Mechanism of Catalytic Water Oxidation by the Ruthenium Blue Dimer Catalyst: Comparative Study in D2O versus H2O

Abstract
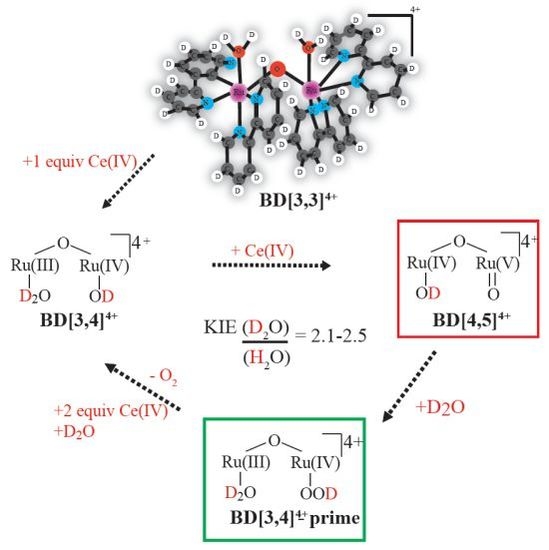
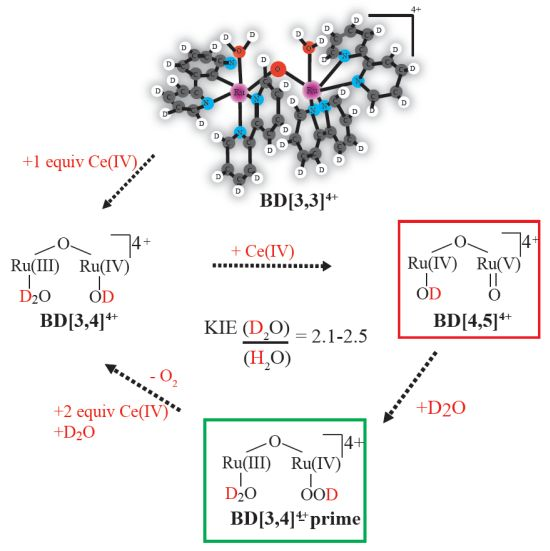
:
1. Introduction
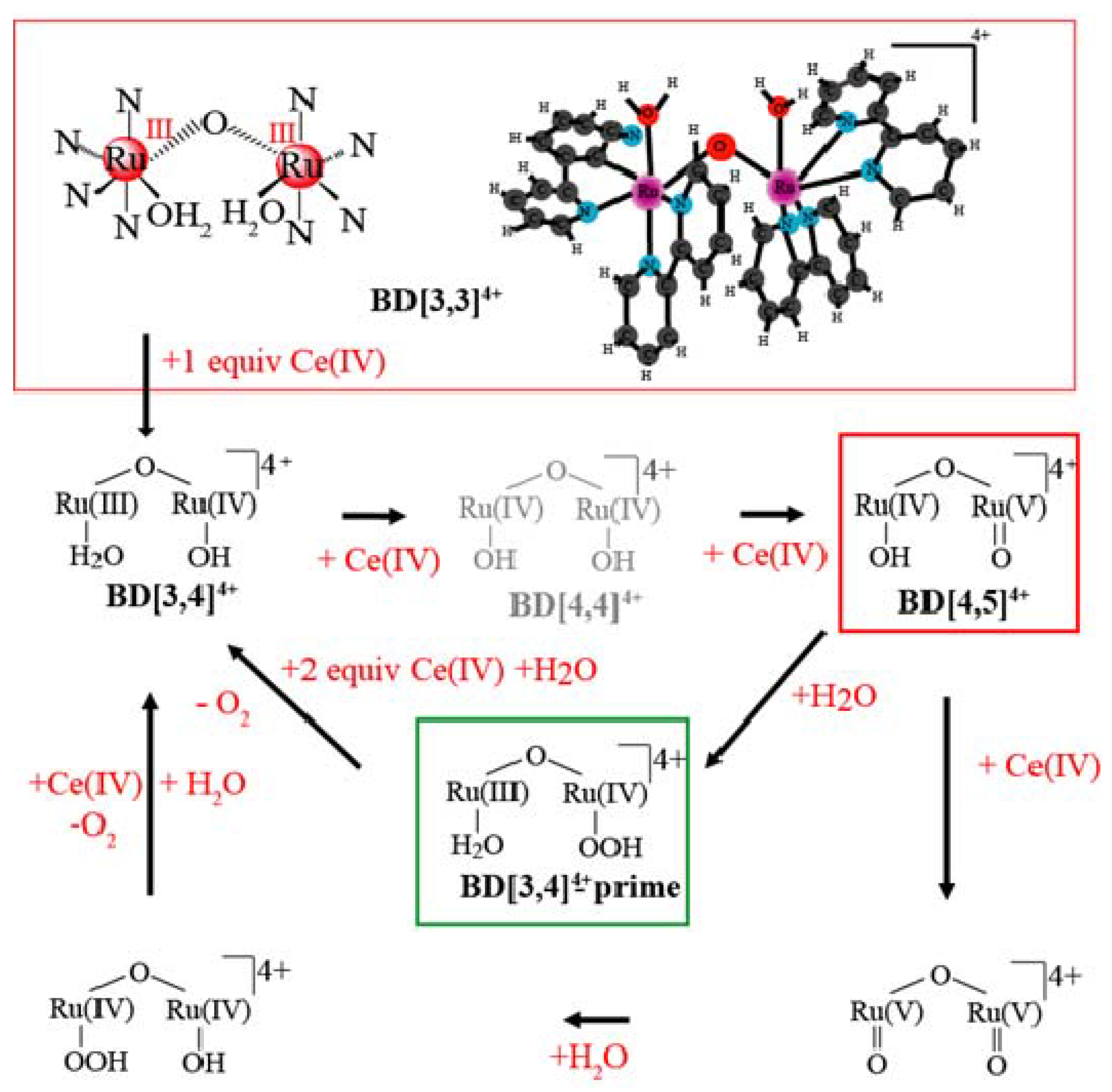
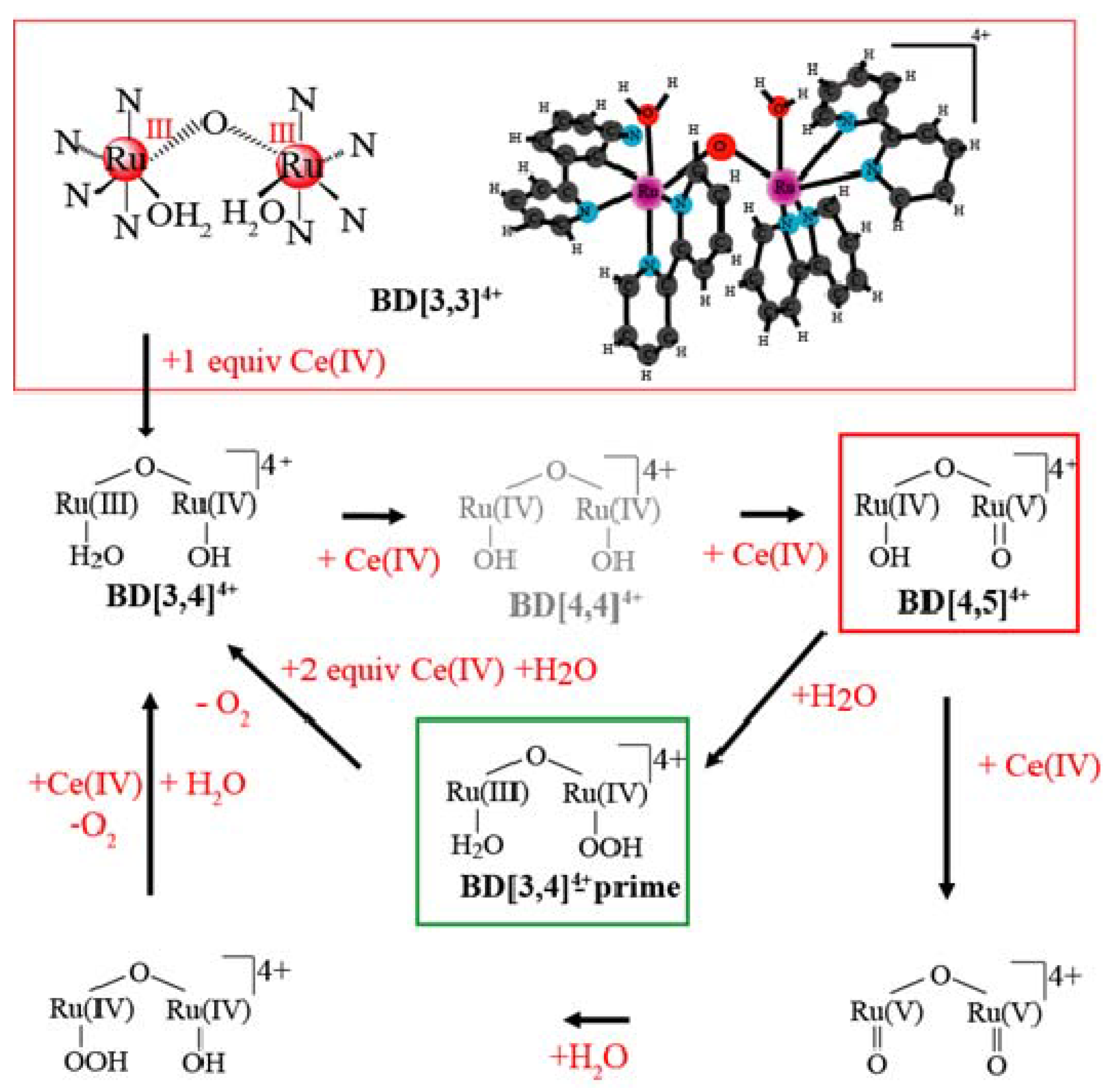
2. Results and Discussion
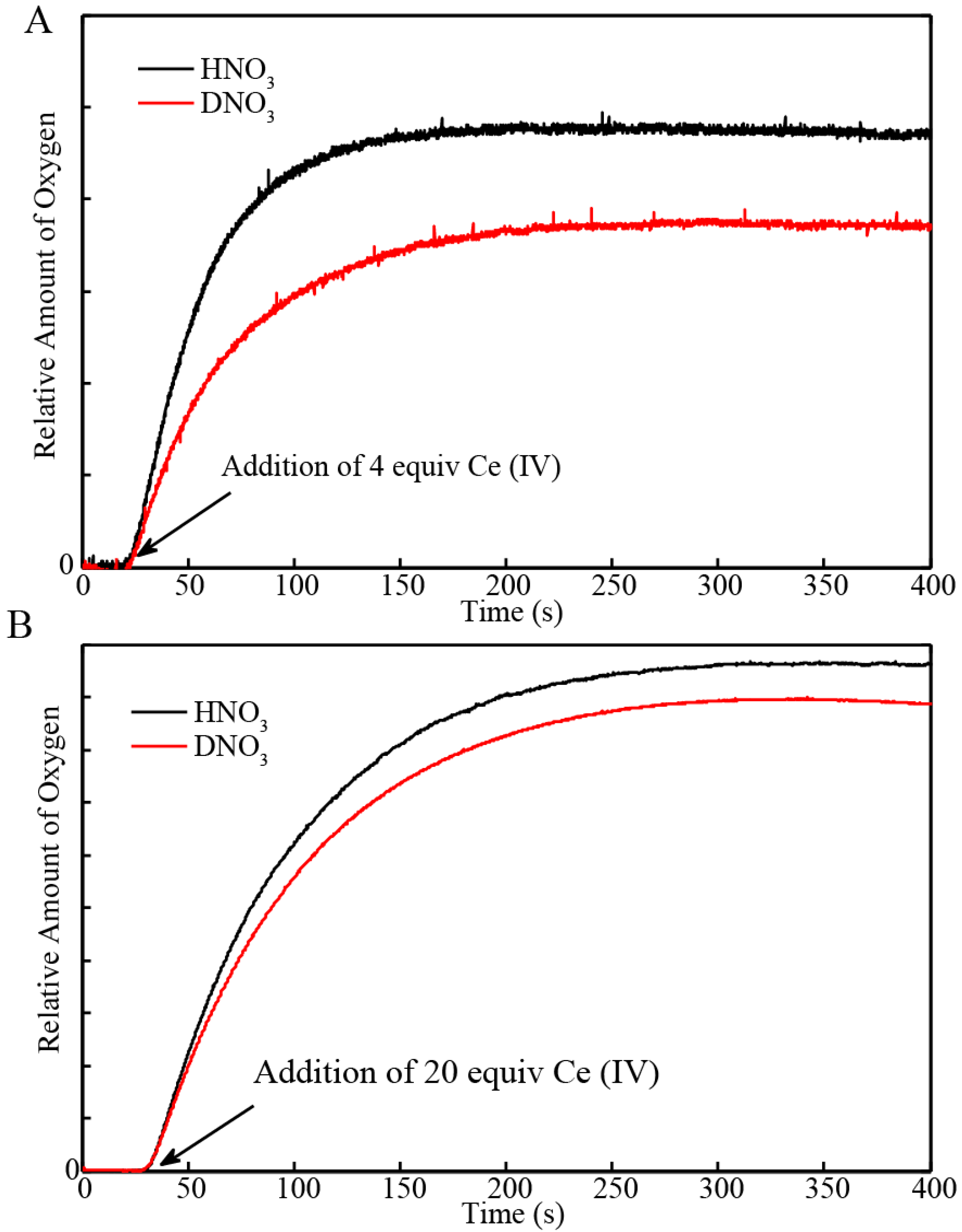
2.1. Determination of the KIE (kH2O/kD2O) for Oxygen Evolution with Blue Dimer Catalyst

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initial Rate of Oxygen Evolution (µmol/s) | Total Oxygen Evolved (µmol) | Turnover Frequency * |
|---|---|---|---|
| BD + 4 equiv H2O | 9.5 × 10−4 | 0.044 | 0.043/s |
| BD + 20 equiv H2O | 4.3 × 10−3 | 0.36 | |
| BD + 4 equiv D2O | 6.1 × 10−4 | 0.034 | 0.038/s |
| BD + 20 equiv D2O | 3.8 × 10−3 | 0.33 |
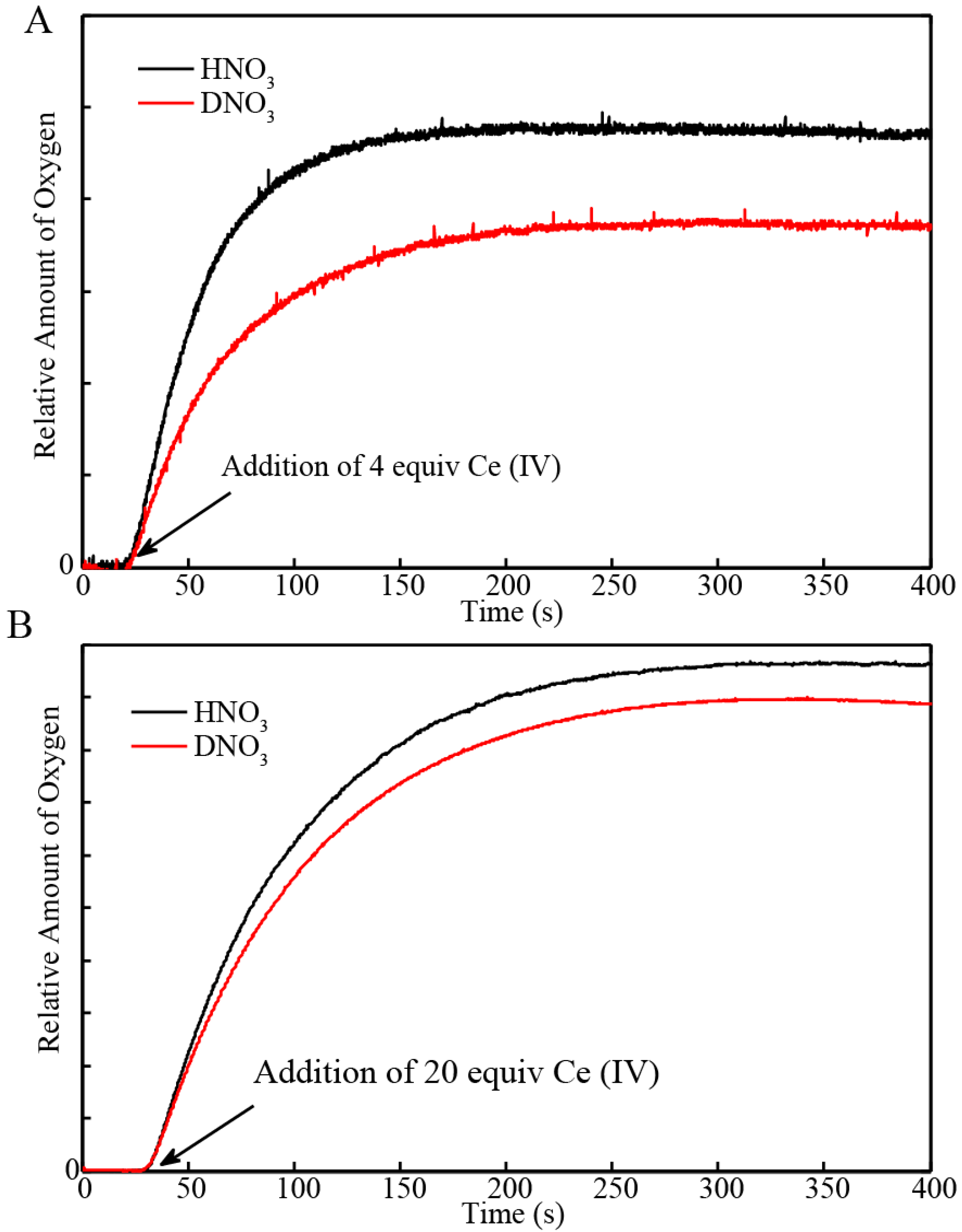
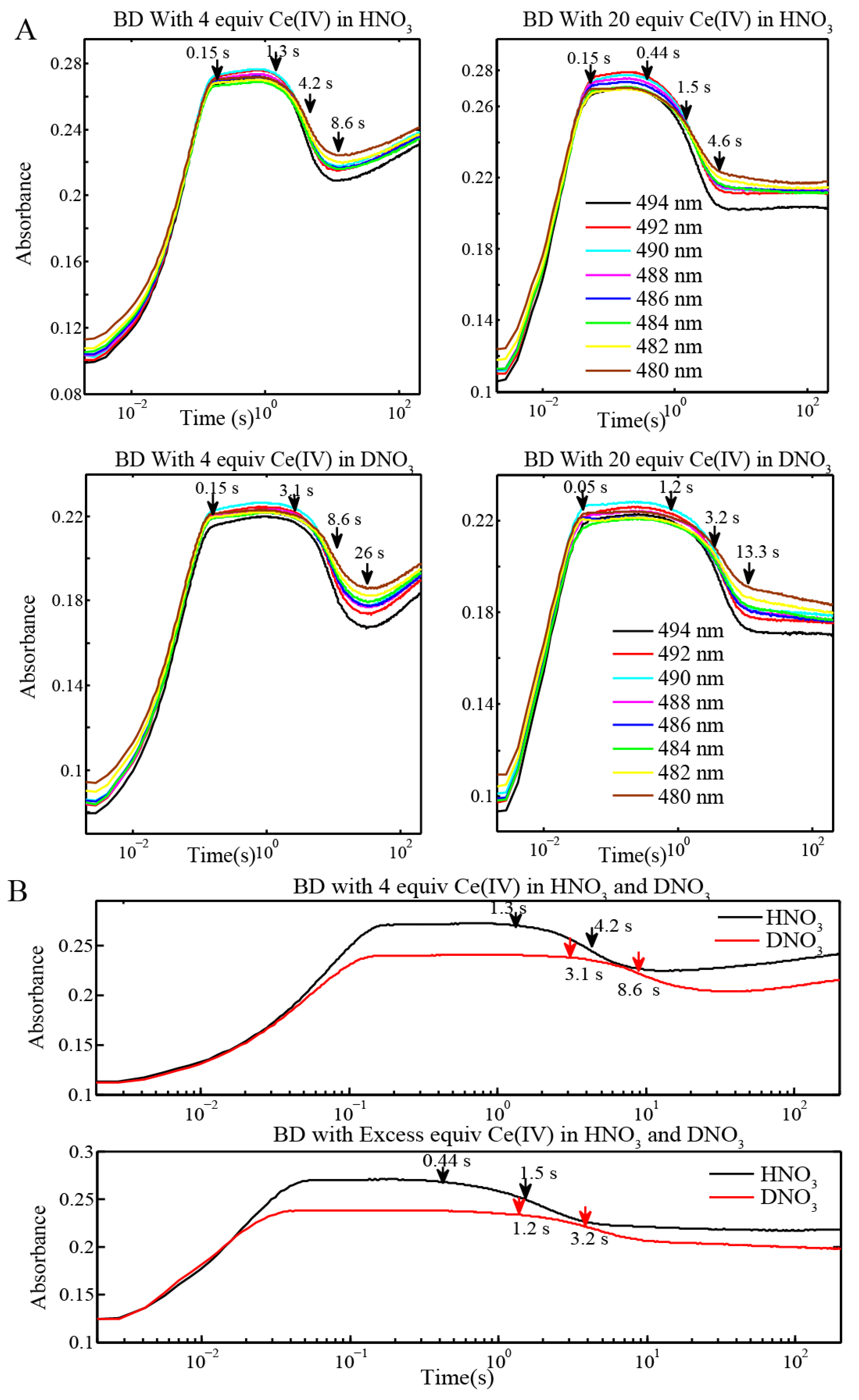
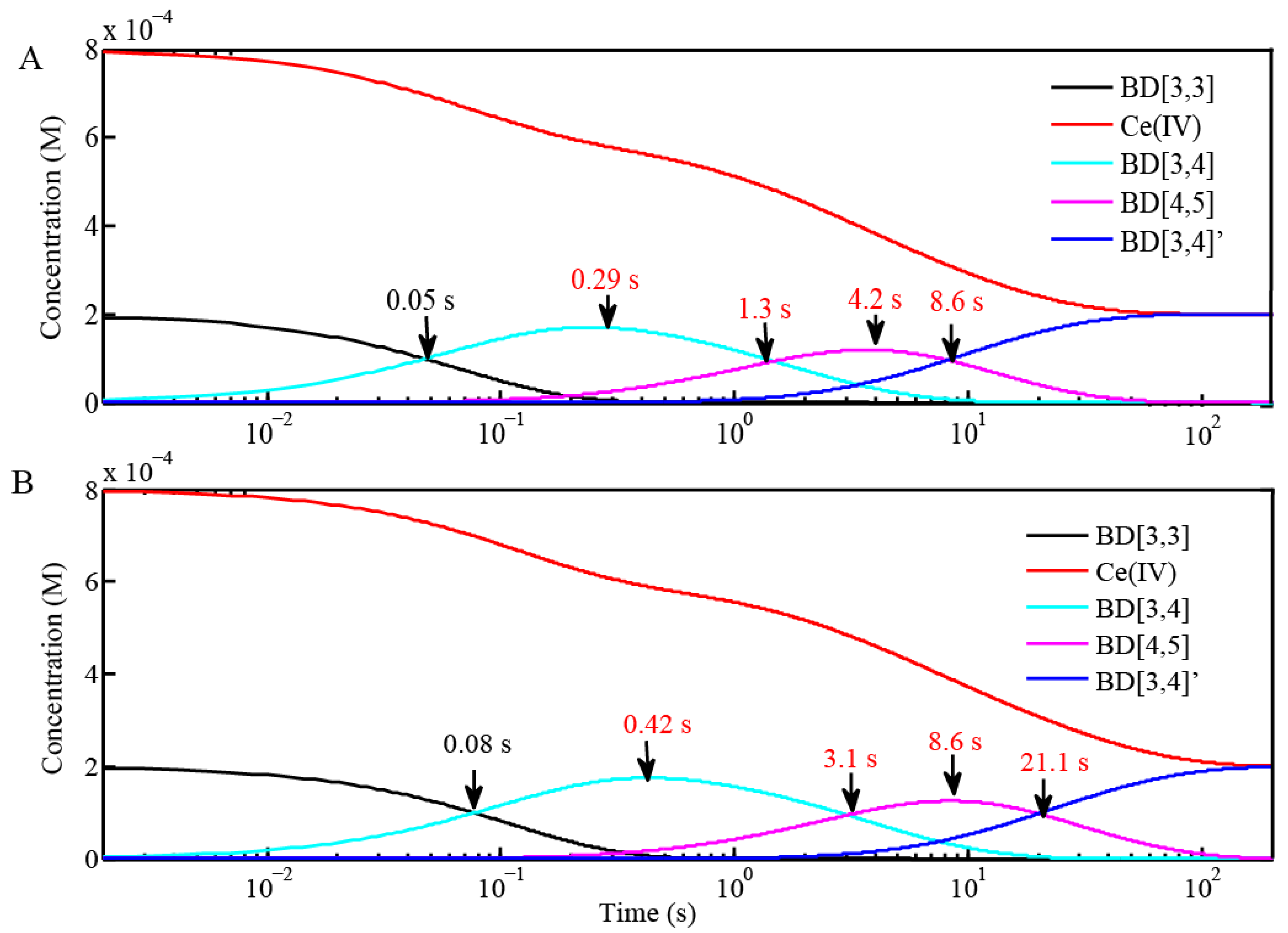
2.2. Stopped-Flow Kinetic Analysis of Blue Dimer Water Oxidation Cycle
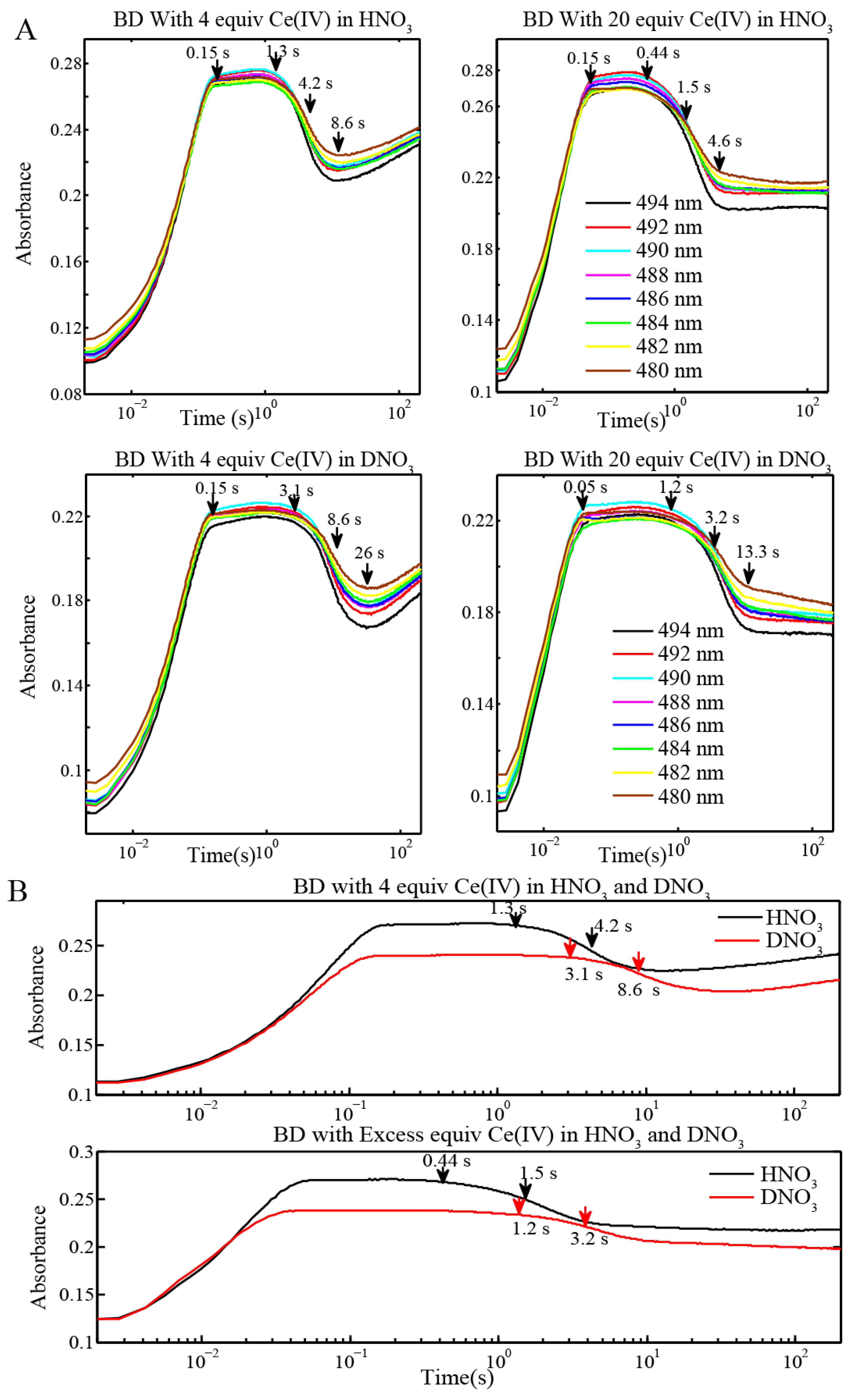
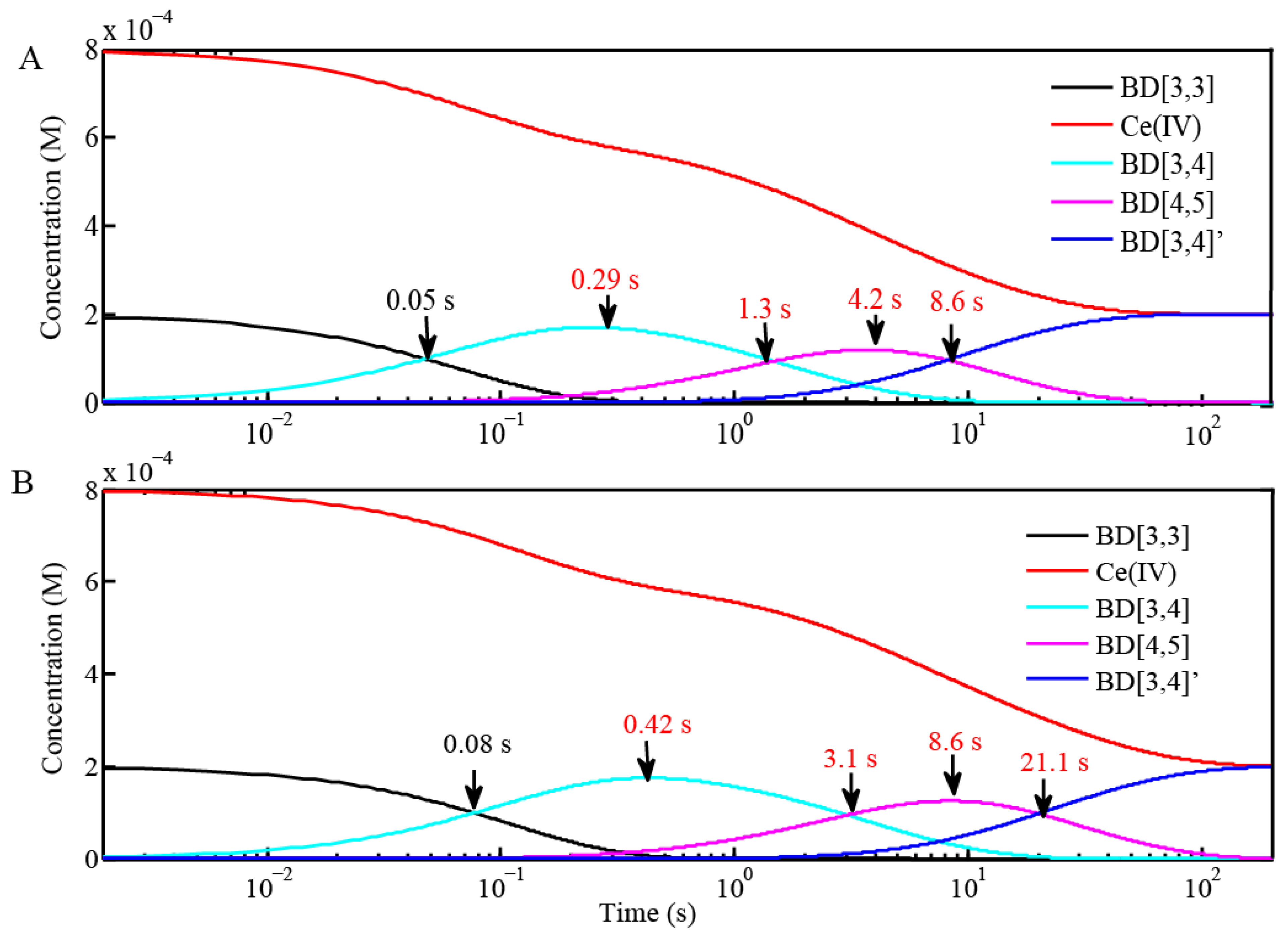
| Reaction (Fits from BD[3,3] + 4 equiv Ce(IV) in H2O and D2O) | Rate Constants in H2O (M−1 s−1) | Rate Constants in D2O (M−1 s−1) |
|---|---|---|
| BD[3,3] + Ce(IV) = BD[3,4] | 2.0 × 104 ± 280 | 1.2 × 104 ± 200 |
| BD[3,4] + Ce(IV) = BD[4,5] | 1000 ± 42 | 470 ± 20 |
| BD[4,5] + Ce(IV) = BD[3,4]′ | 300 ± 10 | 120 ± 12 |
| Reactions | Inflection point for reaction of BD[4,5] in H2O | Inflection point for reaction of BD[4,5] in D2O |
|---|---|---|
| BD + 4 equiv Ce(IV) (0.1 mM for Oxygen Evolution) (Figure 3 and Figure 4) | 4.2 s | 8.6 s |
| BD + 20 equiv Ce(IV) (0.1 mM for Oxygen Evolution) (Figure 3) | 1.5 s | 3.2 s |
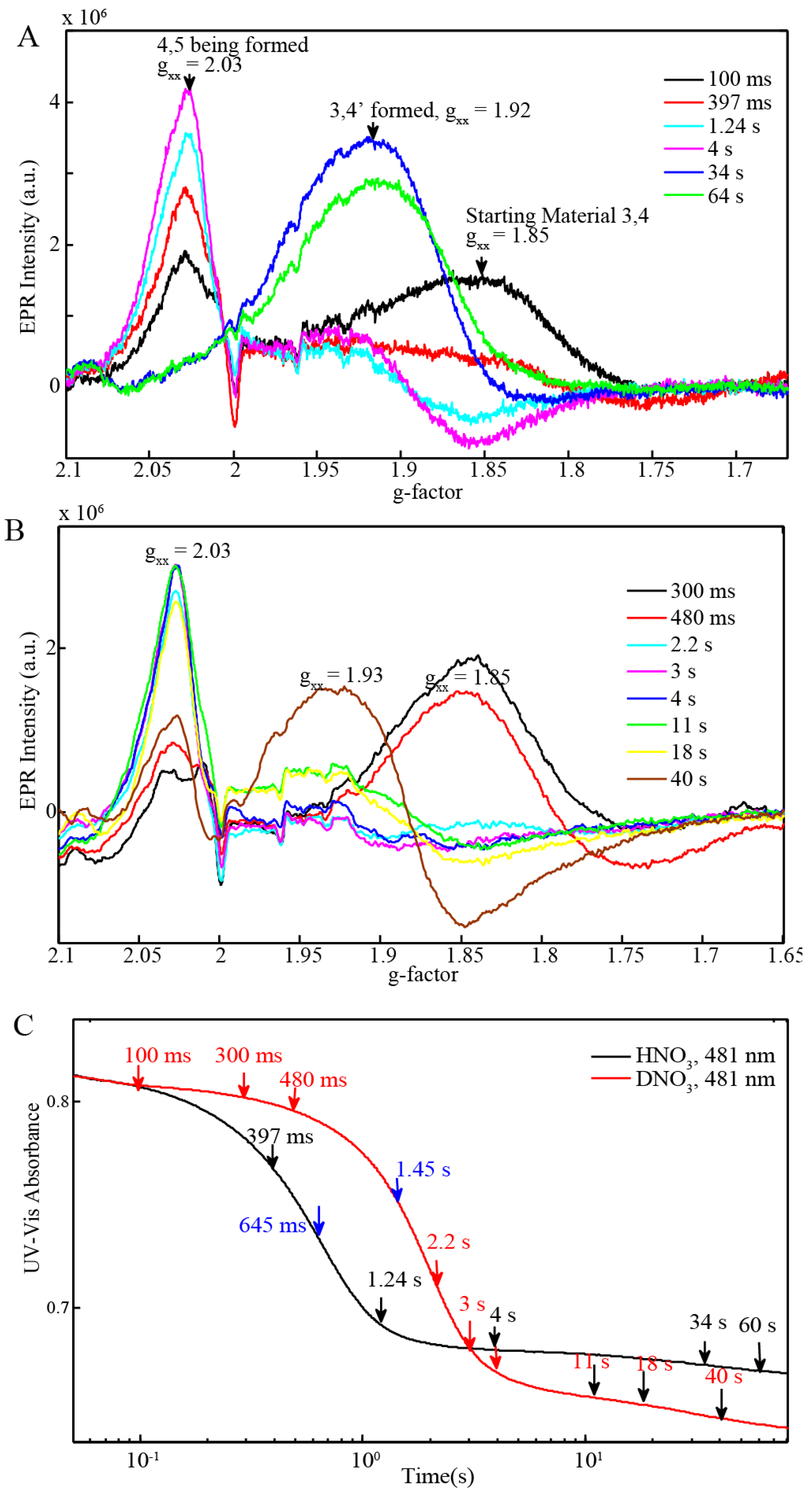
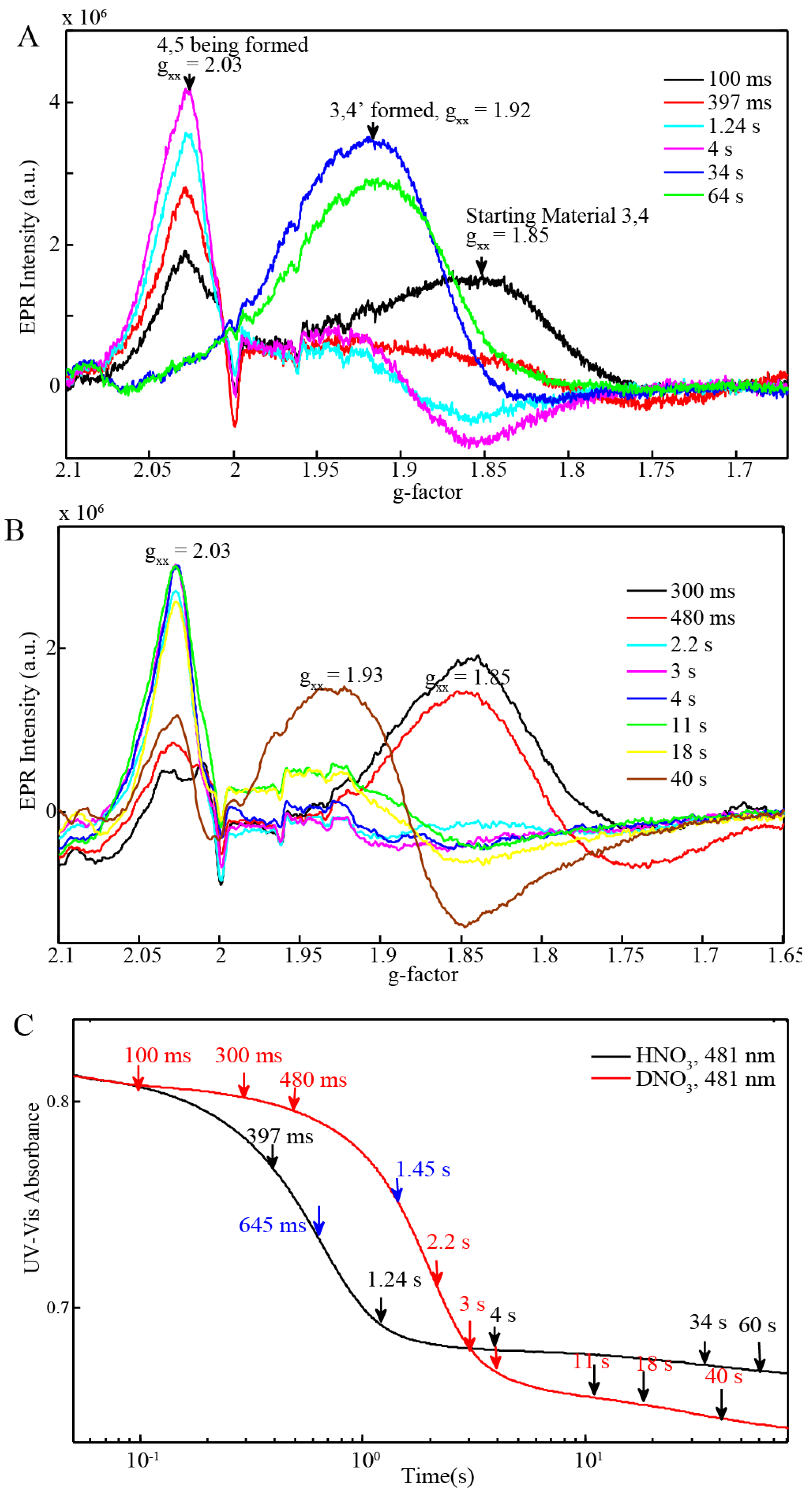
| BD + 20 equiv Ce(IV) (0.25 mM for EPR and resonance Raman) (Figure 6C) | 645 ms | 1.45 s |
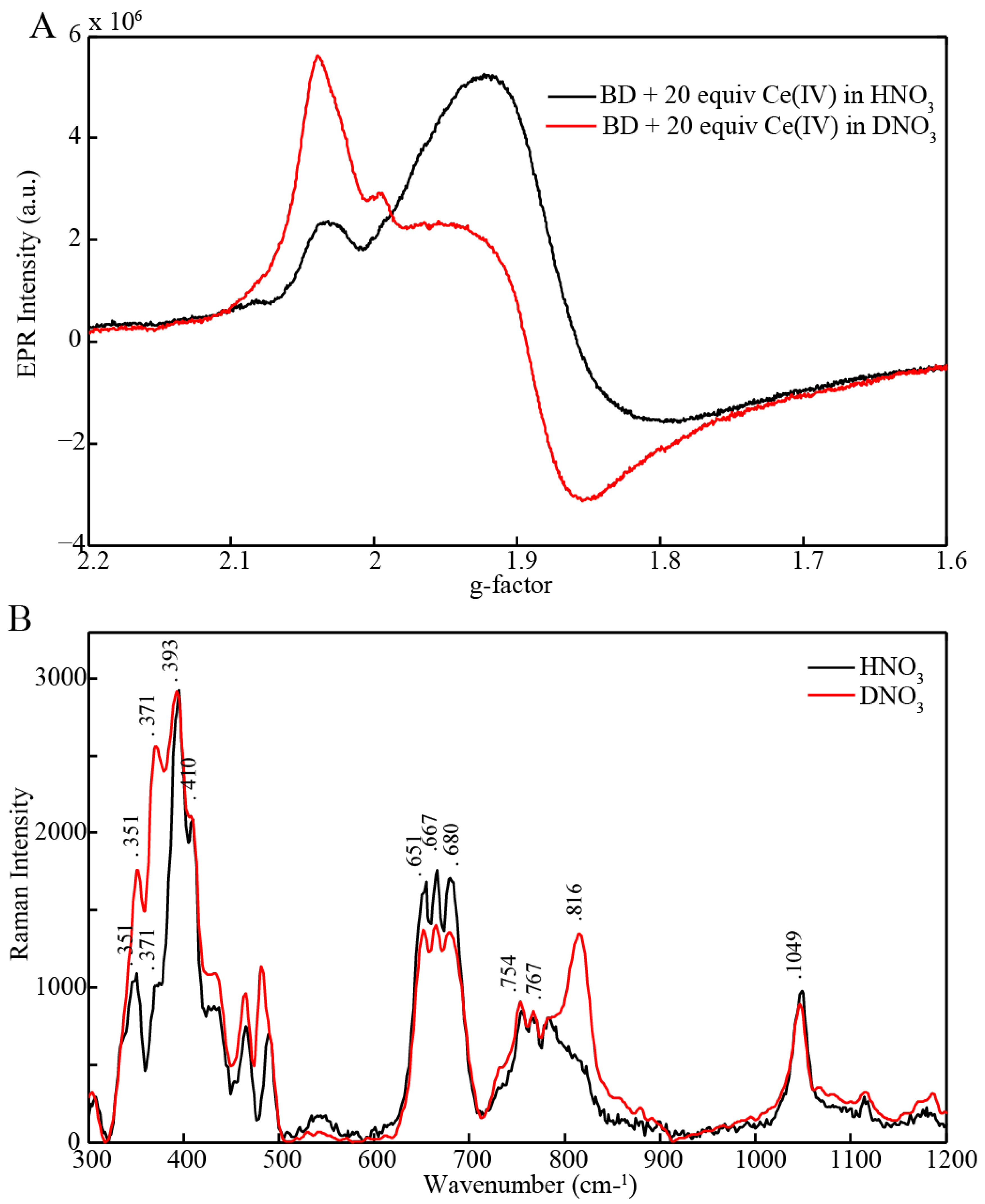
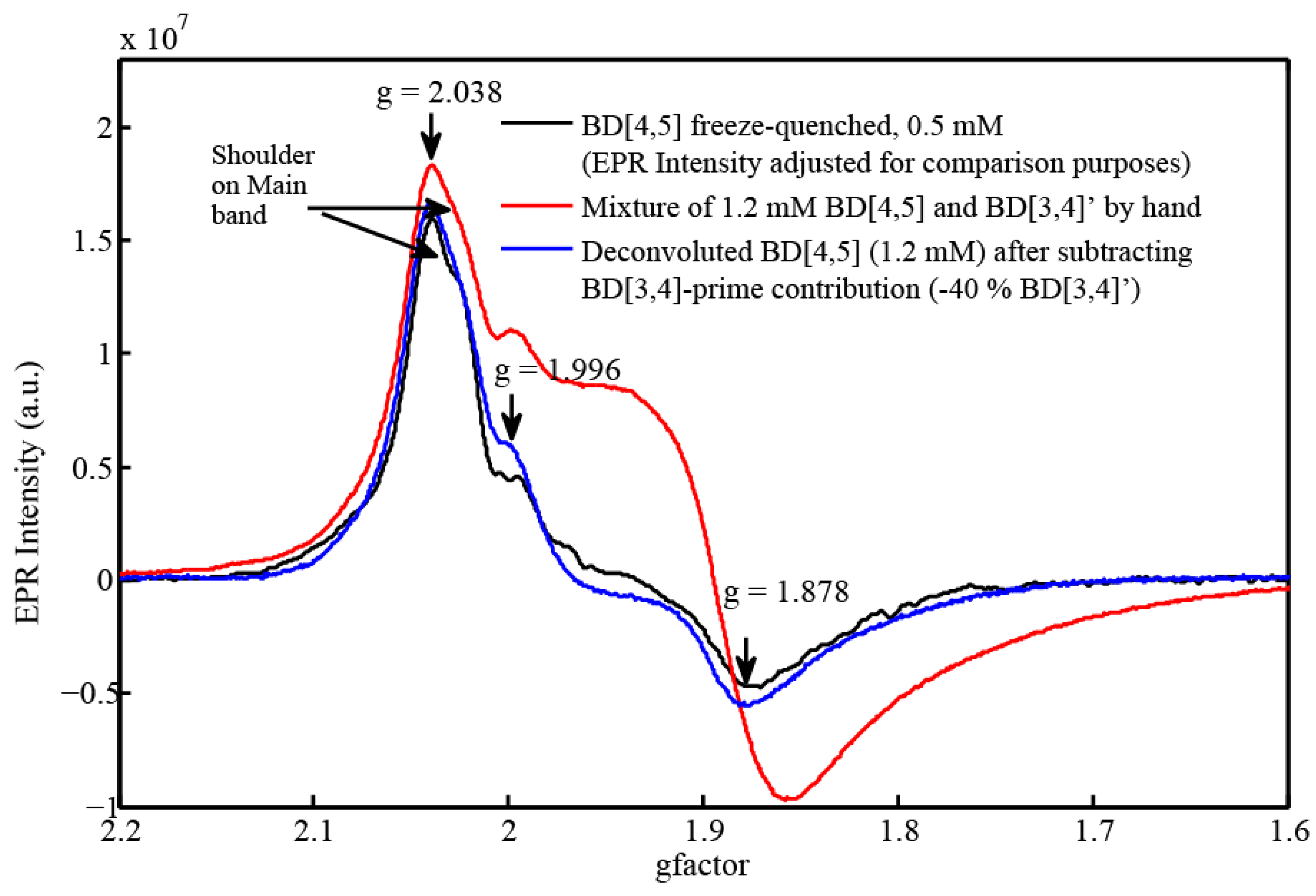
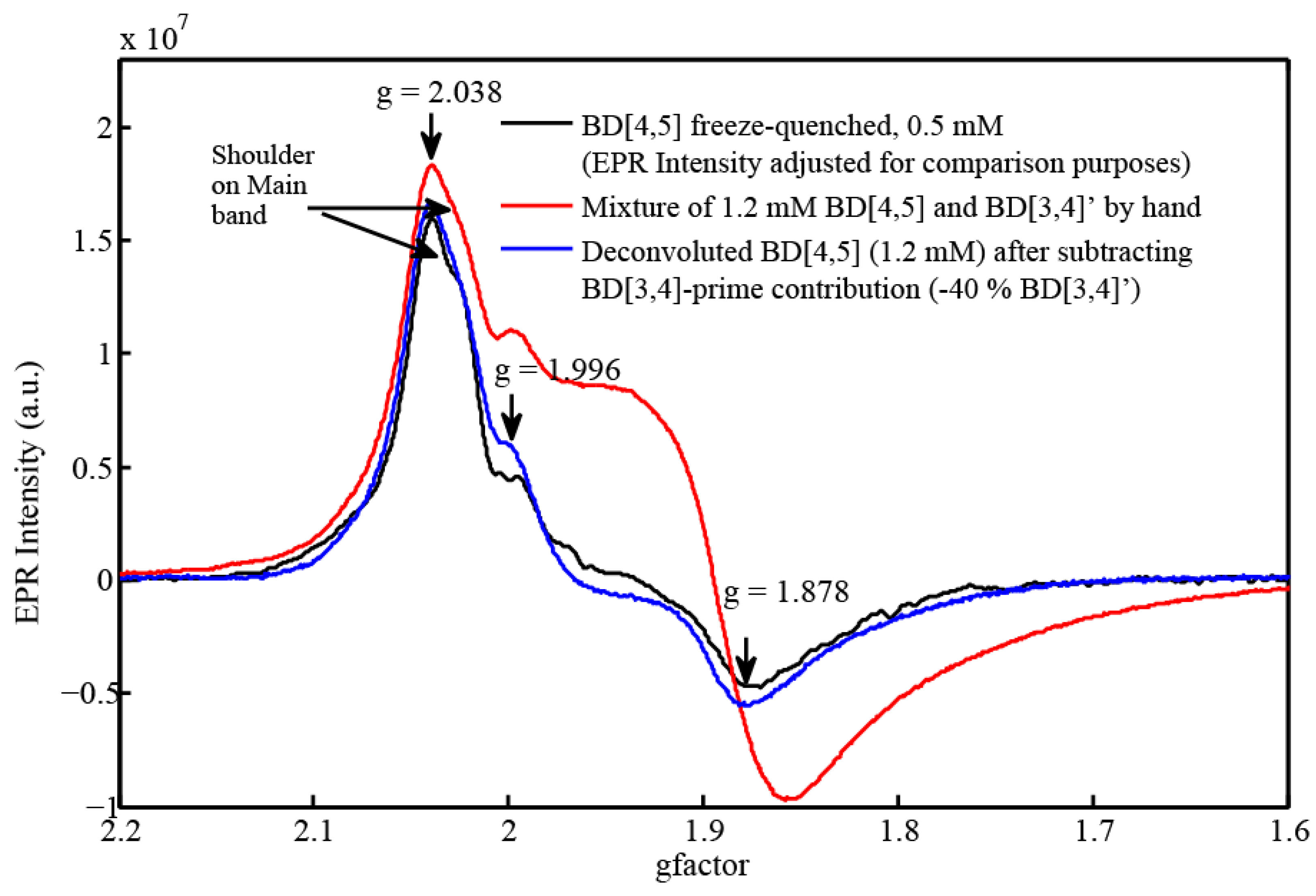
2.3. EPR and Resonance Raman Demonstration of Extended Lifetime of BD[4,5] in D2O
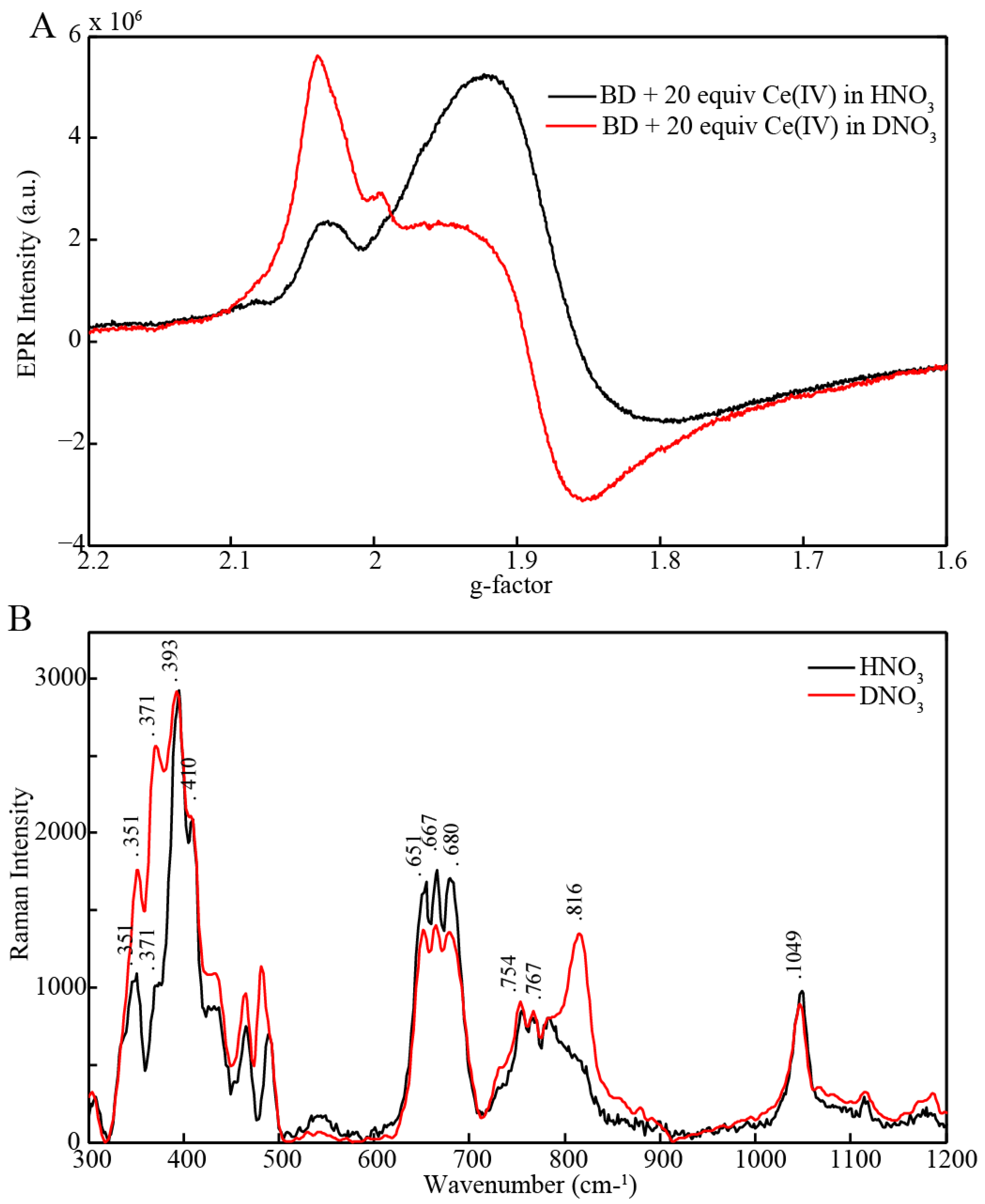
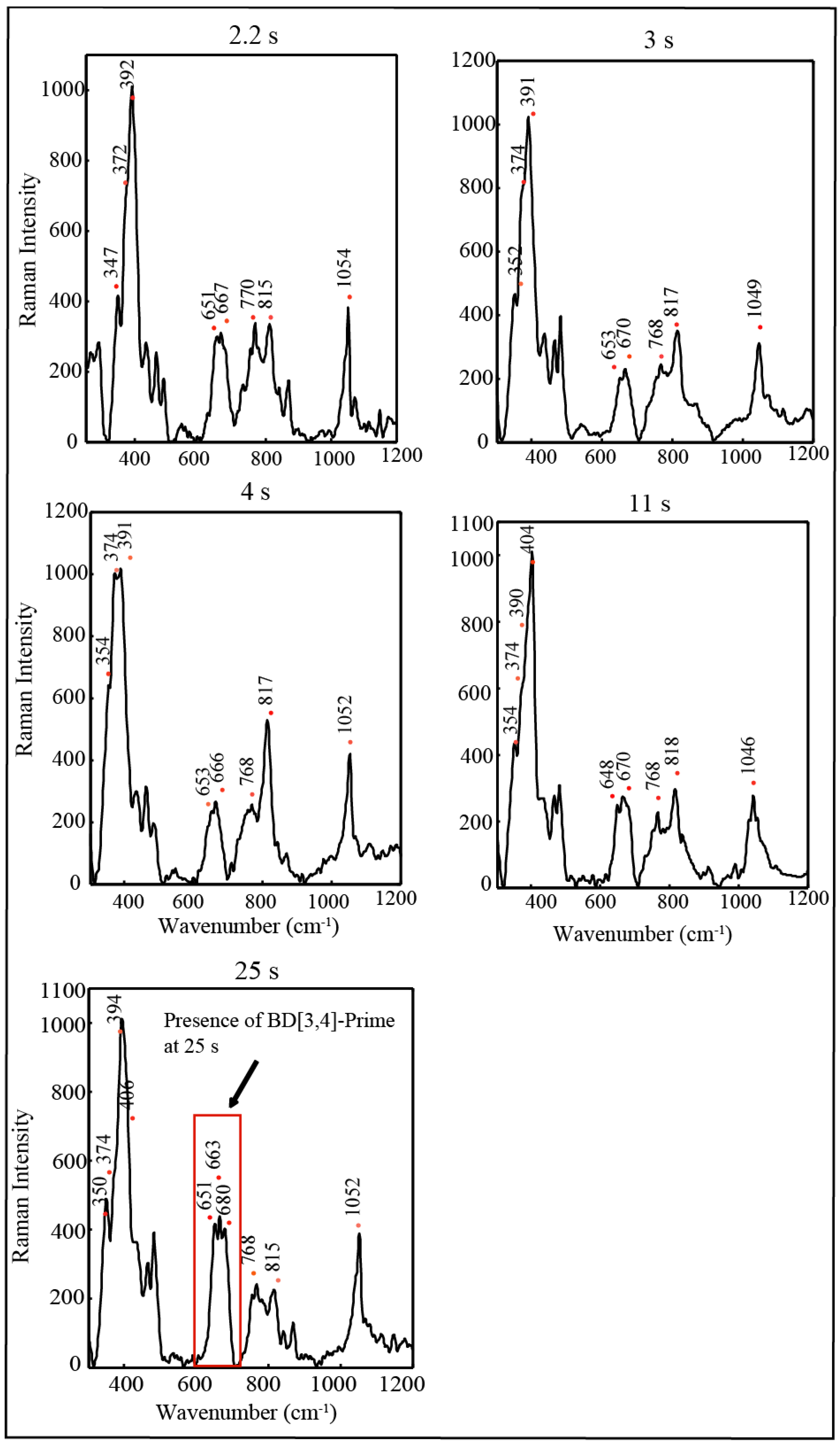
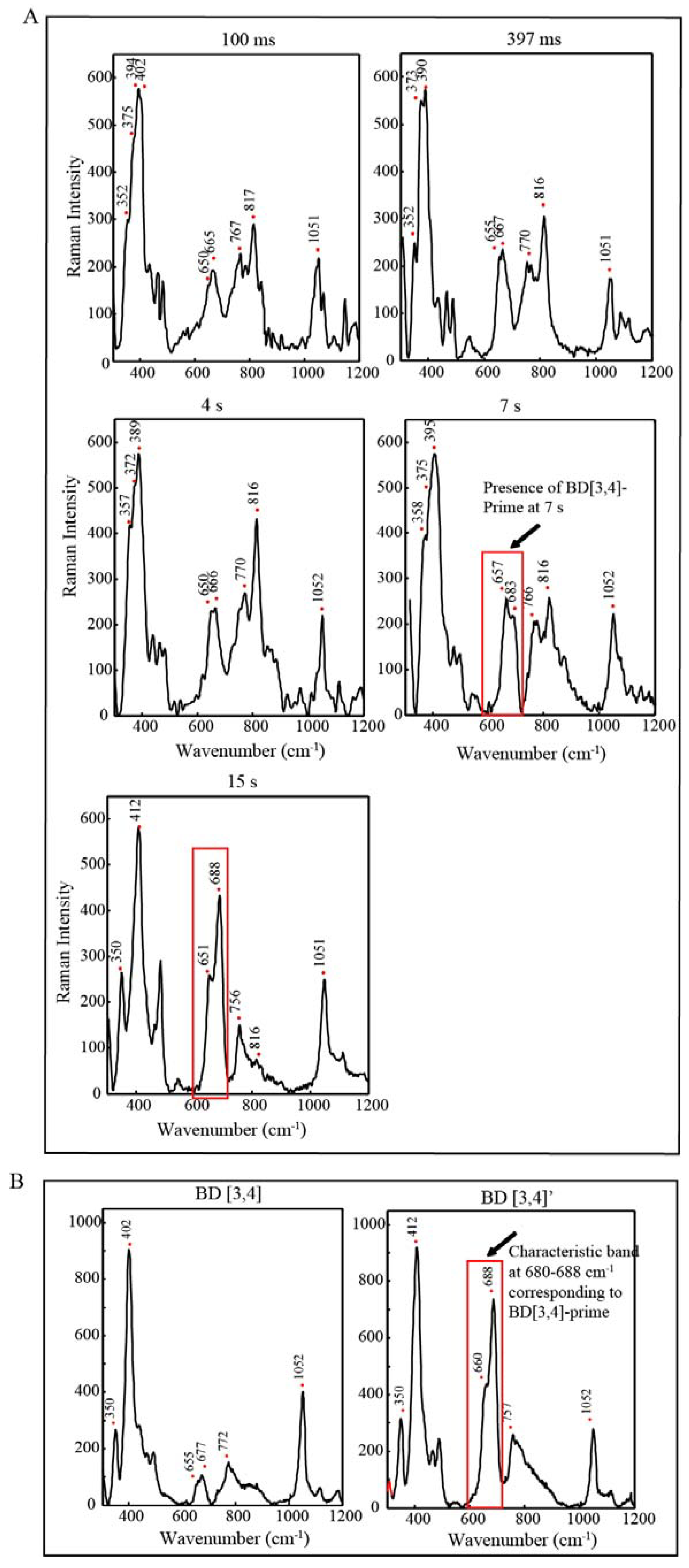
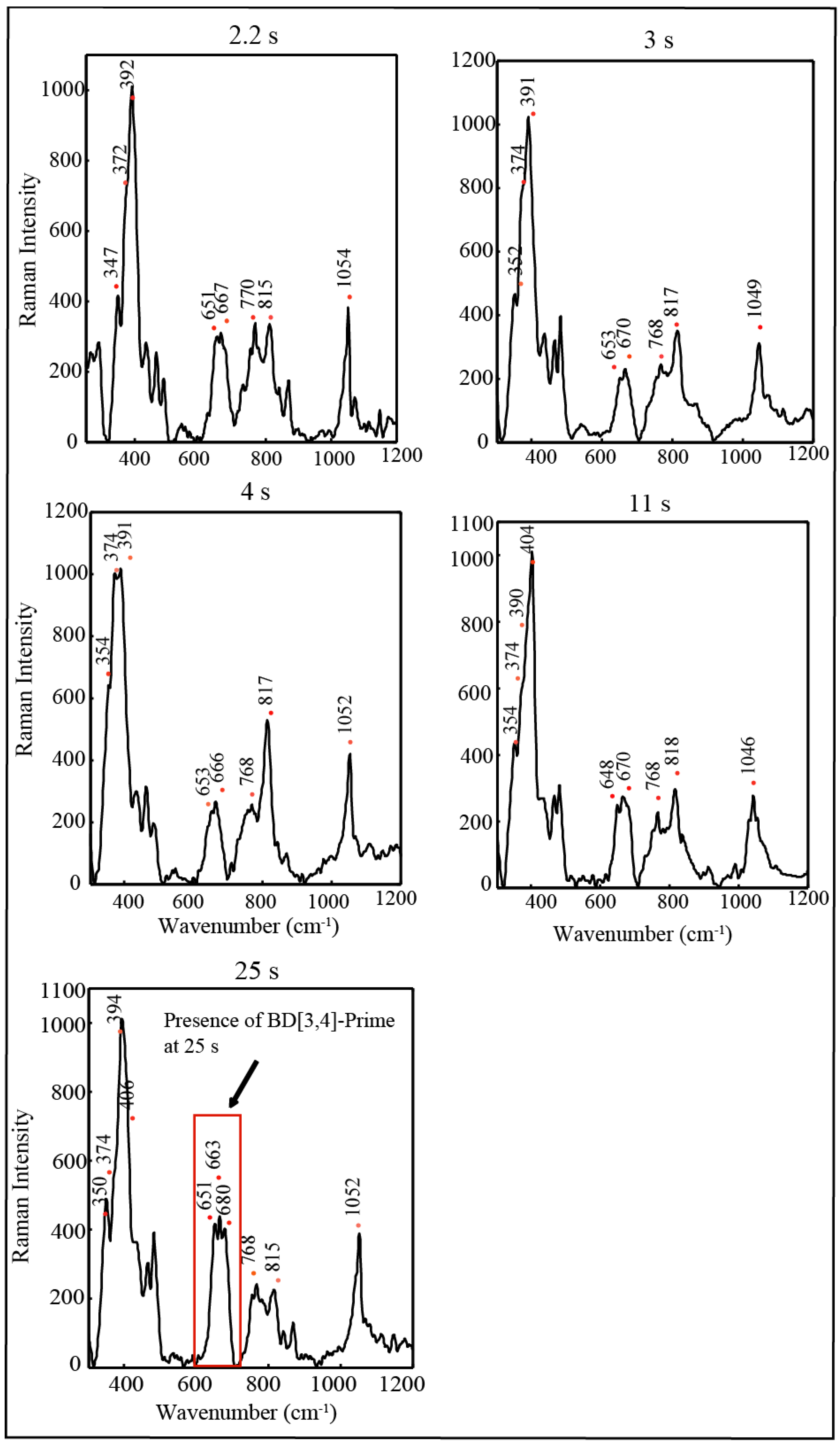
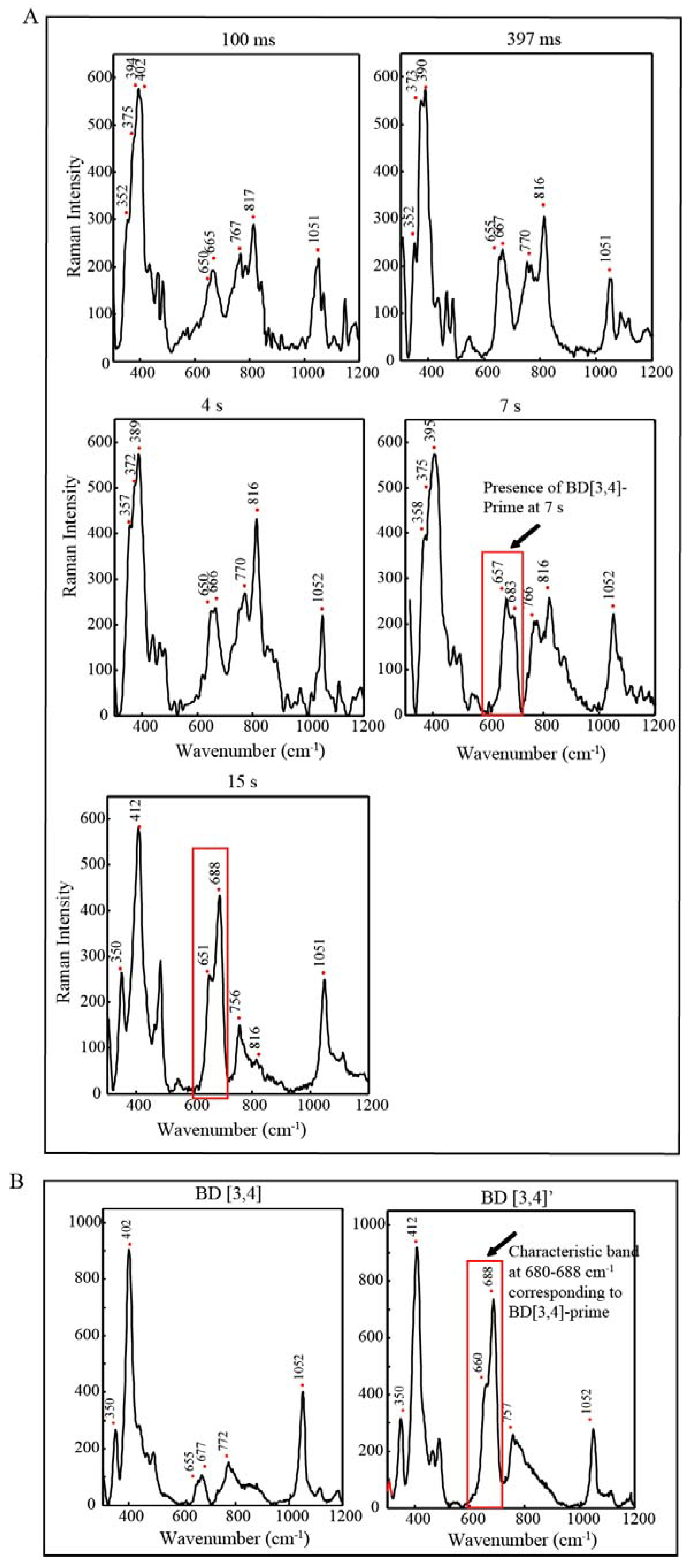
2.4. Time Resolved EPR Spectroscopy and Resonance Raman
3. Experimental Section
3.1. Sample Preparations
3.2. Oxygen (O2) Evolution Measurements
3.3. UV-Vis, Stopped Flow, Freeze Quench
3.4. EPR Measurements
3.5. Resonance Raman Measurements
4. Conclusions
Acknowledgments
Appendix
References
- Wydrzynski, T.; Satoh, S. Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase; Springer: Dordrecht, the Netherlands, 2005. [Google Scholar]
- Esper, B.; Badura, A.; Rogner, M. Photosynthesis as a power supply for (bio) hydrogen production. Trends Plant Sci. 2006, 11, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Barber, J. Photosystem II: An enzyme of global significance. Biochem. Soc. Trans. 2006, 34, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Liu, F.; Concepcion, J.J.; Jurss, J.W.; Cardolaccia, T.; Templeton, J.L.; Meyer, T.J. Mechanisms of water oxidation from the blue dimer to photosystem II. Inorg. Chem. 2008, 47, 1727–1752. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Rodriguez, M.; Sens, C.; Mola, J.; Kollipara, M.R.; Francas, L.; Mas-Marza, E.; Escriche, L.; Llobet, A. Ru complexes that can catalytically oxidize water to molecular dioxygen. Inorg. Chem. 2008, 47, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Zong, R.; Thummel, R.P. A new family of ru complexes for water oxidation. J. Am. Chem Soc. 2005, 127, 12802–12803. [Google Scholar] [CrossRef] [PubMed]
- Cape, J.L.; Lymar, S.V.; Lightbody, T.; Hurst, J.K. Characterization of intermediary redox states of the water oxidation catalyst, [Ru(bpy)2(OH2)]2O4+. Inorg. Chem. 2009, 48, 4400–4410. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Hurst, J.K. Resonance Raman, optical spectroscopic, and EPR characterization of the higher oxidation states of the water oxidation catalyst, cis,cis-[(bpy)2Ru(OH2)]2O4+. J. Am. Chem. Soc. 2000, 122, 5303–5311. [Google Scholar] [CrossRef]
- Moonshiram, D.; Jurss, J.W.; Concepcion, J.J.; Zakharova, T.; Alperovich, I.; Meyer, T.J.; Pushkar, Y. Structure and electronic configurations of the intermediates of water oxidation in blue ruthenium dimer catalysis. J. Am. Chem. Soc. 2012, 134, 4625–4636. [Google Scholar] [CrossRef] [PubMed]
- Moonshiram, D.; Alperovich, I.; Concepcion, J.; Meyer, T.; Pushkar, Y. Experimental demonstration of radicaloid character of RuV=O moiety in intermediate of catalytic water oxidation. Proc. Natl. Acad. Sci. USA 2013, in press. [Google Scholar]
- Jurss, J.W.; Concepcion, J.C.; Norris, M.R.; Templeton, J.L.; Meyer, T.J. Surface catalysis of oxidation by the blue ruthenium dimer. Inorg. Chem. 2010, 49, 3980–3982. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.K.; Zhou, J.; Lei, Y. Pathways for water oxidation catalyzed by the [(bpy)2Ru(OH2)]2O4+ ion. Inorg. Chem. 1992, 31, 1010–1017. [Google Scholar] [CrossRef]
- Binstead, R.A.; Chronister, C.W.; Ni, J.F.; Hartshorn, C.M.; Meyer, T.J. Mechanism of water oxidation by the µ-oxo dimer [(bpy)2(H2O)(RuIIIORuIII)(OH2)(bpy)2]4+. J. Am. Chem. Soc. 2000, 122, 8464–8473. [Google Scholar] [CrossRef]
- Stull, J.A.; Britt, R.D.; McHale, J.L.; Knorr, F.J.; Lymar, S.V.; Hurst, J.K. Anormalous reactivity of ceric nitrate in ruthenium “blue dimer”-catalyzed water oxidation. J. Am. Chem. Soc. 2012, 134, 19973–19976. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.F.; Concepcion, J.J.; Hu, X.Q.; Yang, W.T.; Hoertz, P.G.; Meyer, T.J. Concerted O atom-proton transfer in the O–O bond forming step in water oxidation. Proc. Natl. Acad. Sci. USA 2010, 107, 7225–7229. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Koike, T.; Hurst, J.K. Water exchange rates in the diruthenium mu-oxo ion cis,cis-[(bpy)2Ru(OH2)]2O4+. J. Am. Che. Soc. 2001, 123, 12775–12780. [Google Scholar] [CrossRef]
- Yamada, H.; Siems, W.F.; Koike, T.; Hurst, J.K. Mechanisms of water oxidation catalyzed by the cis,cis-[(bpy)2Ru(OH2)]2O4+ ion. J. Am. Chem. Soc. 2004, 126, 9786–9795. [Google Scholar] [CrossRef] [PubMed]
- Dubrow, R.; Pizer, L.I. Transient kinetic and deuterium isotope effect studies on the catalytic mechanism of phosphoglycerate dehydrogenase. J. Biol. Chem. 1977, 252, 1539–1551. [Google Scholar] [PubMed]
- Gnanamani, M.K.; Jacobs, G.; Shafer, W.D.; Ribeiro, M.C.; Pendyala, V.R.R.; Ma, W.; Davis, B.H. Fischer Tropsch Synthesis: Deuterium isotopic study for the formation of oxygenates over CeO2 supported Pt-Co catalysts. Catal. Commun. 2012, 25, 12–17. [Google Scholar] [CrossRef]
- Nelson, S.D.; Trager, W.F. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab. Dispos. 2003, 31, 1481–1498. [Google Scholar] [CrossRef] [PubMed]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moonshiram, D.; Purohit, V.; Concepcion, J.J.; Meyer, T.J.; Pushkar, Y. Mechanism of Catalytic Water Oxidation by the Ruthenium Blue Dimer Catalyst: Comparative Study in D2O versus H2O. Materials 2013, 6, 392-409. https://doi.org/10.3390/ma6020392
Moonshiram D, Purohit V, Concepcion JJ, Meyer TJ, Pushkar Y. Mechanism of Catalytic Water Oxidation by the Ruthenium Blue Dimer Catalyst: Comparative Study in D2O versus H2O. Materials. 2013; 6(2):392-409. https://doi.org/10.3390/ma6020392
Chicago/Turabian StyleMoonshiram, Dooshaye, Vatsal Purohit, Javier J. Concepcion, Thomas J. Meyer, and Yulia Pushkar. 2013. "Mechanism of Catalytic Water Oxidation by the Ruthenium Blue Dimer Catalyst: Comparative Study in D2O versus H2O" Materials 6, no. 2: 392-409. https://doi.org/10.3390/ma6020392