The Complete Sequence of a Human Parainfluenzavirus 4 Genome

Abstract
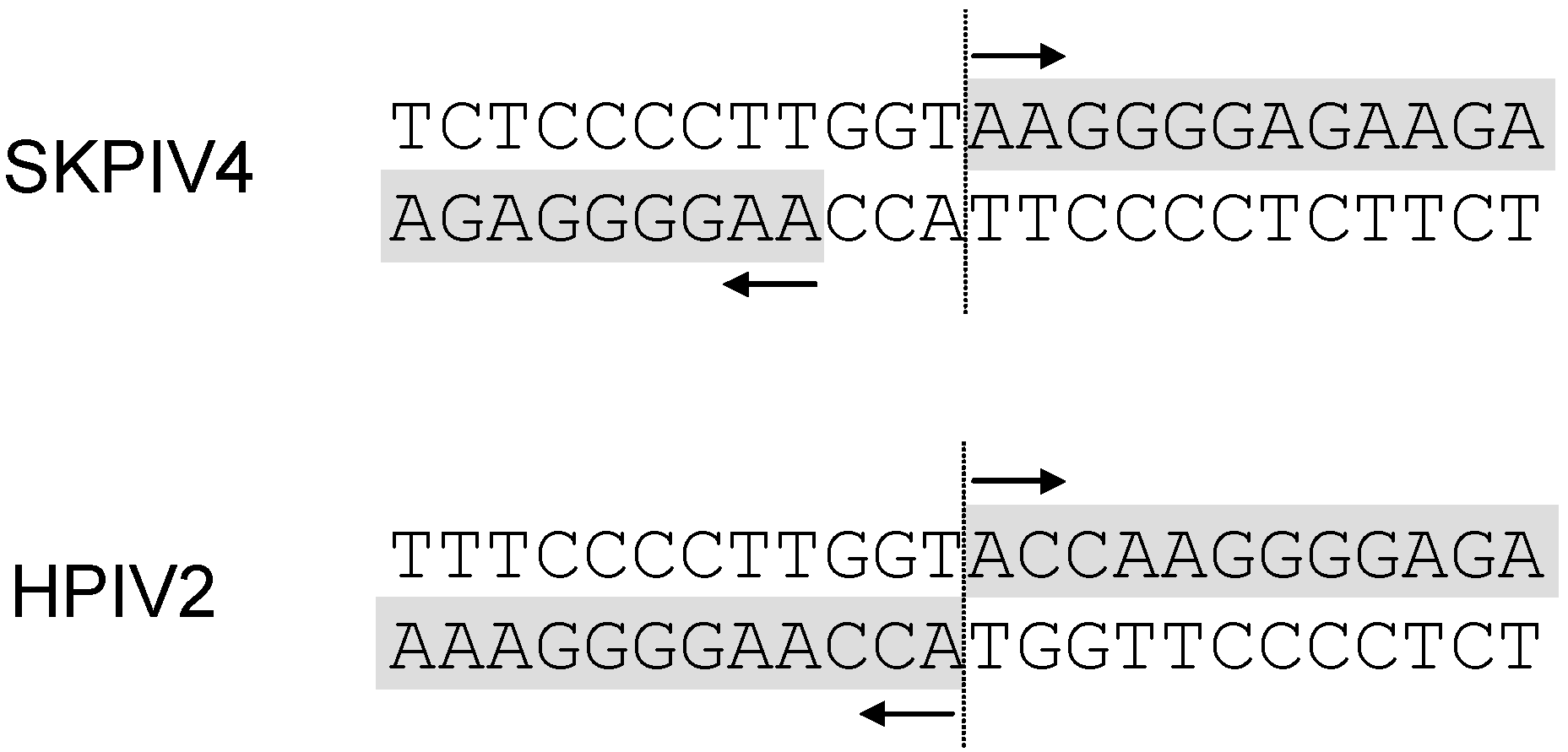
:1. Introduction
2. Results
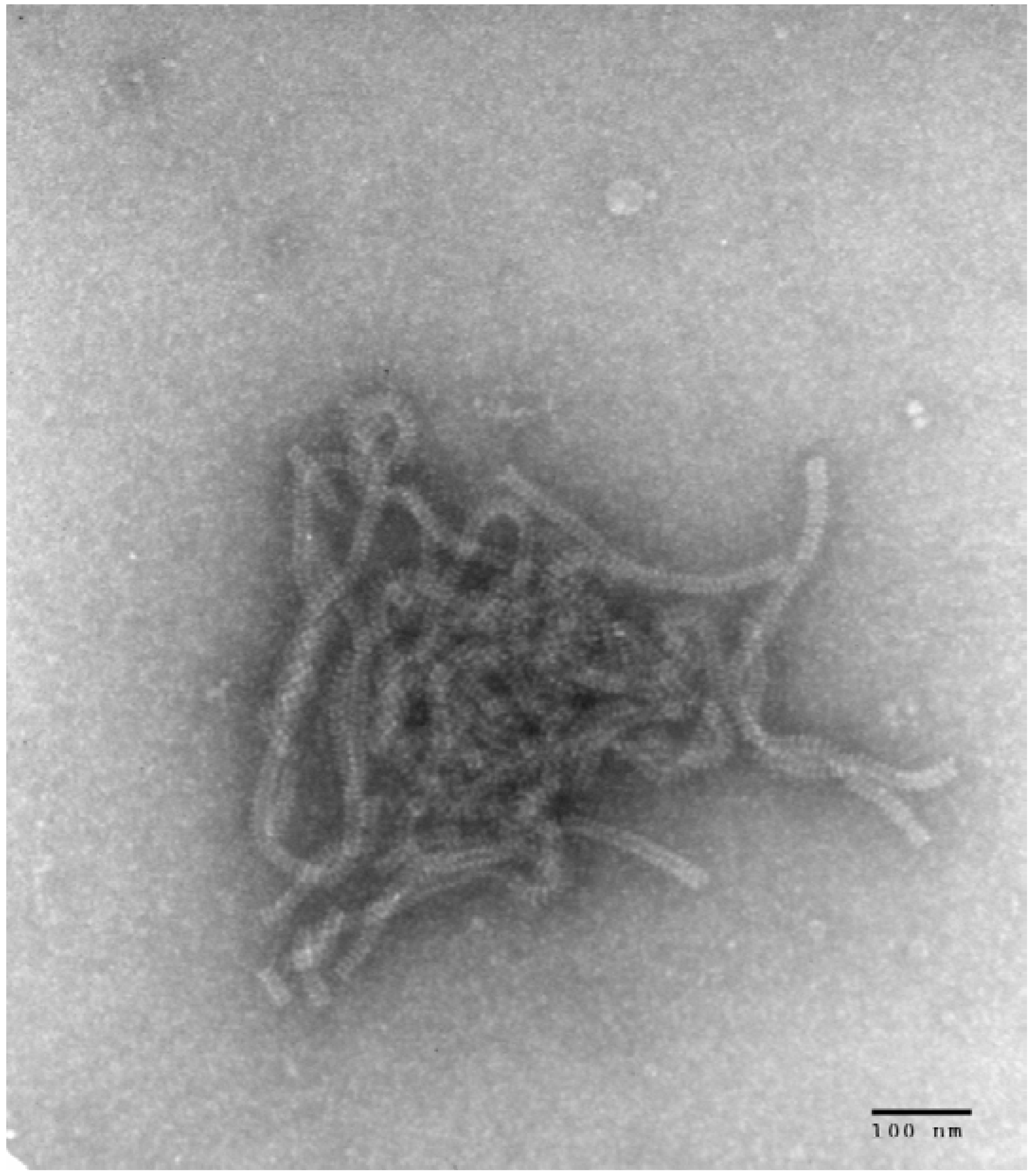
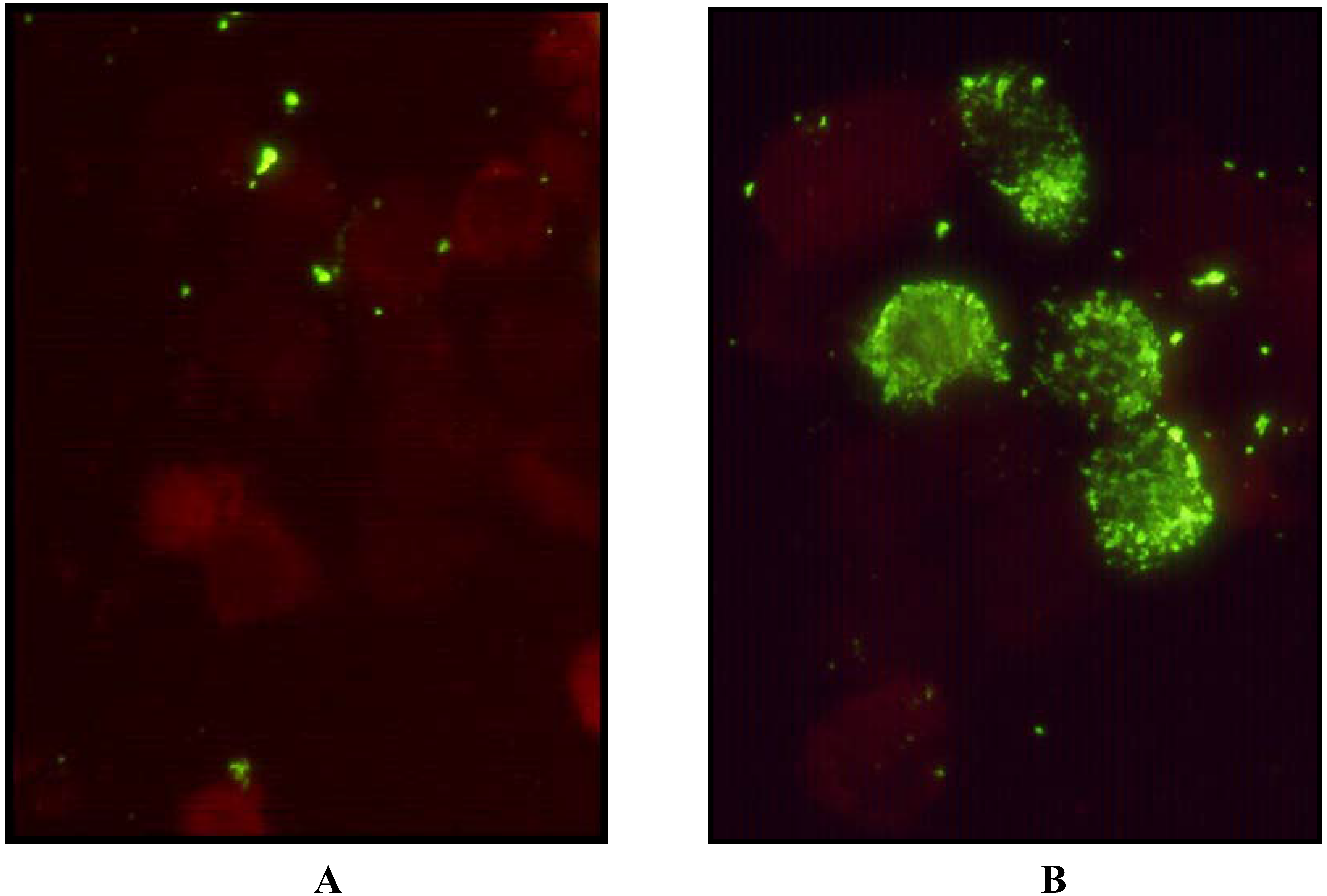
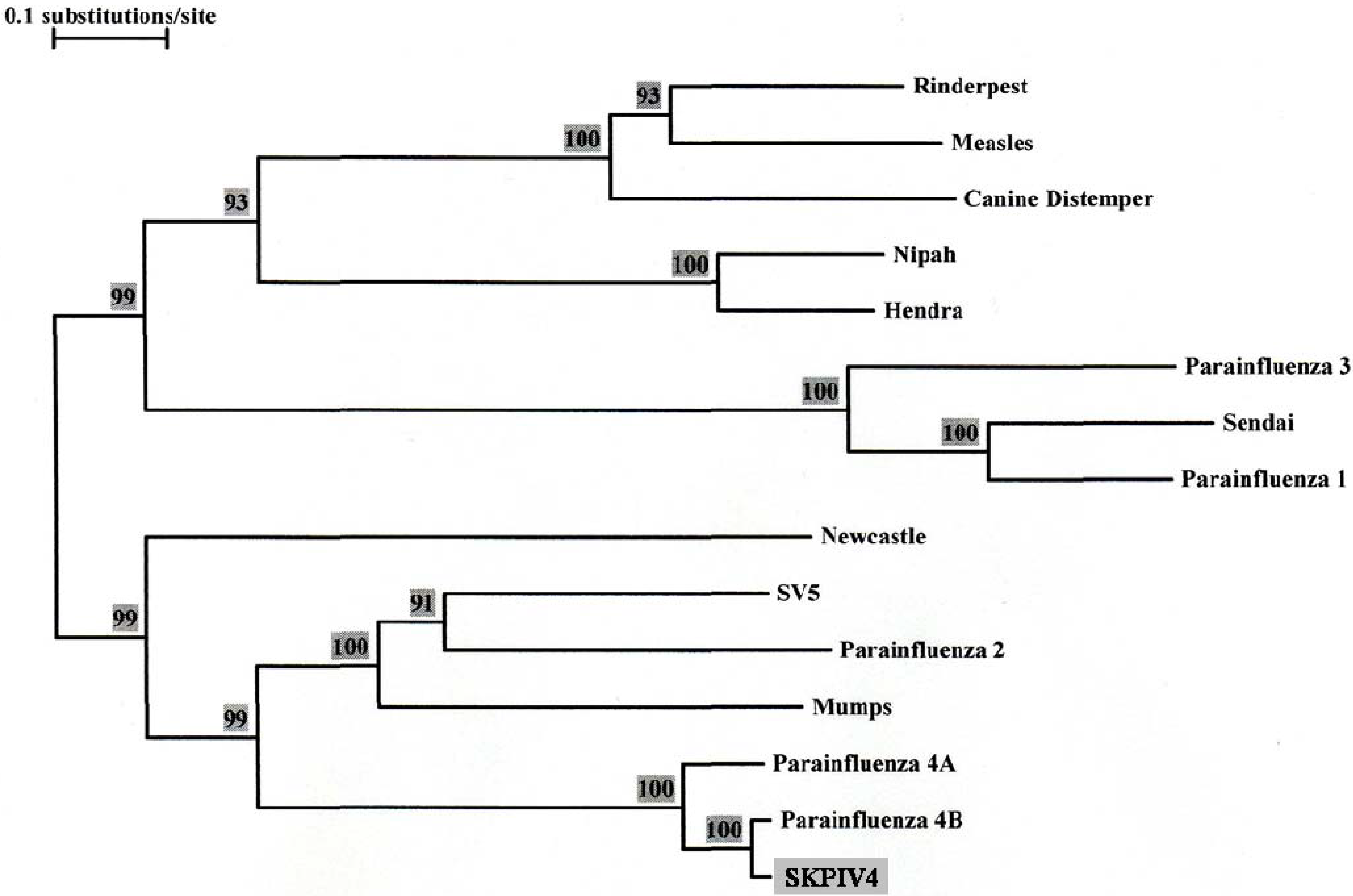
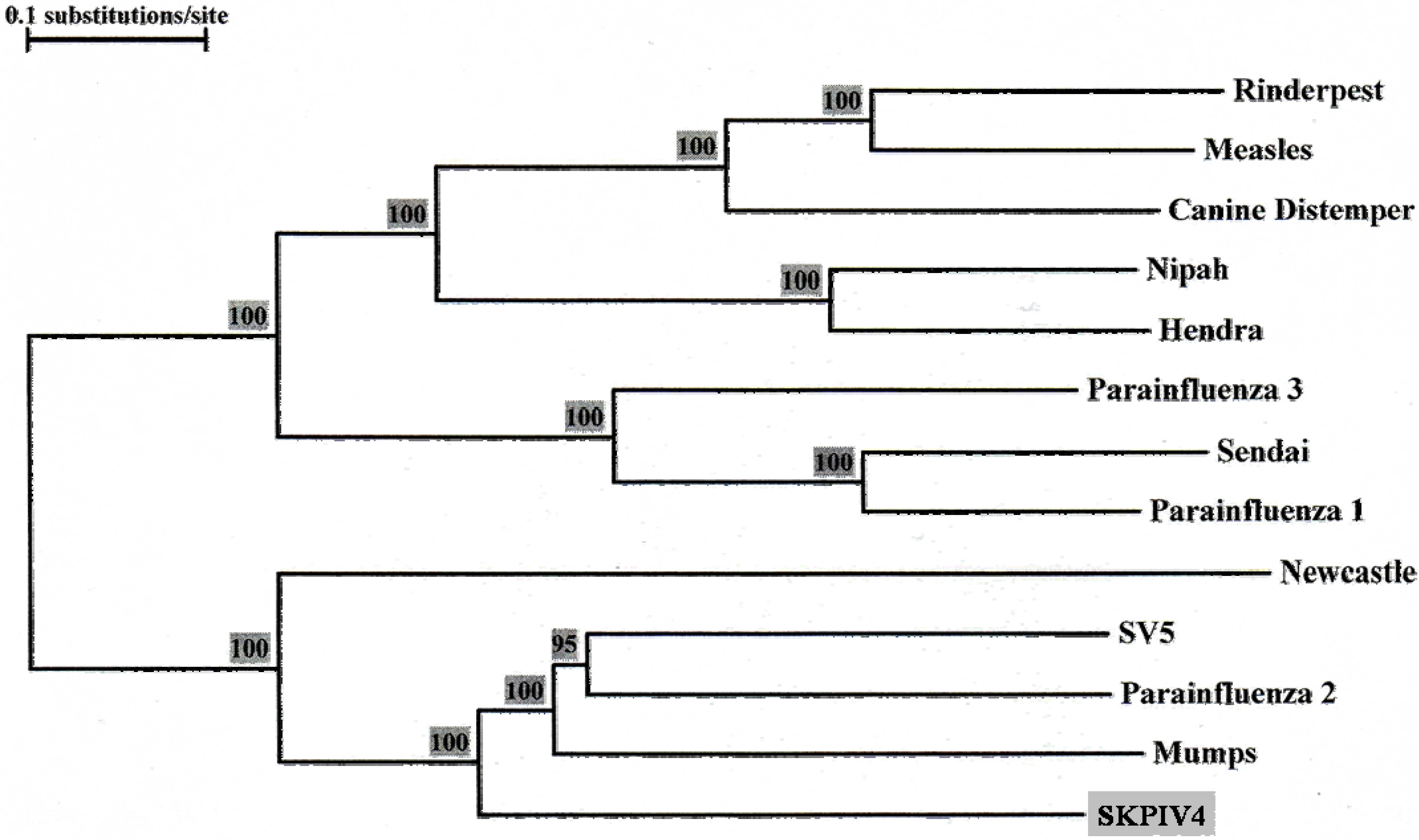
2.1. Identification of the respiratory virus isolate SKPIV4 as a HPIV4
2.3. The Nucleocapsid (N) gene
2.4. The Phosphoprotein/V-protein (P/V) gene
2.5. The Matrix (M) gene
2.6. The Fusion (F) gene
2.7. The Haemagglutinin-Neuraminidase (HN) gene
2.8.The Large (L) gene
2.9. The genomic termini
3. Material and methods
3.1. Source of the HPIV4 strain
3.2. Isolation and culture
3.3. Immunofluorescence microscopy
3.4. Electron microscopy
3.6. Primer design for long RT-PCR
3.7. Long RT-PCR
3.8. RNA ligase mediated amplification of genome ends
3.9. 5’ RACE
3.10. Sequencing of amplicons
3.11. Corroboration of the sequence
3.12. Sequence assembly and analysis
4. Discussion
Acknowledgments
References
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: the viruses and their replication. In Fields Virology 5th Ed. Knipe, D.M., Howley, P. M., Griffin, D. E., Lamb, R. A., Straus, S. E., Martin, M.A., Roizman, B., Eds.; 2007; Wolters Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA. [Google Scholar]
- Kondo, K.; Bando, H.; Kawano, M.; Tsurudome, M.; Komada, H.; Nishio, M.; Ito, Y. Sequencing analyses and comparison of parainfluenza virus type 4A and 4B NP protein genes. Virology 1990, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Bando, H.; Tsurudome, M.; Kawano, M.; Nishio, M.; Ito, Y. Sequence analysis of the phosphoprotein (P) genes of human parainfluenza type 4A and 4B viruses and RNA editing at transcript of the P genes: the number of G residues added is imprecise. Virology 1990, 178, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Fujii, M.; Nakamura, T.; Bando, H.; Kawano, M.; Tsurudome, M.; Komada, H.; Kusakawa, S.; Nishio, M.; Ito, Y. Sequence characterization of the matrix protein genes of parainfluenza virus types 4A and 4B. J. Gen. Virol. 1991, 72, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Komada, H.; Bando, H.; Ito, M.; Ohta, H.; Kawano, M.; Nishio, M.; Tsurudome, M.; Watanabe, N.; Ikemura, N.; Kusagawa, S.; Mao, X.; O'Brien, M.; Ito, Y. Sequence analyses of human parainfluenza virus type 4A and type 4B fusion proteins. J. Gen. Virol. 1995, 76, 3205–3210. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Kondo, K.; Kawano, M.; Komada, H.; Tsurudome, M.; Nishio, M.; Ito, Y. Molecular cloning and sequence analysis of human parainfluenza type 4A virus HN gene: its irregularities on structure and activities. Virology 1990, 175, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Chanock, R.M.; Murphy, B.R.; Collins, P.L. Parainfluenza viruses. In Fields Virology, 4th Ed. Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Eds.; 2001; Lippincott Williams &Wilkins: Philadelphia, USA. [Google Scholar]
- Svenda, M.; Berg, M.; Moreno-Lopez, J.; Linne, T. Analysis of the large (L) protein gene of the porcine rubulavirus LPMV: identification of possible functional domains. Virus Res. 1997, 48, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Gharabaghi, F.; Tellier, R.; Cheung, R.; Collins, C.; Broukhanski, G.; Drews, S.J.; Richardson, S.E. Comparison of a commercial qualitative real-time RT-PCR kit with direct immunofluorescence assay (DFA) and cell culture for detection of influenza A and B in children. J. Clin. Virol. 2008, 42, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Petric, M.; Szymanski, M. Electron Microscopy and Immunoelectron Microscopy. In Clinical Virology Manual , 3rd Ed. Spector, S., Hodinka, R.L., Young, S.A., Eds.; ASM Press: Washington, DC, USA, 2000; pp. 54–65. [Google Scholar]
- Tellier, R.; Bukh, J.; Emerson, S.U.; Purcell, R.H. Long PCR methodology. In PCR Protocols, 2nd Ed. Bartlett, J.M.S., Stirling, D., Eds.; 2003; Humana Press: Totowa, NJ, USA. [Google Scholar]
- Draker, R.; Roper, R.L.; Petric, M.; Tellier, R. The complete sequence of the bovine torovirus genome. Virus Res. 2006, 115, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [PubMed]
- Van de Peer, Y.; De Wachter, R. TREECON for Windows: a software package for the construction drawing of evolutionary trees for the Microsoft Windows environment. Comput. Appl. Biosci. 1994, 10, 569–570. [Google Scholar] [PubMed]
- Forns, X.; Bukh, J.; Purcell, R.H.; Emerson, S.U. How Escherichia coli can bias the results of molecular cloning: preferential selection of defective genomes of hepatitis C virus during the cloning procedure. Proc. Natl. Acad. Sci. USA 1997, 94, 13909–13914. [Google Scholar] [CrossRef]
- Tellier, R.; Bukh, J.; Emerson, S.U.; Purcell, R.H. Long PCR amplification of large fragments of viral genomes: a technical overview. In PCR Protocols 2ndEd. Bartlett, J.M.S., Stirling, D., Eds.; 2003; Humana Press: Totowa, NJ, USA. [Google Scholar]
- Sakaguchi, T.; Toyoda, T.; Gotoh, B.; Inocencio, N.M.; Kuma, K.; Miyata, T.; Nagai, Y. Newcastle disease virus evolution. I. Multiple lineages defined by sequence variability of the hemagglutinin-neuraminidase gene. Virology 1989, 169, 260–272. [Google Scholar]
- Sidhu, M.S.; Menonna, J.P.; Cook, S.D.; Dowling, P.C.; Udem, S.A. Canine distemper virus L gene: sequence and comparison with related viruses. Virology 1993, 193, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Poch, O.; Blumberg, B.M.; Bougueleret, L.; Tordo, N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 1990, 71, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.P.; Siew, J.W.; Murphy, B.R.; Collins, P.L. Minimum protein requirements for transcription and RNA replication of a minigenome of human parainfluenza virus type 3 and evaluation of the rule of six. Virology 1997, 234, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Marcos, F.; Ferreira, L.; Cros, J.; Park, M. S. ; Nakaya, T.; Garcia-Sastre, A.; Villar, E., Mapping of the RNA promoter of Newcastle disease virus. Virology 2005, 331, 396–406. [Google Scholar]
- Murphy, S.K.; Parks, G.D. Genome nucleotide lengths that are divisible by six are not essential but enhance replication of defective interfering RNAs of the paramyxovirus simian virus 5. Virology 1997, 232, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Kaito, M.; Kozuka, Y.; Komada, H.; Noda, N.; Nanba, K.; Tsurudome, M.; Ito, M.; Nishio, M.; Ito, Y. Recovery of infectious human parainfluenza type 2 virus from cDNA clones and properties of the defective virus without V-specific cysteine-rich domain. Virology 2001, 284, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Skiadopoulos, M.H.; Vogel, L.; Riggs, J.M.; Surman, S.R.; Collins, P.L.; Murphy, B.R. The genome length of human parainfluenza virus type 2 follows the rule of six, and recombinant viruses recovered from non-polyhexameric-length antigenomic cDNAs contain a biased distribution of correcting mutations. J. Virol. 2003, 77, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Yu, M.; Hansson, E.; Pritchard, L.I.; Shiell, B.; Michalski, W.P.; Eaton, B.T. The exceptionally large genome of Hendra virus: support for creation of a new genus within the family Paramyxoviridae. J. Virol. 2000, 74, 9972–9979. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Lowe, L.; Tamin, A.; Liu, X.; Bankamp, B.; Bowden, N.; Rollin, P.E.; Comer, J.A.; Ksiazek, T.G.; Hossain, M.J.; Gurley, E.S.; Breiman, R.F.; Bellini, W.J.; Rota, P.A. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis. 2005, 11, 1594–1597. [Google Scholar] [PubMed]
- Jack, P.J.; Boyle, D.B.; Eaton, B.T.; Wang, L.F. The complete genome sequence of J virus reveals a unique genome structure in the family Paramyxoviridae. J. Virol. 2005, 79, 10690–10700. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, M.; Zhang, H.; Magoffin, D. E.; Jack, P.J.; Hyatt, A.; Wang, H.Y.; Wang, L.F. Beilong virus, a novel paramyxovirus with the largest genome of non-segmented negative-stranded RNA viruses. Virology 2006, 346, 219–228. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR amplicon | Primer | Primer Sequence (5' → 3') | Size (bp) |
|---|---|---|---|
| a | Para4‐1 | CGAACAATTTCTTCAAACAACTGAAGATCG | 1,438 |
| Para4‐2 | CTGTTCATTCTGATAGTTGGAGTCTGGTGTG | ||
| b | LongPara‐P1 | GGTTGCATTCAGGTTTCTCAATCGTTCAGGC | 3,138 |
| LongPara‐M2 | GCCCCCATAGATCACTGATGCCTACGCTTAAC | ||
| c | LongPara‐M1 | CCGATCCACACGAATGAGGGGTATACATCTAGAG | 4,266 |
| LongPara‐HN2 | CGTAAGGAGTGACGAATGTGAGTGGGTAAGACGAAC | ||
| d | LongPara‐HN1 | GCTCAGTGGTTGCTGTCCTTGACGGATGTTTAC | 3,545 |
| LongPara‐2 | GGAGTGATTTCGTCAACTTAAGCTGATCAAGAACTACACCG | ||
| e | LongPara‐L1 | GGAGATACCAAGCAATAATACCCTTTGCTAGAAC | 3,013 |
| LongPara‐L4 | GCTGTTACATGGATAAGGATGTATATTTGGGTTTG | ||
| f | LongPara‐L5 | TAGCTGTGCAATGTCTCATGTGGGGCGTTAAAACC | 1,733 |
| LongPara‐L6 | GTCGTACAGTATCCCGGATTGAACTGCGTAAAACTCACC |
| Primer | Primer Sequence (5' → 3') |
|---|---|
| ParaGRacer‐LS | CTGATAATCAAAAGATCCTACAAGCAGGTGG |
| ParaGRacer‐NRS | CAGATGATGATACGGCAAGTCGGAGG |
| LEnd‐1 | CTTTAGAAATGAATGAGCAAGTAGTCG |
| LEnd‐2 | CAGATTTGTCTAGTGAGGATGTTGTC |
| GSP1‐v.2 | GAAAGATACGGAGACGAGACAAC |
| GSP2‐v.2 | CAACATCCTCACTAGACAAATCTG |
| ORF | SKPIV4 | HPIV4B | HPIV4A | Mumps | HPIV2 |
|---|---|---|---|---|---|
| N | 1656 | 1656 | 1656 | 1650 | 1629 |
| (‐551) | (‐551) | (‐551) | (‐549) | (‐542) | |
| P/V | 1198 | 1198 | 1198 | 1174 | 1186 |
| P1 | (‐399) | (‐399) | (‐399) | (‐391) | (‐395) |
| V | (‐229) | (‐229) | (‐229) | (225) | (226) |
| M | 1149 | 1149 | 1149 | 1128 | 1134 |
| (‐382) | (‐382) | (‐382) | (‐375) | (‐377) | |
| F | 1632 | 1632 | 1632 | 1617 | 1656 |
| (‐543) | (‐543) | (‐543) | (‐538) | (‐551) | |
| HN | 1740 | 1725 | 1722 | 1749 | 1716 |
| (‐579) | (‐574) | (‐573) | (‐582) | (‐571) | |
| L | 6840 | N/A | N/A | 6786 | 6789 |
| (‐2279) | (‐2261) | (‐2262) |
| Non‐coding interval | SKPIV4 | HPIV2 | HPIV4B | HPIV4A |
|---|---|---|---|---|
| 5' NC | 154 | 156 | ≥ 33 | ≥ 33 |
| N‐P/V | 285 | 207 | ≥ 284 | 284 |
| P/V‐M | 295 | 300 | 294 | 294 |
| M‐F | 494 | 176 | ≥ 483 | ≥ 483 |
| F‐HN | 699 | 372 | 699 | 720 |
| HN‐L | 722 | 260 | ≥ 527 | ≥ 529 |
| 3' NC | 497 | 65 | N/A | N/A |
© 2009 by Molecular Diversity Preservation International, Basel, Switzerland
Share and Cite
Yea, C.; Cheung, R.; Collins, C.; Adachi, D.; Nishikawa, J.; Tellier, R. The Complete Sequence of a Human Parainfluenzavirus 4 Genome. Viruses 2009, 1, 26-41. https://doi.org/10.3390/v1010026
Yea C, Cheung R, Collins C, Adachi D, Nishikawa J, Tellier R. The Complete Sequence of a Human Parainfluenzavirus 4 Genome. Viruses. 2009; 1(1):26-41. https://doi.org/10.3390/v1010026
Chicago/Turabian StyleYea, Carmen, Rose Cheung, Carol Collins, Dena Adachi, John Nishikawa, and Raymond Tellier. 2009. "The Complete Sequence of a Human Parainfluenzavirus 4 Genome" Viruses 1, no. 1: 26-41. https://doi.org/10.3390/v1010026
APA StyleYea, C., Cheung, R., Collins, C., Adachi, D., Nishikawa, J., & Tellier, R. (2009). The Complete Sequence of a Human Parainfluenzavirus 4 Genome. Viruses, 1(1), 26-41. https://doi.org/10.3390/v1010026