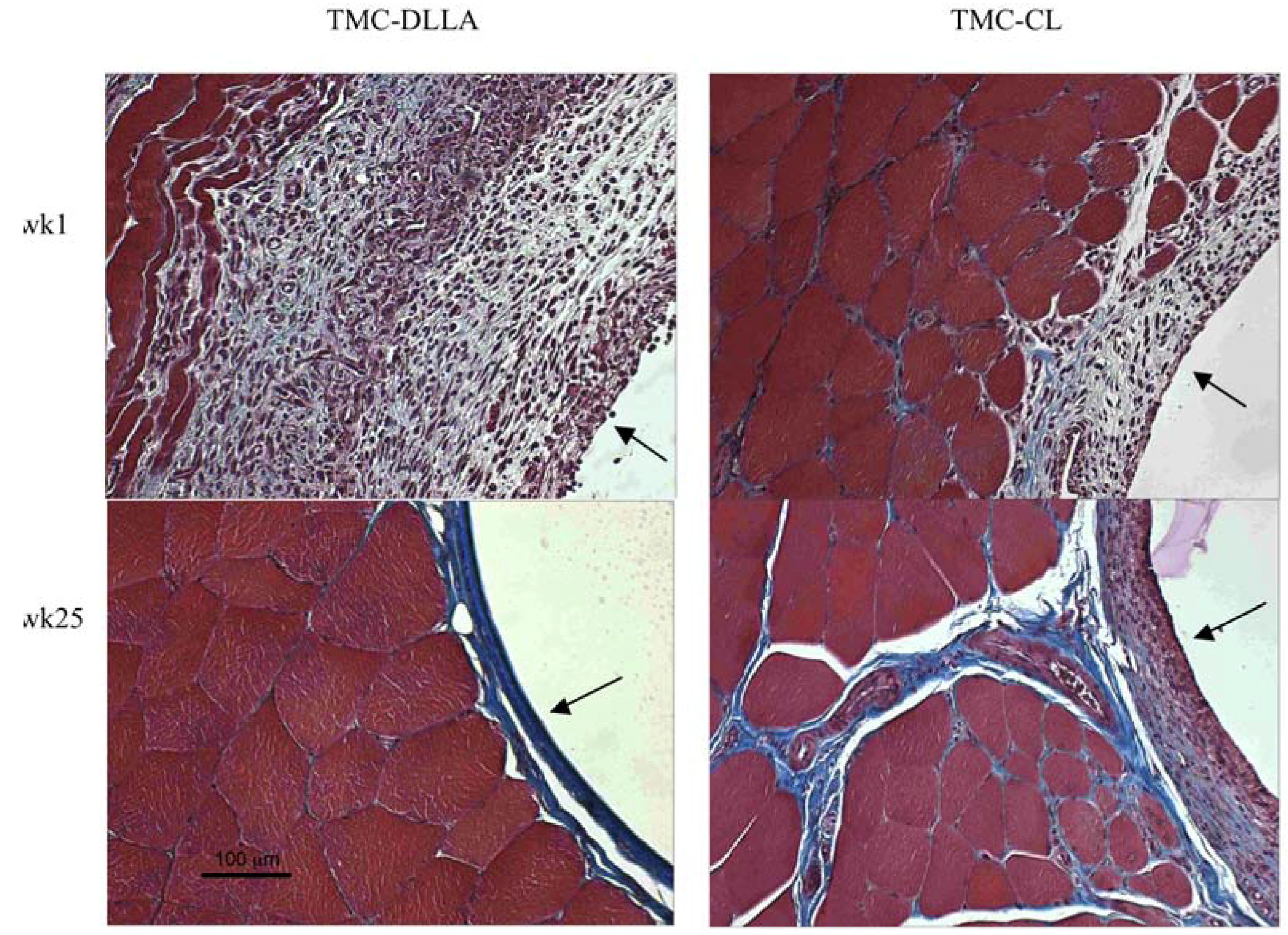
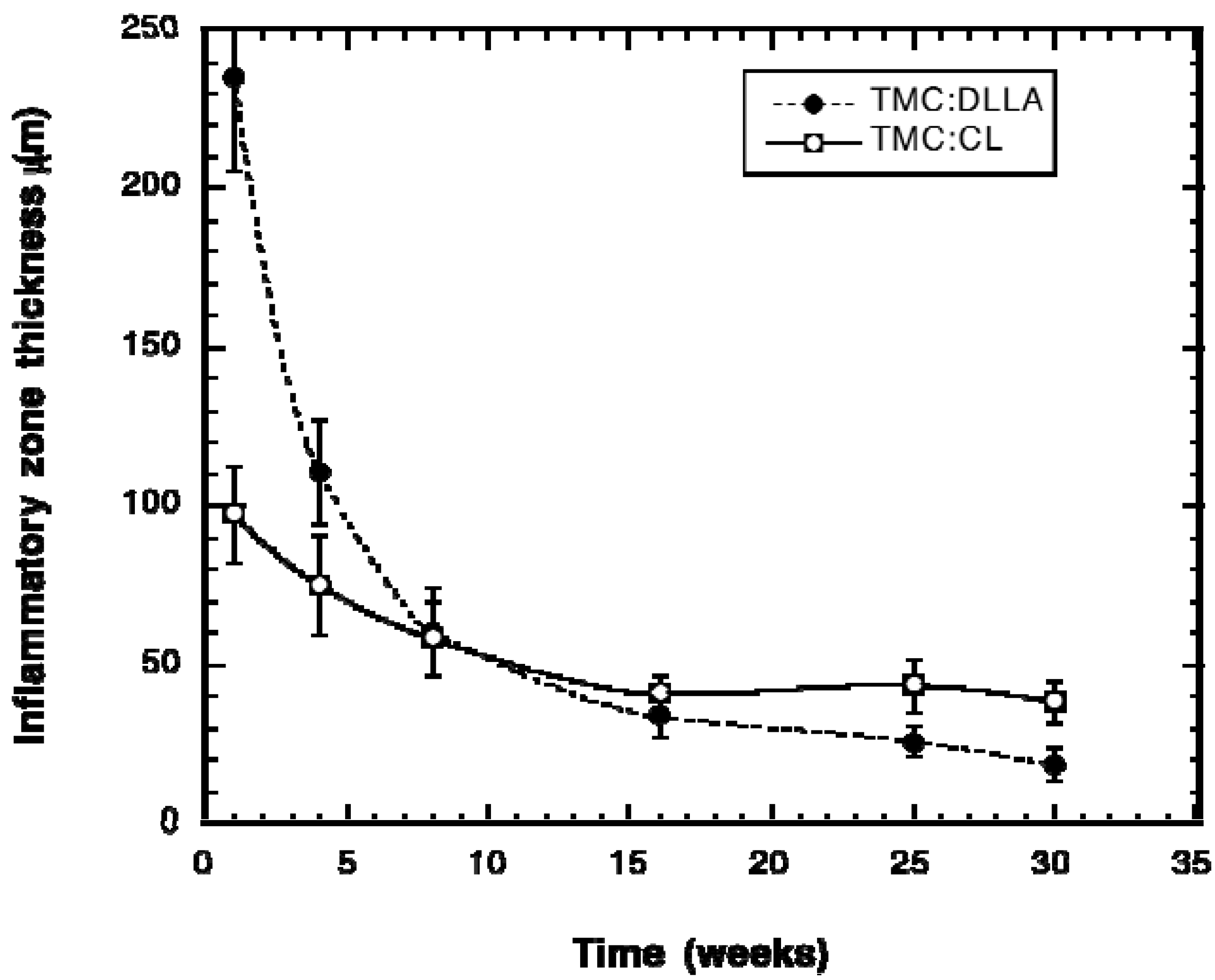
3. Experimental Section
3.1. Materials
3.2. Synthesis of Star-Copolymers
3.3. Acrylation of the Star-Copolymers
3.4. UV Initiated Cross-Linking of the Acrylated Star-Copolymers
3.5. Polymer Characterization
3.6. In Vivo Implantation
3.7. Statistics
4. Conclusions
Acknowledgements
References
- Webb, A.R.; Yang, J.; Ameer, G.A. Biodegradable polyester elastomers in tissue engineering. Expert Opin. Biol. Th. 2004, 4, 801. [Google Scholar] [CrossRef]
- Kang, Y.; Yang, J.; Khan, S.; Anissian, L.; Ameer, G.A. A new biodegradable polyester elastomer for cartilage tissue engineering. J. Biomed. Mater. Res. A 2006, 77A, 331. [Google Scholar] [CrossRef]
- Wang, T.; Ameer, G.A.; Sheppard, B.J.; Langer, R. A tough biodegradable elastomer. Nat. Biotechnol. 2002, 20, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Neufeld, R.J.; Amsden, B. Osmotic driven release kinetics of bioactive therapeutic proteins from a biodegradable elastomer are linear, constant, similar and adjustable. Pharm. Res. 2006, 23, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Younes, H.M.; El-Kadi, A.O.S.; Neufeld, R.J.; Amsden, B.G. Sustained interferon-gamma delivery from a photocrosslinked biodegradable elastomer. J. Control. Release 2005, 102, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B.G. Biodegradable elastomers in drug delivery. Expert Opin. Drug Delivery 2008, 5, 175–187. [Google Scholar] [CrossRef]
- Ifkovits, J.L.; Burdick, J.A. Review: Photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Eng. 2007, 13, 2369–2385. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Neufeld, R.; Amsden, B. Sustained release of bioactive therapeutic proteins from a biodegradable elastomeric device. J. Control. Release 2007, 117, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Neufeld, R.; Amsden, B. Maintenance of vascular endothelial growth factor and potentially other therapeutic proteins bioactivity during a photo-initiated free radical cross-linking reaction forming biodegradable elastomers. Eur. J. Pharm. Biopharm. 2007, 66, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Nijst, C.L.E.; Bruggeman, J.P.; Karp, J.M.; Ferreira, L.; Zumbuehl, A.; Bettinger, C.; Langer, R. Synthesis and characterization of photocurable elastomers from poly(glycerol-co-sebacate). Biomacromolecules 2007, 8, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B.; Misra, G.; Gu, F.; Younes, H. Synthesis and characterization of a photocrosslinked biodegradable elastomer. Biomacromolecules 2004, 5, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Lendlein, A.; Langer, R. Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 2002, 296, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Chapanian, R.; Tse, M.Y.; Pang, S.C.; Amsden, B.G. The role of oxidation and enzymatic hydrolysis on the in vivo degradation of trimethylene carbonate based photocrosslinkable elastomers. Biomaterials 2009, 30, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Kwon, I.K.; Kidoaki, S. Photocurable biodegradable liquid copolymers: Synthesis of acrylate-end-capped trimethylene carbonate-based prepolymers, photocuring, and hydrolysis. Biomacromolecules 2004, 5, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Arnold, S.C.; Matsuda, T. Liquid, phenylazide-end-capped copolymers of epsilon-caprolactone and trimethylene carbonate: Preparation, photocuring characteristics, and surface layering. Biomacromolecules 2002, 3, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Grijpma, D.W.; Hou, Q.P.; Feijen, J. Preparation of biodegradable networks by photo-crosslinking lactide, epsilon-caprolactone and trimethylene carbonate-based oligomers functionalized with fumaric acid monoethyl ester. Biomaterials 2005, 26, 2795–2802. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Schappacher, M.; Bareille, R.; Dupuy, B.; Soum, A.; Bertrand-Barat, J.; Baquey, C. Study of a (trimethylenecarbonate-co-epsilon-caprolactone) polymer—Part 2: In vitro cytocompatibility analysis and in vivo ED1 cell response of a new nerve guide. Biomaterials 2001, 22, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Pego, A.P.; Van Luyn, M.J.A.; Brouwer, L.A.; van Wachem, P.B.; Poot, A.A.; Grijpma, D.W.; Feijen, J. In vivo behavior of poly(1,3-trimethylene carbonate) and copolymers of 1,3-trimethylene carbonate with d,l-lactide or epsilon-caprolactone: Degradation and tissue response. J. Biomed. Mater. Res. A 2003, 67A, 1044–1054. [Google Scholar] [CrossRef]
- Mizutani, M.; Matsuda, T. Liquid photocurable biodegradable copolymers: In vivo degradation of photocured poly(epsilon-caprolactone-co-trimethylene carbonate). J. Biomed. Mater. Res. 2002, 61, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B.; Wang, S.; Wyss, U. Synthesis and characterization of thermoset biodegradable elastomers based on star-poly(-caprolactone-co-d,l-lactide). Biomacromolecules 2004, 54, 1399–1404. [Google Scholar] [CrossRef]
- Zhu, K.J.; Hendren, R.W.; Jensen, K.; Pitt, C.G. Synthesis, properties, and biodegradation of poly(1,3-trimethylene carbonate). Macromolecules 1991, 24, 1736–1740. [Google Scholar] [CrossRef]
- Brode, G.L.; Koleske, J.V. Lactone polymerization and polymer properties. J. Macromol. Sci. Chem. A 1972, 6, 1109–1144. [Google Scholar] [CrossRef]
- Chapanian, R.; Tse, M.Y.; Pang, S.C.; Amsden, B. In vivo degradation mechanism of, and tissue response to, photo-cross-linked elastomers prepared from star shaped prepolymers of poly(ε-caprolactone-co-d,l-lactide). J. Biomed. Mater. Res. A 2009, in press. [Google Scholar]
- Gent, A.N. Strength of elastomers. In Science and Technology of Rubber; Academic Press: New York, NY, USA, 1978; pp. 419–454. [Google Scholar]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. Mechanical properties of substances of high molecular weight .19. The temperature dependence of relaxation mechanisms in amorphous polymers and other glass-forming liquids. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar]
- Wiggins, J.S.; Hassan, M.K.; Mauritz, K.A.; Storey, R.F. Hydrolytic degradation of poly(d,l-lactide) as a function of end group: Carboxylic acid vs. hydroxyl. Polymer 2006, 47, 1960–1969. [Google Scholar] [CrossRef]
- Li, S.M.; McCarthy, S. Further investigations on the hydrolytic degradation of poly(d,l-lactide). Biomaterials 1999, 20, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.; Mader, K.; Gopferich, A. pH and osmotic pressure inside biodegradable microspheres during erosion. Pharm. Res. 1999, 16, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B.G.; Tse, M.Y.; Turner, N.D.; Knight, D.K.; Pang, S.C. In vivo degradation behavior of photo-cross-linked star-poly(epsilon-caprolactone-co-d,l-lactide) elastomers. Biomacromolecules 2006, 7, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Timmins, M.; Liebmann-Vinson, A. Biodegradable Polymers: Degradation Mechanisms Part I. In Introduction to Polymeric Biomaterials; Arshady, R., Ed.; Citus Books: London, UK, 2003; Volume 2, pp. 285–328. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Timbart, L.; Tse, M.Y.; Pang, S.C.; Amsden, B.G. Tissue Response to, and Degradation Rate of, Photocrosslinked Trimethylene Carbonate-Based Elastomers Following Intramuscular Implantation. Materials 2010, 3, 1156-1171. https://doi.org/10.3390/ma3021156
Timbart L, Tse MY, Pang SC, Amsden BG. Tissue Response to, and Degradation Rate of, Photocrosslinked Trimethylene Carbonate-Based Elastomers Following Intramuscular Implantation. Materials. 2010; 3(2):1156-1171. https://doi.org/10.3390/ma3021156
Chicago/Turabian StyleTimbart, Laurianne, Man Yat Tse, Stephen C. Pang, and Brian G. Amsden. 2010. "Tissue Response to, and Degradation Rate of, Photocrosslinked Trimethylene Carbonate-Based Elastomers Following Intramuscular Implantation" Materials 3, no. 2: 1156-1171. https://doi.org/10.3390/ma3021156